

Abstract
Background:
Congenital myasthenic syndromes (CMS) are a heterogeneous group of diseases involving neuromuscular transmission. The classification of these syndromes is based on the localization of the defect (pre-synaptic, post-synaptic, and neuromuscular junction) and on the molecular analysis.
Aim:
To report on a series of 7 patients affected by post-synaptic CMS.
Patients and Methods:
We examined sex, familiarity, age of onset, clinical symptoms, and response to tensilon test, patellar and pupillary reflexes, presence of cranial nerve involvement, Gowers′ sign, presence of ptosis, grade of muscular weakness, and response to the treatment and gene deletions.
Results:
Ptosis, muscular hypotonia, and light variability in muscular weakness were the main clinical signs. Cholinergic receptor, nicotinic, epsilon (CHRNE) gene mutations were mainly reported.
Conclusions:
The study points out that the clinical and molecular pattern reported in our patients do not differentiate from the data reported in the literature. Treatment with pyridostigmine and modulation of the therapy allows a good quality of life.
Keywords: Cholinergic receptor, nicotinic, epsilon gene, hypotonic syndromes, myasthenic syndromes, neuromuscular diseases, receptor-associated protein of the synapse gene
Introduction
Congenital myasthenic syndromes (CMS) are a heterogeneous group of diseases characterized by an anomalous neuromuscular transmission. Prevalence is estimated in 1–2/500,000 newborns. CMS are due to enzymatic deficit of acetylcholinesterase. The first clinical manifestations may appear at different times, from the neonatal period to adulthood.[1–3] The classification is principally based on the localization of the deficiency which can be pre-synaptic, synaptic or post-synaptic.[1] Mutations were first identified in the subunits of the nicotinic acetylcholine receptor (AchR) but now mutations in ten different genes, encoding post, pre or synaptic proteins are known to cause CMS. Post-synaptic CMS, which are the most frequent, are caused by mutations that determine a quantitative or qualitative diminution of the receptor of acetylcholine (slow channel syndrome and fast channel syndrome).[1] Receptor-associated protein of the synapse (RAPSN) gene mutations (involving protein in the clustering of the AchR), Muscle-specific kinase receptor (MuSK), Docking Protein 7 (Dok7), a cytoplasm activator of the tyrosin-kinasic receptor specific muscle, and the gene of sodium channel, voltage-gated, type 4A are the most involved genes. Pre-synaptic CMS are rare forms due to mutation of the gene that codifies for the choline acetyltransferase. Autosomic recessive, except for slow channel syndrome, and dominant autosomic inheritance have been reported.[2,3]
This is a single center report on seven patients, prevalently young, who suffered of post-synaptic congenital myasthenia.
Patients and Methods
During the period from September 2008 to June 2011 seven patients were admitted to the Pediatric Hospital Vittorio Emanuele, Catania, Italy, four males, and three females, presenting as main symptoms hypotonia, ptosis and easy fatigability. Diagnosis of CMS was performed on the basis of the clinical features and response to cholinesterase inhibitor. Electrophysiological examination including electromyography and single fiber electrophysiological test (range of normal value 18–32 microns) performed in all the patients do not give sufficient diagnostic information (except for jitter resulting higher in patients 1 and 6) as was for muscle biopsy, performed in patients 2, 6, and 7 to exclude other muscular disorders.
The following parameters have been taken into consideration in CMS patients: Sex, familiarity, current age, age of onset of symptoms, age at the diagnosis, clinical signs at the onset, response to tensilon test and gene deletions. Clinical signs at the present age, i.e., patellar and pupillary reflex, cranial nerve involvement, Gower′s sign, ocular ptosis, grade of muscular involvement and response to treatment, were evaluated. Genetic analysis mainly involved in myasthenic syndromes (Cholinergic receptor, nicotinic, alpha 1 (CHRNA1), CHRNE; RAPSN; DOK7; MuSK and Choline acetyltransferase (ChAT) have been performed in all the patients.
Results
The seven patients affected by CMS showed the following characteristic [Table 1]. Ratio female/male was 3/4. Familiarity was present in 4 patients (mother in patients 1, 3, and 6; first cousin in patient 2). The onset of the affection was presumed to be at the age from neonatal age to 5 years; and the diagnosis was performed between the age of 1 and 6 years. Main clinical signs were ptosis, muscular hypotonia, and in two patients (patients 2 and 6) dysphonia and swallowing disturbance. The jitter was revealed in patients 1 and 6. Treatment with pyridostigmine was satisfying in 4 patients, good in 2 and excellent in one patient. Mutations in CHRNE gene was reported in four patients (patients 3, 5, 6, 7), in RAPSN in patients 1 and 2 and CHRNA1 in patient 4.
