

Abstract
NADH:ubiquinone oxidoreductase (complex I) is a complicated respiratory enzyme that conserves the energy from NADH oxidation, coupled to ubiquinone reduction, as a proton motive force across the mitochondrial inner membrane. During catalysis, NADH oxidation by a flavin mononucleotide is followed by electron transfer to a chain of iron–sulfur clusters. Alternatively, the flavin may be reoxidized by hydrophilic electron acceptors, by artificial electron acceptors in kinetic studies, or by oxygen and redox-cycling molecules to produce reactive oxygen species. Here, we study two steps in the mechanism of NADH oxidation by complex I. First, molecular fragments of NAD(H), tested as flavin-site inhibitors or substrates, reveal that the adenosine moiety is crucial for binding. Nicotinamide-containing fragments that lack the adenosine do not bind, and ADP-ribose binds more strongly than NAD+, suggesting that the nicotinamide is detrimental to binding. Second, the primary kinetic isotope effects from deuterated nicotinamide nucleotides confirm that hydride transfer is from the pro-S position and reveal that hydride transfer, along with NAD+ dissociation, is partially rate-limiting. Thus, the transition state energies are balanced so that no single step in NADH oxidation is completely rate-limiting. Only at very low NADH concentrations does weak NADH binding limit NADH:ubiquinone oxidoreduction, and at the high nucleotide concentrations of the mitochondrial matrix, weak nucleotide binding constants assist product dissociation. Using fast nucleotide reactions and a balance between the nucleotide binding constants and concentrations, complex I combines fast and energy-conserving NADH oxidation with minimal superoxide production from the nucleotide-free site.
NADH:ubiquinone oxidoreductase (complex I) is the largest and most complicated enzyme of the respiratory chains of mammalian mitochondria and many other aerobic organisms.1−3 It catalyzes the oxidation of NADH by a noncovalently bound flavin mononucleotide, the reduction of ubiquinone to ubiquinol, and couples the redox reaction to proton translocation across the mitochondrial inner membrane in eukaryotes, or the cytoplasmic membrane in prokaryotes. NADH oxidation, to generate the fully reduced flavin, most likely occurs by direct hydride transfer from the nicotinamide ring of the bound nucleotide to the flavin,4 with the nicotinamide ring juxtaposed above the isoalloxazine ring system.5 During catalysis, the reduced flavin is reoxidized by electron transfer to the chain of iron–sulfur clusters leading to the ubiquinone-binding site,1,2 but it can also undergo side reactions to produce reactive oxygen species6−8 that are implicated in the pathologies of many diseases.9,10
The flavin site in complex I is mechanistically versatile: it uses at least three different mechanisms to catalyze NADH oxidation coupled to the reduction of numerous different electron acceptors.8,11 The simplest mechanism is for NADH:ubiquinone oxidoreduction: the flavin is reduced by NADH and then reoxidized by the Fe–S clusters, and the flavin can be reoxidized regardless of whether a nucleotide is bound. Alternatively, a number of electron acceptors, including molecular O2,6 hydrophilic quinones,7 hexacyanoferrate {FeCN, ferricyanide, [Fe(CN)6]3–},11,12 and oxidized nucleotides such as NAD+ and APAD+,13 react directly with the reduced flavin only when no nucleotide is bound. A second class of electron acceptors, including hexaammineruthenium III {HAR, [Ru(NH3)6]3+} and paraquat, are positively charged and react only when a nucleotide is bound, most likely by interacting with the negatively charged nucleotide phosphates.8 Both classes include molecules that can be reoxidized by molecular O2 in redox-cycling reactions that stimulate significantly the rate of production of reactive oxygen species.7,8,14 The mechanisms are summarized in Scheme 1.
Scheme 1. Mechanisms of NADH Oxidation by the Flavin in Complex I.
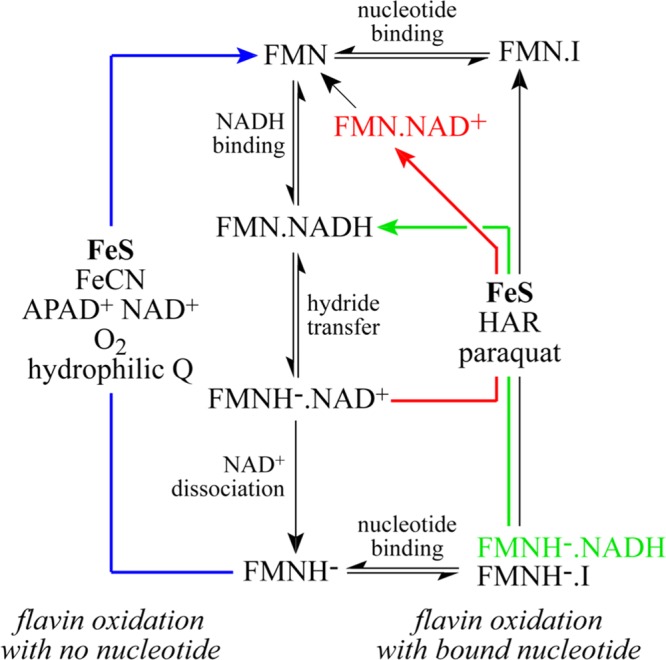
The oxidation states of the flavin are denoted FMN for the oxidized flavin and FMNH– for the reduced flavin, the NADH-bound states by FMN.NADH and FMNH–.NADH, the NAD+-bound states by FMN.NAD+ and FMNH–.NAD+, and the inhibitor-bound states by FMN.I and FMNH–.I (where I is a nucleotide that inhibits NADH or NAD+ binding). The blue arrow on the left (FMNH– to FMN) shows the oxidation of the flavin by electron acceptors that react when no nucleotide is bound. The green, red, and black arrows on the right show the oxidation of the flavin by electron acceptors that react with the nucleotide-bound state. The reduced flavin can be oxidized by the Fe–S clusters, to transfer the electrons to bound ubiquinone, regardless of whether a nucleotide is bound.
