

Abstract
Three opioid receptors (ORs) are known: μ opioid receptors (MORs), δ opioid receptors (DORs), and κ opioid receptors (KORs). Each is encoded by a distinct gene, and the three OR genes share a highly conserved genomic structure and promoter features, including an absence of TATA boxes and sensitivity to extracellular stimuli and epigenetic regulation. However, each of the genes is differentially expressed. Transcriptional regulation engages both basal and regulated transcriptional machineries and employs activating and silencing mechanisms. In retinoic acid–induced neuronal differentiation, the opioid receptor genes undergo drastically different chromatin remodeling processes and display varied patterns of epigenetic marks. Regulation of KOR expression is distinctly complex, and KOR exerts a unique function in neurite extension, indicating that KOR is not simply a pharmacologic cousin of MOR and DOR. As the expression of OR proteins is ultimately controlled by extensive posttranscriptional processing, the pharmacological implication of OR gene regulation at the transcriptional level remains to be determined.
Keywords: gene regulation, transcription factor, chromatin remodeling, neuronal differentiation, retinoic acid
1. INTRODUCTION
Opioids remain the most widely prescribed analgesics and among the most widely abused categories of recreational drugs. The specific biological targets of opioids, opioid receptors (ORs), were first detected by use of radioactive ligand binding assays in the early 1970s (1–3). Use of these assays defined three pharmacologically distinct opioid receptors, based on their different binding profiles with respect to specific types of ligands: μ opioid receptor (MOR), δ opioid receptor (DOR), and κ opioid receptor (KOR) (4, 5). Almost two decades after the initial detection of ORs, the first OR complementary deoxyribonucleic acid (cDNA), which encodes DOR, was cloned through functional screening by Kieffer et al. (6) and Evans et al. (7). With the DOR cDNA as the probe, multiple groups raced to obtain the MOR and KOR cDNAs (8–10) and the genes encoding MOR, DOR, and KOR from various animal species in a short period of time (11–16). Cloning the cDNAs and genomic DNAs of ORs marked the beginning of a new era of intensive molecular studies. These include studies of the mechanisms of action of opioids, ligand-receptor interactions, effectors, genetics, and regulatory pathways, along with revelations of certain physiological actions of the endogenous opioids and ORs. Several key questions have been answered since the initial phase of molecular studies was launched.
First, sequencing of the OR cDNA and genomic DNA clones indicated that the three ORs are encoded by three distinct genes and that ORs belong to the superfamily of 7-transmembrane, G protein–coupled receptors (GPCRs). This finding shed light on the mechanism of opioids’ action and propelled extensive studies of their signal transduction, specific pharmacological effects, and downstream effectors (17). Second, amino acid and nucleotide sequence comparisons have established that the MOR, DOR, and KOR genes are highly (73–100%) conserved in their homologous coding exons, which are located in the center of each gene. These exons encode the 7-transmembrane domain, which suggests that all three OR genes evolved from one ancestral gene that initially spanned this domain. However, whereas the OR genes are highly homologous within their 7-transmembrane domain, they diverge dramatically (with only 9–20% conservation) in their amino termini that protrude outside the cell surface as well as in their carboxyl termini that extend into the intracellular space (18, 19). The evolutionary divergence of their termini underlines the very different ligand binding patterns (primarily through their amino termini), pharmacological effects, and signal transduction pathways (primarily through their carboxyl termini) of the three ORs. Molecular modeling has provided some insight regarding the relationship of the various OR ligands to their corresponding receptors or receptor pairs (20–23). Third, transcriptional and epigenetic regulation studies have established that, despite the conservation in their structures and certain common regulatory mechanisms, each OR gene is also subjected to unique regulatory pathways and exhibits a distinct pattern of expression (24, 25). The studies of these gene regulatory mechanisms constitute another major effort in the field and is the focus of this review. Fourth, alignments of cDNA with genomic DNA and studies of messenger RNA (mRNA) processing have established that multiple mRNA isoforms, or variants, are produced from each OR gene (24, 26, 27). Initial excitement generated from this discovery prompted an examination of the variants’ potential role in relation to the pharmacological subtypes of ORs—which proved a disappointment, because none of the mRNA variants corresponded to any pharmacologically defined subtype. Instead, recent reports have suggested that pharmacological OR subtypes probably correspond to certain specific heterodimeric receptor units (22, 23) and that in the