Table 1.
Clinical signs and molecular evaluation
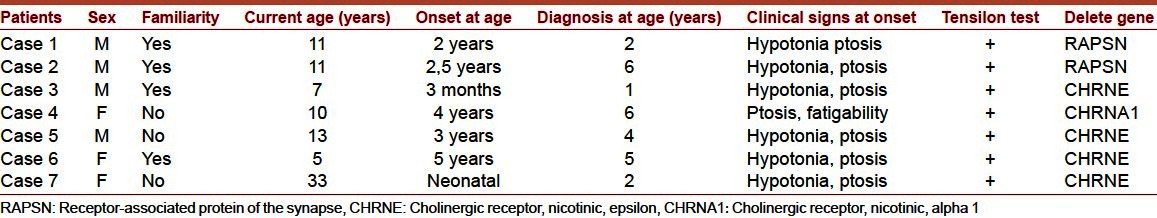
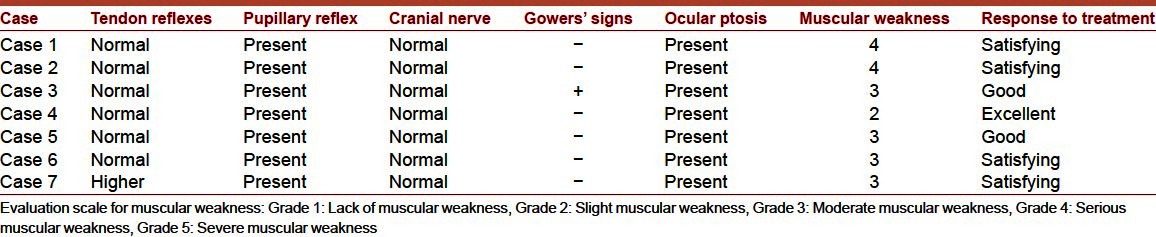
Clinical signs of the patients are resumed in Table 2. Patellar and pupillary reflexes were normal and no cranial involvement was observed. Gowers′ sign was present in patient 3. Muscular weakness evaluation ranged between grade 2 (patient 4), 3 (in patients 3, 5, 6, 7) and 4 (in patients 1 and 2).
Table 2.
Clinical signs at the present day
Discussion
The most frequent forms of CMS are the post-synaptic. They are caused by a reduction or absence of expression of AchR at the post-synaptic membrane level, with main mutations involving RAPSN, CHRNA1 and CHRNE genes. Depending on the variants, CMS may have an extremely variable clinic onset, as they may appear at birth, during childhood or in adulthood.[3–5]
In the neonatal form the difficulty in sucking, in swallowing and in breathing and general hypotonia may be present. The later onset can be characterized by a delay in the motor stages with frequent falls.[5]
During childhood or in adulthood age, this affection can also appear with ophthalmoplegia, mono and bilateral ptosis, strabismus, facial muscles deficit and asthenia which may worsen with physical exercise.[6] The dynamic signs are the most difficult to recognize since they undergo daily fluctuations with a worsening during the evening.[3,6]
Electrophysiological evaluation with repetitive stimulation of each fiber (jitter) may be diagnostically helpful.[7–9] Another important test is the use of tensilon, which, reducing acetylcholine hydrolysis improves the amount of substratum and so determining, for short periods, an improvement of muscular strength.[8] Genetic test, when positive, allows to confirm the diagnosis.[10,11]
The therapy is based on the use of cholinesterase-inhibiting drugs, which are effective in the majority of CMS. Diaminopyrimidine (which increases the release of acetylcholine from terminal nerve) can be used alone or in combination with the acetylcholinesterase therapy. The clinical manifestations of the patient generally tend to remain relatively good if the patient follows an adequate pharmacological treatment and a normal life-style.[12]
From our data and from what we found in literature it can be inferred that CMS represent an important moment of challenge for the pediatrician in the identification and timely diagnosis of the disease. Predisposing factors are not frequently found with the exception of the familiarity when present. In our series, familiar factors have been observed in 4 on 7 patients. Both sexes can be affected: in our series with a mild prevalence of male (4/7). The onset of the clinical signs is wide, in fact in our group it ranged from neonatal period to 5 years, with an average of 4.5 years.