Several studies have investigated how rates of NADH oxidation by complex I depend on NADH, electron acceptor, and flavin-site inhibitor concentrations, to improve our understanding of the thermodynamics and kinetics of catalysis.8,11,13,15−17 However, knowledge of the dissociation constants for NADH and NAD+ bound to the oxidized and reduced flavin states remains very limited. Values of KMNADH and KINAD+ have been measured during NADH oxidation and values of KMNAD+ and KINADH during NAD+ reduction by coupled submitochondrial particles,15−17 but these values are not thermodynamic constants; they comprise many different contributions and can be particularly misleading when the rate of the reaction is not limited by the substrate being titrated. The NADH-dependent steps in catalysis are most accessible when NADH oxidation is coupled to reduction of an “artificial” electron acceptor, because flavin reoxidation is faster than when the reaction is limited by ubiquinone reduction. Previously, we modeled data from the NADH:FeCN, APAD+, O2, and HAR reactions, using realistic reaction mechanisms and common parameters for steps that are common to each reaction, to try to define values for the nucleotide dissociation constants; however, the complexity of the mechanisms endows them with considerable flexibility, and only the dissociation constant for NADH bound to the reduced flavin state could be defined with any accuracy.11 In contrast, the dissociation constant of ADP-ribose (see Figure 1), a competitive inhibitor of NADH oxidation,18 could be determined to be 24–30 μM when the flavin is oxidized and ∼10-fold higher when it is reduced.11,16 However, ADP-ribose lacks the nicotinamide moiety that interacts most directly with the flavin, so the reason for the selectivity is not clear. ADP and ATP, but not adenosine or nicotinamide mononucleotide [NMN+ (see Figure 1)], have been reported to inhibit the FeCN reaction,19 and ADP and ATP were reported to bind to the reduced flavin site and to stimulate the reduction of HAR,8 suggesting that the nucleotide binding affinities are conferred by specific parts of the nucleotide molecules. Here, we use a comprehensive set of nucleotide analogues (see Figure 1) to elucidate how different parts of the NAD+ and NADH molecules determine their binding affinities. Our results contribute to our understanding of the mechanism of catalysis by complex I, to our understanding of how the metabolic status of the mitochondrion affects oxidative stress, and to the future design of molecules that bind specifically to the reduced flavin site to block superoxide production.
Figure 1.

Molecular structures of the nucleotides and nucleotide fragments used in this study. The structure of NADH is shown in the center, indicating the S and R protons that were substituted with deuterium to form [4S-2H]NADH (S-NADD) and [4R-2H]NADH (R-NADD), respectively. The different nicotinamide groups of APADH and NAD+ are shown at the left, and the bars at the right indicate the compositions of the nucleotides and fragments used.
At saturating NADH concentrations (where increasing the NADH concentration further does not increase the rate), NADH binding does not limit the rate of NADH oxidation, so hydride transfer or NAD+ dissociation may do so. NAD+ could be added as an inhibitor to probe whether its dissociation from the reduced flavin is rate-limiting, but it reacts with the reduced flavin precluding a clear interpretation of the data. Alternatively, the possibility that hydride transfer is rate-limiting can be probed by substituting the hydrogen with deuterium, to determine whether a primary kinetic isotope effect (KIE, the ratio of rates for the protonated and deuterated forms) is observed.20 The magnitude of the observed KIE will depend on the kinetic complexity of the reaction (the extent to which hydride transfer and other steps limit the rate) as well as on the intrinsic KIE of the hydride transfer reaction itself (which reflects the contributions of quantum tunneling and protein dynamics to the reaction mechanism).21−24 Here, we reveal the KIEs exhibited for NADH oxidation by complex I, by studying the oxidation of deuterated forms of NADH coupled to the reduction of artificial electron acceptors by known mechanisms. Thus, we confirm the stereospecificity of NADH oxidation5,25 and elucidate the role of hydride transfer in controlling its rate. Finally, we discuss which steps are rate-limiting during NADH oxidation by complex I and describe how nucleotide concentrations, both in vitro and in vivo, control the rates of catalytic NADH:ubiquinone oxidoreduction and reactive oxygen species production.
Materials and Methods
All chemicals were from Sigma-Aldrich Ltd. All nucleotides were of the highest purity available (at least 95%). AMP, ADP, ATP, NMN+, and NAD+ were prepared at stock concentrations of 50 mM in 20 mM Tris-HCl buffer and corrected to pH 7.5 with concentrated Tris base. Adenosine was prepared at a stock concentration of 16 mM in 20 mM Tris-HCl buffer.
Preparation of Complex I from Bovine Heart Mitochondria
Complex I was prepared as described previously,26,27 concentrated to approximately 10 mg/mL (determined by the Pierce bicinchoninic acid assay), snap-frozen in liquid nitrogen, and stored at −80 °C.
Preparation of Modified Nicotinamide Nucleotides
[4R-2H]NADH, [4S-2H]NADH, [4R-2H]APADH, and [4S-2H]APADH (see Figure 1) were produced from NAD+ and APAD+ using the stereospecific NAD+-linked dehydrogenase enzymes horse liver alcohol dehydrogenase (4R stereoisomers)28 and glucose dehydrogenase from Bacillus megaterium (4S stereoisomers).29 Ten units of enzyme per milliliter were added to a solution of 10 mM NAD+ or APAD+ in 0.1 M Tris-HCl (pH 8) containing either 100 mM d-glucose-d1 or 6% (v/v) 2-propanol-d8 and the mixture incubated at room temperature until the absorbance at 340 nm stopped increasing (approximately 30 min). Then, the mixture was applied to a Q-Sepharose column pre-equilibrated in 20 mM Tris-HCl (pH 7.7) and the deuterated nucleotide eluted with 20 mM Tris-HCl (pH 7.7) containing 0.5 M KCl.30 The nucleotide product was then desalted, using a Sephadex G-10 column with water as the eluent, lyophilized, and stored at −20 °C. Nondeuterated NADH and APADH were prepared using glucose dehydrogenase and nondeuterated glucose and purified using the same method. The nucleotide chemical purity was confirmed by high-performance liquid chromatography (HPLC) (see Figures S1 and S2 of the Supporting Information); typically, the products are 85–90% pure, and the small levels of contaminants present were shown, by comparing in-house and commercially supplied samples, to have a negligible effect on the kinetic measurements. Nucleotide isotopic purity (stereospecific labeling) was confirmed by nuclear magnetic resonance (NMR) spectroscopy (see Figures S3 and S4 of the Supporting Information); typically, the products are 85–95% pure, and the level of contamination with nondeuterated NADH/APADH may lead to a small underestimate of the KIE. NMNH was prepared by chemical reduction of NMN+.31 NMN+ (25 mg, 75 μmol) was dissolved in 4 mL of an aqueous solution of 1.3% sodium hydrogen carbonate, and 25 mg of sodium hydrosulfite was added. The mixture was transferred to an anaerobic glovebox and incubated for 2 h at 25 °C and then bubbled with air for 15 min to remove excess hydrosulfite. As a control for the formation of alternative reduced nicotinamides, and for the presence of the products of hydrosulfite oxidation, NADH was prepared using the same protocol and confirmed to be oxidized by complex I.
Kinetic Measurements
Kinetic activity measurements were conducted at 32 °C, in 20 mM Tris-HCl (pH 7.5), in 96-well plates in a Molecular Devices microtiter plate reader. The concentration of complex I was varied to give an appropriate rate for each reaction. NADH and other nucleotides, potassium hexacyanoferrate(III) (ferricyanide, FeCN), and hexaammineruthenium(III) chloride (HAR) were added from concentrated stock solutions in the assay buffer, and all reactions were initiated with complex I. Initial rates were calculated using linear regression (typically over 15 s), and background rates (without complex I) were subtracted. NADH:APAD+ transhydrogenation (“the APAD+ reaction”) was monitored at 400–450 nm (ε = 3.16 mM–1 cm–1),13 NADH:FeCN oxidoreduction (“the FeCN reaction”) at 420–500 nm (ε = 1.02 mM–1 cm–1),11 and NADH:HAR oxidoreduction (“the HAR reaction”) at 340–380 nm (ε = 4.81 mM–1 cm–1).8 Superoxide was detected by the reduction of acetylated cytochrome c at 550–541 nm (ε = 18.0 mM–1 cm–1).6 Each data point is the mean average of at least three independent measurements; standard errors were calculated for each data point and were always equal to <10% of the average value.
Results
Inhibition of Flavin-Site Reactions by NADH Analogues and Fragments
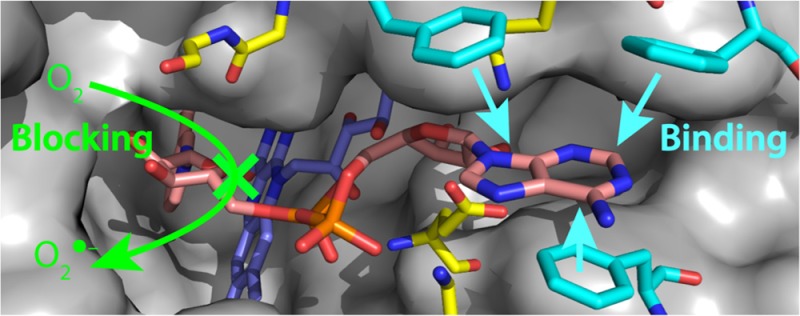
Adenosine shows weak inhibition of the APAD+ (Figure 2A) and HAR (Figure 2C) reactions, with IC50 values of 10 and 7 mM, respectively (Table 1), under the conditions investigated, and no discernible inhibition of the FeCN reaction (Figure 2B). It shows a weak stimulatory effect on superoxide production, measured by the reduction of acetylated cytochrome c (Figure 2D). Although adenosine probably competes with NADH for the oxidized flavin site, Figure 2 shows that the dominant effect is from it competing with NADH for the reduced flavin site. In the APAD+ reaction,13 adenosine inhibits by competing with APAD+ for the reduced flavin (adenosine and APAD+ cannot bind together, and APAD+ reduction is much slower than NADH oxidation). Like APAD+, both FeCN and O2 react with the nucleotide-free reduced flavin (see Scheme 1),11 but because adenosine lacks the nicotinamide riboside moiety and both of the phosphates (see Figure 1), it may bind without occluding the flavin site from them (adenosine and FeCN or O2 can bind together) (see Figure 3). Adenosine actually increases the rate of the O2 reaction (Figure 2D), and it increases the rate of the FeCN reaction at high NADH concentrations also, because it relieves the inhibition by high NADH concentrations that results when NADH occludes the reduced flavin.11 Finally, HAR requires a nucleotide with negatively charged phosphates to be bound to the reduced flavin site before it can react;8 therefore, the adenosine-bound reduced flavin is unreactive, and the observed rate of the HAR reaction represents the fraction of the enzyme with either NADH or NAD+ bound.