Analyzing our data we have found that there is a great difficulty in making a diagnosis in children due to difficulty in recognizing the signs and symptoms, as seen in our patients where the diagnosis was performed relatively late between 2 and 6 years of age. Motor delay and intolerance of physical exercise are relevant signs, while ptosis remains, in our experience, the most frequent clinical signs, being present in all of our patients. Patellar reflexes are usually normal; in one of our patients we have observed a hyperexcitability of tendon reflexes with increase of reflexogenous area, probably due to perinatal pathological events.
Electrophysiological study gave inconclusive diagnostic results (jitter was higher only in two children).
The tensilon test confirmed to be the best diagnostic test, resulting positive in all of our patients. None of the patients showed severe muscle impairment, thus resulting in a normal life.
Molecular analysis displayed anomaly of genes RASPN, CHRNA1 and CHRNE with the last one more frequently involved. No difference of severity of the disease we have found according to the gene anomaly reported.
However in agreement with the literature, we have found that in post-synaptic CMS, CHRNE gene is the most frequently involved.
Treatment with pyridostigmine gives good results, even if in some cases the response to treatment may be irregular in the course of time and the therapeutic answer is reached only after some months. We must also specify that it is necessary to modulate the posology according to the clinical answer and in case of no-responder it is useful to add pyridostigmine and 3,4 diaminopyridine. Therapy with pyridostigmine seems to be more effective in the subjects that have anomaly involved in subunit ε of AchR compared to subjects who have other gene anomalies.[5]
This study points out that CMS is an uncommon disease not easily recognizable in childhood. Fatigability and ptosis are relevant clinical signs and molecular analysis represents an excellent way of diagnostic confirmation.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Harper CM. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111–23. doi: 10.1055/s-2004-829592. [DOI] [PubMed] [Google Scholar]
- 2.Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: Recent advances. Arch Neurol. 1999;56:163–7. doi: 10.1001/archneur.56.2.163. [DOI] [PubMed] [Google Scholar]
- 3.Engel AG, Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol. 2005;5:308–21. doi: 10.1016/j.coph.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Vincent A, Newsom-Davis J, Wray D, Shillito P, Harrison J, Betty M, et al. Clinical and experimental observations in patients with congenital myasthenic syndromes. Ann N Y Acad Sci. 1993;681:451–60. doi: 10.1111/j.1749-6632.1993.tb22929.x. [DOI] [PubMed] [Google Scholar]
- 5.Beeson D, Hantaï D, Lochmüller H, Engel AG. 126th International Workshop: Congenital myasthenic syndromes, 24–26 September 2004, Naarden, the Netherlands. Neuromuscul Disord. 2005;15:498–512. doi: 10.1016/j.nmd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Milone M, Shen XM, Selcen D, Ohno K, Brengman J, Iannaccone ST, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228–35. doi: 10.1212/WNL.0b013e3181ae7cbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gurnett CA, Bodnar JA, Neil J, Connolly AM. Congenital myasthenic syndrome: Presentation, electrodiagnosis, and muscle biopsy. J Child Neurol. 2004;19:175–82. [PubMed] [Google Scholar]
- 8.Pitt M. Neurophysiological strategies for the diagnosis of disorders of the neuromuscular junction in children. Dev Med Child Neurol. 2008;50:328–33. doi: 10.1111/j.1469-8749.2008.02038.x. [DOI] [PubMed] [Google Scholar]
- 9.Gilchrist JM, Massey JM, Sanders DB. Single fiber EMG and repetitive stimulation of the same muscle in myasthenia gravis. Muscle Nerve. 1994;17:171–5. doi: 10.1002/mus.880170207. [DOI] [PubMed] [Google Scholar]
- 10.Müller JS, Mildner G, Müller-Felber W, Schara U, Krampfl K, Petersen B, et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology. 2003;60:1805–10. doi: 10.1212/01.wnl.0000072262.14931.80. [DOI] [PubMed] [Google Scholar]
- 11.Gaudon K, Pénisson-Besnier I, Chabrol B, Bouhour F, Demay L, Ben Ammar A, et al. Multiexon deletions account for 15% of congenital myasthenic syndromes with RAPSN mutations after negative DNA sequencing. J Med Genet. 2010;47:795–6. doi: 10.1136/jmg.2010.081034. [DOI] [PubMed] [Google Scholar]
- 12.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–7. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]