Figure 2.

Inhibition of the APAD+, FeCN, HAR, and O2 reactions by NAD(H) fragments. The rates of (A) NADH:APAD+ oxidoreduction (the APAD+ reaction), (B) NADH:FeCN oxidoreduction (the FeCN reaction), (C) NADH:HAR oxidoreduction (the HAR reaction), and (D) NADH:O2 oxidoreduction (superoxide production) are presented as a function of the concentration of an added nucleotide. The added nucleotides are adenosine (●), AMP (○), ADP (◆), ADP-ribose (◇), NAD+ (■), and NMN+ (□). Conditions: pH 7.5, 32 °C, atmospheric O2, and 0.1 mM NADH for panels A–C and 0.03 mM NADH for panel D; 1 mM APAD+ for panel A; 1 mM FeCN for panel B; and 3.5 mM HAR for panel C.
Table 1. IC50 Values for the Inhibition of the APAD+, FeCN, HAR, and O2 Reactions by NAD(H) Fragmentsa.
| adenosine | AMP | ADP | ADP-ribose | NAD+ | NMN+ | |
|---|---|---|---|---|---|---|
| APAD+ | 10 | 7 | 7 | 0.5 | 0.7 | no inhibition |
| FeCN | no inhibitionb | 12 | 7 | 0.6 | 4 | no inhibition |
| HAR | 7 | 7b | stimulated | 0.2b | 4 | no inhibition |
| O2 | stimulated | 7 | 7 | 0.05 | – | – |
All values are reported in millimolar and are derived from the data presented in Figure 2. The inhibition of the O2 reaction by NAD+ and NMN+ is not reported because these data are confounded by effects on the reduction potential of the nucleotide pool.
Stimulated under some conditions.
Figure 3.
Occlusion of the flavin by the binding of adenosine, AMP, ADP, ADP-ribose, and NAD(H). (A) NAD(H)-bound active site in complex I from Thermus thermophilus (Protein Data Bank entry 3IAM(5)) in surface representation. The bound NAD(H) molecule is colored with the adenosine moiety (including the second ribose) in red, the two phosphates in cyan and purple, the first ribose in green, and the nicotinamide in pink. The flavin is colored blue, and the protein is colored gray. (B) The flavin and bound nucleotide are shown as sticks from the same viewpoint, using the same color scheme. Amino acids that interact with the bound nucleotide are colored yellow, with hydrogen bonding interactions shown by dashed black lines, hydrophobic stacking interactions by dashed gray lines, and the hydride transfer between C4 of the nicotinamide ring and N5 of the flavin isoalloxazine ring by a dashed red line.
AMP and ADP both inhibit the APAD+ reaction (Figure 2A) with IC50 values of 7 mM (Table 1); therefore, they bind slightly more tightly than adenosine, and the phosphate groups make only a small contribution to the nucleotide binding affinity. AMP and ADP inhibit the FeCN reaction (Figure 2B) with IC50 values of 12 and 7 mM, respectively, and they both inhibit superoxide production also, with IC50 values of 7 mM (Table 1). In order of adenosine, AMP, ADP, the increased level of inhibition represents a combination of increased binding affinity, increased negative charge from the phosphates (repelling the negative FeCN), and increased size [increased occlusion of the reduced flavin (see Figure 3)]. Figure 2C shows that the HAR reaction is stimulated significantly by intermediate concentrations of ADP (∼6 mM) and inhibited at high concentrations, consistent with ADP binding to the reduced flavin promoting interaction of the positively charged HAR molecule by increasing the local negative charge, and by ADP inhibiting NADH oxidation at high concentrations.8 AMP behaves in a similar way, but the activation is less marked because AMP has only one negatively charged phosphate.
ADP-ribose, as shown previously,11,18 is a strong inhibitor of the APAD+ (Figure 2A), FeCN (Figure 2B), and superoxide (Figure 2D) reactions, with IC50 values of 0.5, 0.2, and 0.05 mM, respectively (Table 1). Thus, it has a stronger binding affinity than adenosine, AMP, or ADP. It is a similarly effective inhibitor of the HAR reaction [IC50 = 0.6 mM (Figure 2C)] through the inhibition of NADH oxidation, with a slight “lag” in the onset of the inhibition indicating the reactivity of the inhibitor-bound state toward HAR reduction.8 Detailed kinetic analyses have shown previously that ADP-ribose binds more strongly when the flavin is oxidized than when it is reduced,11,16 consistent with the observed strong inhibition of all four reactions discussed.
Finally, the influence of the nicotinamide ring on nucleotide binding is revealed through the effects of NAD+ and NMN+ (see Figure 1). NAD+ inhibits the APAD+ reaction (Figure 2A) with an IC50 value of 0.7 mM (similar to ADP-ribose), but it inhibits the FeCN (Figure 2B) and HAR (Figure 2C) reactions more weakly than ADP-ribose, with IC50 values of 4 mM for both. Thus, the nicotinamide headgroup appears either to have little effect on binding affinity or even to weaken the binding of NAD+, relative to that of ADP-ribose. However, NAD+ is a competitive substrate as well as an inhibitor, complicating the interpretation of our observations. Interestingly, NMN+ does not inhibit any of the flavin-coupled NADH oxidation reactions (Figure 2); therefore, the adenosine moiety is crucial for nucleotide binding, and very little binding affinity is afforded by the nicotinamide, ribose, or phosphates alone. It is unlikely that binding of the adenosine moiety alters the conformation of the flavin site, to enhance the affinity for NMN+, because including 10 mM adenosine with the NMN+ did not enhance its inhibition. The lack of binding affinity of NMN+ is not specific to the oxidized nicotinamide, because (as observed previously19 and further tested here) NMNH is not oxidized by complex I in the presence of either APAD+ or FeCN.
Kinetic Isotope Effect (KIE) in NADH Oxidation by Complex I
Panels A–C of Figure 4 show the NADH/NADD concentration dependencies of the FeCN, HAR, and APAD+ reactions using NADH, [4R-2H]NADH (R-NADD), and [4S-2H]NADH (S-NADD) (see Figure 1). In both the FeCN and HAR reactions, the rates with R-NADD are, within error, the same as those with NADH, suggesting that there is no KIE for R-NADD. In contrast, both rates with S-NADD are significantly decreased, providing clear evidence of a primary KIE that both confirms the stereochemistry of NADH oxidation (hydride transfer of the 4S proton to the flavin5,25) and indicates that hydride transfer contributes to determining the overall rate of the two reactions. The primary KIE of S-NADD in the HAR reaction is ∼2 (Figure 4B); it is similar at lower concentrations of NADH(D) in the FeCN reaction (up to ∼50 μM NADH/S-NADD) and then decreases to 1 (no KIE) at higher NADH concentrations. At higher concentrations, the reaction is inhibited by the binding of NADH to the reduced flavin site,11 so the rate of reaction becomes less dependent on hydride transfer and more dependent on the FeCN half-reaction. Similarly, only a small KIE is observed with S-NADD in the APAD+ reaction, because the rate of the reaction is limited by APAD+ binding and reduction.11,13 Finally, Figure 4D shows how the KIE for the HAR reaction increases at higher pH values, because the rate with NADH increases more than the rate with S-NADD. This suggests that a different step, probably dissociation of NAD+ from the oxidized flavin site, becomes faster at high pH and so restricts the rate less. For S-NADD, hydride transfer is comparatively slow, and the effects of accelerating NAD+ dissociation are less evident. The maximal observed KIE of 2.6 for NADH oxidation puts a lower limit on the intrinsic value of the KIE of the hydride transfer step alone but does not reveal its true value (which may be much higher).
Figure 4.

KIEs in the NAD(H/D):FeCN, NAD(H/D):HAR, NAD(H/D):APAD+, APAD(H/D):FeCN, and APAD(H/D):HAR oxidoreduction reactions. The rates of NAD(H/D) oxidation (A–C) are shown as a function of the concentration of NADH (●), R-NADD (○), or S-NADD (◆) for (A) the FeCN reaction, (B) the HAR reaction, and (C) the APAD+ reaction. (D) Dependence of the rates for NADH (●) and S-NADD (○) in the HAR reaction [0.1 mM NAD(H/D) concentration] on pH. (E and F) The rates of APAD(H/D) oxidation are shown as a function of the concentration of APADH (●) and S-APADD (○) for (E) the FeCN reaction and (F) the HAR reaction. Conditions: pH 7.5 (except for panel D), 32 °C, and 1 mM FeCN for panels A and E, 1 mM APAD+ for panel C, and 3.5 mM HAR for panels B, D, and F.
Panels E and F of Figure 4 show the APADH and APADD concentration dependencies of the FeCN and HAR reactions using APADH and [4S-2H]APADH (S-APADD) as substrates. [4R-2H]APADH (R-APADD) could not be prepared with high stereospecific purity. The KIE from S-APADD reaches ∼6.5 in the FeCN reaction and ∼3.5 in the HAR reaction, suggesting that the hydride transfer step is more strongly rate-limiting in the APADH oxidation reactions than in the NADH oxidation reactions. The fact that APADH:FeCN and APADH:HAR oxidoreductions are already 10-fold slower than NADH:FeCN and NADH:HAR oxidoreductions suggests that the hydride transfer step with APADH is more than 10 times slower than with NADH. Finally, comparison of panels A and E of Figure 4 shows that, in contrast to the NADH:FeCN reaction, the APADH:FeCN reaction is not inhibited significantly by the highest APADH concentrations tested. This may be because the binding affinity of APADH for the reduced flavin is weaker than that of NADH or because highly rate-limiting hydride transfer obscures the inhibition (as observed for S-NADD in Figure 4A).
Discussion
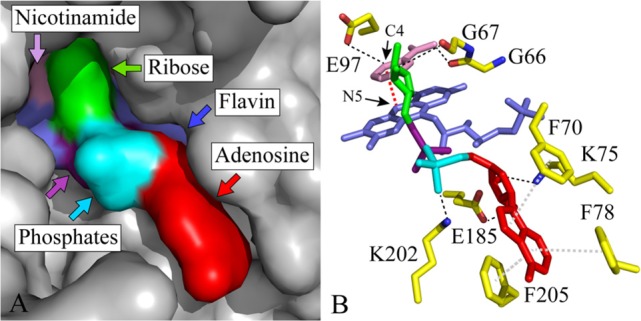
It is clear that the adenosine moiety contributes significantly to the binding affinity of NADH (and other nucleotides) for the active site of complex I. The NAD(H)-bound structure of the hydrophilic domain of complex I from Thermus thermophilus (Figure 3)5 showed that its aromatic adenine ring stacks among three conserved phenylalanines (F70, F78, and F205 in the Nqo1 subunit of T. thermophilus) and that its ribose moiety forms hydrogen bonds with a conserved glutamate (E185) and a conserved lysine (K75). It is possible that the three phenylalanine residues assist in “trapping” the nucleotide to form an encounter complex and facilitate progression to the fully bound state via a multistep binding process.32
The data described here (summarized in Table 1) suggest that AMP and ADP do not bind more strongly than adenosine, but the structure revealed interactions between the “AMP” phosphate and a conserved lysine (K202) and between the “ADP” phosphate and the flavin. As AMP and ADP are both highly charged (predominantly AMP2– and ADP3– 33), it is possible that these interactions are offset by electrostatic effects or by entropic contributions from nucleotide binding or solvent ordering around the highly solvated phosphates; in the future, these effects may be distinguished by nucleotide analogues with the negative phosphates replaced with neutral sulfonamides.34 Furthermore, (unlike adenosine) AMP and ADP (and ATP) are large enough to obstruct the access of O2 to the reduced flavin; as they are present at high concentrations in the mitochondrial matrix (the concentration of the total adenosine nucleotide pool has been estimated to be 10 mM35), they compete with nicotinamide nucleotides for the flavin site of complex I and may contribute to determining the rate of superoxide production.
The data presented here show clearly that ADP-ribose has a much stronger binding affinity than adenosine, AMP, and ADP, yet only one extra hydrogen bond, from the ribose to the backbone carbonyl of G67, is apparent in the T. thermophilus structure.5 ADP-ribose is predominantly ADP-ribose2–, so it has the same charge as AMP but forms two additional hydrogen bonds. The balance between charge and hydrogen bonding may thus explain the sharply increased binding affinity of ADP-ribose. In comparison, the weaker inhibition by NAD+ (and lack of inhibition by NMN+) suggests that the oxidized nicotinamide group is detrimental to nucleotide binding; although the structure revealed interactions between the nicotinamide and the backbone carbonyls of G66 and E97,5 the oxidation states of both flavin and nicotinamide to which these pertain are unclear. Alternatively, the apparently detrimental effect of the oxidized nicotinamide group may be a false indication if the ADP-ribose molecule binds in a conformation different from that of the ADP-ribose moiety of a bound nucleotide.
It is difficult to extrapolate from NAD+ binding to the oxidized flavin to NADH binding. The nicotinamide ring in NADH is uncharged and puckered,36 whereas in NAD+, it is positively charged, aromatic, and planar (more suited to stacking against the flavin isoalloxazine). Although the constants for binding of ADP-ribose and NADH to the reduced flavin are similar, and ADP-ribose binds ∼10-fold more strongly when the flavin is oxidized than reduced,11,16 whether the oxidation state selectivity and similarity extend to NADH binding to the oxidized flavin (suggesting a binding constant of 25 μM) is unclear. Previously, we modeled data from the FeCN, HAR, APAD+, and O2 reactions together, using a realistic mechanism for each reaction, and found that all our kinetic data could be modeled accurately with any NADH binding constant of <140 μM.8 Thus, we infer that NADH binds only weakly to the oxidized (and reduced) flavin site. Indeed, the only strong-binding nucleotide inhibitor known for the flavin site is “NADH-OH”,37 but both NADH-OH binding and dissociation are very slow (rate constant for dissociation of ∼0.001 s–1,16 more than 6 orders of magnitude slower than that for NAD+ dissociation), suggesting a different mode of binding, perhaps even covalent bond formation. Finally, it is known that binding of NADH to the oxidized flavin is both fast and reversible, because the rate of NADH oxidation approaches NADH-diffusion control at low NADH concentrations,11 and because oxidation of NADH by the complex I flavin is thermodynamically efficient.38 Thus, we conclude that nucleotide binding by complex I is fast but weak. These properties minimize product inhibition, even in the mitochondrial matrix where the NAD+ concentration is high (the total NADH and NAD+ concentration is ∼3 mM, of which ∼10% is present as NADH39−42). Furthermore, weak NADH binding is unlikely to limit the rate of catalysis in vivo, because the maximal rate of ubiquinone reduction by complex I is slower than the maximal rate of NADH oxidation, and because the NADH concentration in the mitochondrial matrix is high. Low NADH concentrations limit the rate of NADH oxidation in vitro because the flavin site is only partially occupied, a thermodynamic, rather than kinetic, limitation.
Binding of NADH to the oxidized flavin is usually considered the first step in the NADH oxidation reaction. Once the oxidized flavin–NADH complex has formed, oxidation of NADH by the flavin occurs by transfer of hydride from nicotinamide C4 to flavin N5. The primary KIE observed here agrees with previous conclusions that NADH oxidation is stereospecific, transferring the hydride from the B-face (pro-S position) of its nicotinamide ring.5,25 However, while the primary KIE suggests that the hydride transfer step is rate-limiting in NADH oxidation, the relatively low values observed (∼2 for S-NADD oxidation coupled to FeCN or HAR, increasing to 2.6 at high pH) indicate that hydride transfer20−24 is not the only rate-limiting step. Larger KIEs were observed for the oxidation of APADH (up to ∼6.5), suggesting that the transfer of hydride from APADH to the flavin is slower, and more rate-limiting. In both cases, however, the observed KIEs represent only the lower limit of the intrinsic isotope effect; the actual value may be much higher. By extension, the intrinsic rate of hydride transfer may also be much higher than the observed rates of reaction; previously, we estimated that hydride transfer and NAD+ dissociation both occur at >5000 s–1 in complex I.11 Notably, in mitochondrial transhydrogenase, the rate of transfer of hydride from NADH to NADP+ is ∼21000 s–1, and the KIE is 2–3, similar to that observed here.43 Thus, our data suggest that hydride transfer and NAD+ dissociation are both partially rate-limiting for oxidation of NADH by complex I; the enzyme balances out the transition state energies, flattening the free energy profile of the reaction so that no single step is fully rate-limiting.
Finally, because nucleotide binding by complex I is fast and reversible, the flavin site exists in a distribution of empty and occupied states, as defined by the nucleotide concentrations, and in a distribution of oxidized and reduced states, as defined primarily by the NADH and NAD+ ratio and influenced by the rate of ubiquinone reduction. A high occupancy of the reduced flavin site is advantageous for the minimization of superoxide production, because bound nucleotides block the access of O2, and it is promoted by the high nucleotide concentrations present in the matrix. Furthermore, during catalysis, the pathway taken for NADH oxidation (the order of the hydride transfer, NAD+ dissociation, flavin oxidation, and NADH binding steps, which varies with the nucleotide concentration and rate of ubiquinone reduction) is relevant to determining the rate of superoxide production. Ideally, NAD+ would be exchanged for NADH with the flavin in the oxidized state, so that the reduced flavin is not exposed to O2. Alternatively, the lifetime of the solvent-exposed reduced flavin can be minimized by fast nucleotide exchange. Through balancing weak nucleotide binding constants with high nucleotide concentrations, and through fast nucleotide exchange, the complex I flavin site thus combines fast and energy-conserving NADH oxidation with only minimal superoxide production.
Acknowledgments
We thank Drs. Ji-Chun Yang and David Neuhaus (MRC Laboratory of Molecular Biology) and Drs. Masaru Kato and Erwin Reisner (University of Cambridge, Cambridge, U.K.) for NMR analyses of deuterated nucleotides.
Glossary
Abbreviations
- APAD and APADH
reduced and oxidized forms of 3-acetylpyridine adenine dinucleotide, respectively
- FeCN
ferricyanide or hexacyanoferrate(III)
- HAR
hexaammineruthenium(III)
- KIE
kinetic isotope effect
- NMN+ and NMNH
reduced and oxidized forms of nicotinamide mononucleotide, respectively
- R-APADD and S-APADD
[4R-2H]APADH and [4S-2H]APADH, respectively
- R-NADD and S-NADD
[4R-2H]NADH and [4S-2H]NADH, respectively.
Supporting Information Available
HPLC and NMR analyses of the deuterated nucleotides prepared for this study are reported in Figures S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This research was funded by The Medical Research Council.
Supplementary Material
References
- Hirst J. (2010) Towards the molecular mechanism of respiratory complex I. Biochem. J. 425, 327–339. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A.; Hinchliffe P. (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436. [DOI] [PubMed] [Google Scholar]
- Baradaran R.; Berrisford J. M.; Minhas G. S.; Sazanov L. A. (2013) Crystal structure of the entire respiratory complex I. Nature 494, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraaije M. W.; Mattevi A. (2000) Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 25, 126–132. [DOI] [PubMed] [Google Scholar]
- Berrisford J. M.; Sazanov L. A. (2009) Structural basis for the mechanism of respiratory complex I. J. Biol. Chem. 284, 29773–29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussmaul L.; Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. S.; Sharpley M. S.; Hirst J. (2009) Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I) and production of reactive oxygen species. Biochemistry 48, 2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell J. A.; King M. S.; Hirst J. (2011) A ternary mechanism for NADH oxidation by positively charged electron acceptors, catalyzed at the flavin site in respiratory complex I. FEBS Lett. 585, 2318–2322. [DOI] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell J. A.; Yakovlev G.; Hirst J. (2009) Reactions of the flavin mononucleotide in complex I: A combined mechanism describes NADH oxidation coupled to the reduction of APAD+, ferricyanide or molecular oxygen. Biochemistry 48, 12005–12013. [DOI] [PubMed] [Google Scholar]
- Dooijewaard G.; Slater E. C. (1976) Steady-state kinetics of high molecular weight (type I) NADH dehydrogenase. Biochim. Biophys. Acta 440, 1–15. [DOI] [PubMed] [Google Scholar]
- Yakovlev G.; Hirst J. (2007) Transhydrogenation reactions catalyzed by mitochondrial NADH-ubiquinone oxidoreductase (complex I). Biochemistry 46, 14250–14258. [DOI] [PubMed] [Google Scholar]
- Cochemé H. M.; Murphy M. P. (2008) Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 283, 1786–1798. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. D. (1993) Kinetics, control and mechanism of ubiquinone reduction by mammalian respiratory chain-linked NADH-ubiquinone reductase. J. Bioenerg. Biomembr. 25, 367–375. [DOI] [PubMed] [Google Scholar]
- Grivennikova V. G.; Kotlyar A. B.; Karliner J. S.; Cecchini G.; Vinogradov A. D. (2007) Redox-dependent change of nucleotide affinity to the active site of the mammalian complex I. Biochemistry 46, 10971–10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A. D. (2008) NADH/NAD+ interaction with NADH:ubiquinone oxidoreductase (complex I). Biochim. Biophys. Acta 1777, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharova T. V.; Vinogradov A. D. (1997) A competitive inhibition of the mitochondrial NADH-ubiquinone oxidoreductase (complex I) by ADP-ribose. Biochim. Biophys. Acta 1320, 256–264. [DOI] [PubMed] [Google Scholar]
- Hatefi Y.; Stempel K. E.; Hanstein W. G. (1969) Inhibitors and activators of the mitochondrial reduced diphosphopyridine nucleotide dehydrogenase. J. Biol. Chem. 244, 2358–2365. [PubMed] [Google Scholar]
- Bell R. P. (1980) The Tunnel Effect in Chemistry, pp 77–105, Chapman Hall, New York. [Google Scholar]
- Sen A.; Kohen A. (2010) Enzymatic tunneling and kinetic isotope effects: Chemistry at the crossroads. J. Phys. Org. Chem. 23, 613–619. [Google Scholar]
- Nagel Z. D.; Klinman J. P. (2010) Update 1 of: Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 110, PR41–PR67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. (2012) Good vibrations in enzyme-catalysed reactions. Nat. Chem. 4, 161–168. [DOI] [PubMed] [Google Scholar]
- Klinman J. P. (2013) Importance of protein dynamics during enzymatic C–H bond cleavage catalysis. Biochemistry 52, 2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernster L.; Hoberman H. D.; Howard R. L.; King T. E.; Lee C.-P.; Mackler B.; Sottocasa G. (1965) Stereospecificity of certain soluble and particulate preparations of mitochondrial reduced nicotinamide-adenine dinucleotide dehydrogenase from beef heart. Nature 207, 940–941. [DOI] [PubMed] [Google Scholar]
- Sharpley M. S.; Shannon R. J.; Draghi F.; Hirst J. (2006) Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248. [DOI] [PubMed] [Google Scholar]
- Sherwood S.; Hirst J. (2006) Investigation of the mechanism of proton translocation by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria: Does the enzyme operate by a Q-cycle mechanism?. Biochem. J. 400, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Karadaghi S.; Cedergren-Zeppezauer E. S.; Hövmoller S.; Petratos K.; Terry H.; Wilson K. S. (1994) Refined crystal structure of liver alcohol dehydrogenase-NADH complex at 1.8 Å resolution. Acta Crystallogr. D50, 793–807. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Kurisu G.; Kusunoki M.; Tabata S.; Urabe I.; Osaki S. (2001) Crystal structure of glucose dehydrogenase from Bacillus megaterium IWG3 at 1.7 Å resolution. J. Biochem. 129, 303–312. [DOI] [PubMed] [Google Scholar]
- Orr G. A.; Blanchard J. S. (1984) High-performance ion-exchange separation of oxidized and reduced nicotinamide adenine dinucleotides. Anal. Biochem. 142, 232–234. [DOI] [PubMed] [Google Scholar]
- Lehninger A. L. (1957) Preparation of reduced DPN (chemical method). Methods Enzymol. 3, 885–887. [Google Scholar]
- McClendon S.; Zhadin N.; Callender R. (2005) The approach to the Michaelis complex in lactate dehydrogenase: The substrate binding pathway. Biophys. J. 89, 2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberty R. A.; Smith R. M.; Bock R. M. (1951) The apparent ionization constants of the adenosinephosphates and related compounds. J. Biol. Chem. 193, 425–434. [PubMed] [Google Scholar]
- Chen L.; Gao G.; Bonnac L.; Wilson D. J.; Bennett E. M.; Jayaram H. N.; Pankiewicz K. W. (2007) Methylenebis(sulfonamide) linked nicotinamide adenine dinucleotide analogue as an inosine monophosphate dehydrogenase inhibitor. Bioorg. Med. Chem. Lett. 17, 3152–3155. [DOI] [PubMed] [Google Scholar]
- Schild L.; Blair P. V.; Davis W. I.; Baugh S. (1999) Effect of adenine nucleotide pool size in mitochondria on intramitochondrial ATP levels. Biochim. Biophys. Acta 1413, 14–20. [DOI] [PubMed] [Google Scholar]
- Oppenheimer N. J.; Arnold L. J.; Kaplan N. O. (1971) A structure of pyridine nucleotides in solution. Proc. Natl. Acad. Sci. U.S.A. 68, 3200–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyar A. B.; Karliner J. S.; Cecchini G. (2005) A novel strong competitive inhibitor of complex I. FEBS Lett. 579, 4861–4866. [DOI] [PubMed] [Google Scholar]
- Zu Y.; Shannon R. J.; Hirst J. (2003) Reversible, electrochemical interconversion of NADH and NAD+ by the catalytic (Iλ) subcomplex of mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Am. Chem. Soc. 125, 6020–6021. [DOI] [PubMed] [Google Scholar]
- Bücher T., and Sies H. (1976) Mitochondrial and cytosolic redox states in perfused rat liver: Methods and problems in metabolic compartmentation. In Use of isolated liver cells and kidney tubules in metabolic studies (Tager J. M., Söling H. D., and Williamson J. R., Eds.) pp 41–64, North-Holland Publishing Co., Amsterdam. [Google Scholar]
- Tischler M. E.; Friedrichs D.; Coll K.; Williamson J. R. (1977) Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 184, 222–236. [DOI] [PubMed] [Google Scholar]
- Williamson J. R.; Corkey B. E. (1979) Assay of citric acid cycle intermediates and related compounds: Update with tissue metabolite levels and intracellular distribution. Methods Enzymol. 53, 200–222. [DOI] [PubMed] [Google Scholar]
- Sies H. (1982) Nicotinamide nucleotide compartmentation. In Metabolic compartmentation (Sies H., Ed.) pp 205–231, Academic Press, London. [Google Scholar]
- Pinheiro T. J. T.; Venning J. D.; Jackson J. B. (2001) Fast hydride transfer in proton-translocating transhydrogenase revealed in a rapid mixing continuous flow device. J. Biol. Chem. 276, 44757–44761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.