case of MOR mRNA, such variants may play roles in modulating animal behavior or pain sensation (27–29). Moreover, molecular studies of KOR mRNA variants show that extensive posttran-scriptional regulation, which occurs via varied untranslated regions (UTRs), controls the time, location, and signal specificity of neuronal production of KOR protein (26). The significance of the UTR-based regulation is underscored by recent findings of specific physiological functions of KOR derived from transcripts carrying various UTRs, because the KOR proteins produced from UTR-carrying transcripts contribute to neurite extension and cellular response to neuronal activities (30–33). Fifth, studies of OR signal transduction have established that the number of receptor molecules and the microdomains where receptors exist, in addition to the availability of the downstream effectors, are important for ligands to elicit specific biological effects (34). A recently emerged view is that different ligands of a specific type of OR can induce different modes of ligand-receptor interaction (21), tempting one to speculate that distinct shapes of ligands with receptors at different spatial locales and/or of different local densities coordinate such different modes of interaction. The density (number), spatial distribution, and timing of OR expression in a cell can be regulated at multiple levels, starting from the transcription of the gene and continuing through multiple posttranscriptional processes. However, transcription initiates the control over cellular production of certain types of OR. Since the first genomic information about ORs became available in 1994, numerous labs have extensively examined transcriptional control of all three OR genes. Here we review data that have elucidated how these three genes are regulated at the level of transcription, including studies of OR genomic structures, transcription factors (TFs), regulatory signals, and epigenetic regulation. We also discuss the implications of these data and suggest directions for future research.
2. GENOMIC STRUCTURES OF OPIOID RECEPTOR GENES
2.1. Genomic Structure and Alternative Promoter/Splicing
The cDNA sequences of the three ORs are homologous in the 7-transmembrane domain, which is encoded by three highly conserved coding exons. The similarity of the genomic organization among the ORs is obvious from the alignment of the mouse MOR (11), DOR (13, 14), and KOR (12) cDNAs (Figure 1). The degree of conservation is remarkable not only because their DNA and amino acid sequences are homologous but also because their exon-intron boundaries (splicing junctions) are absolutely conserved with respect to the specific amino acid residues where splicing occurs.
Figure 1.

The alignment of the mouse μ opioid receptor (mMOR), mouse δ opioid receptor (mDOR), and mouse κ opioid receptor (mKOR) complementary DNAs (cDNAs). The major cDNAs of MOR, DOR, and KOR are aligned according to the translation initiation codon (number 1 above each cDNA). Alternatively spliced forms are not shown. Corresponding exons are numbered above each cDNA with the approximate sizes of introns indicated above the amino acid residues where splicing occurs. In the coding region (dark blue boxes), numbers above the horizontal bars represent the amino acid positions. The broken, light blue bars represent the 5′ and 3′ untranslated regions (UTRs) of each cDNA. Abbreviation: TM, transmembrane. Adapted from Reference 24, copyright © 2002, Elsevier Press.
However, outside the 7-transmembrane region, the sequences and genomic arrangements of these three genes substantially differ. Divergent alternative splicing occurs in the regions upstream or downstream of the conserved 7-transmembrane domain for the MOR and KOR genes—both of which also use alternative promoters (27, 29, 35, 36). MOR and KOR mRNA variants that are altered in their 5′ UTRs are differentially regulated at the level of translation (37–40), and KOR variants with different 3′ UTRs are differentially regulated at the level of RNA stability or transport (30, 41). Although mRNA variants of mouse MOR have been associated with differential behavior and drug response of animals (27, 29), in a carefully conducted study attempting to clarify the mature mRNAs for the reported mouse MOR variants, the data show only one mature mRNA species in the mouse brain that can be detected by Northern blot, as a fragment roughly 11.5–12 kb in length (42). Human MOR variants produced by extensive splicing in both the upstream and downstream regions of its 7-transmembrane domain have also been detected by reverse transcription–polymerase chain reaction (RT-PCR) (28, 29). The discrepancy between the Northern blot data (revealing a single band of MOR mRNA, or a group of MOR-hybridizing transcripts migrating at a similar position on the gel) and the RT-PCR data (indicating numerous transcripts with widely varied lengths) may arise from different sensitivities of the detection methods. It is also possible that cDNAs produced by extensive splicing on the Oprm locus may represent certain immature forms of MOR mRNA, or even mRNAs of some hitherto uncharacterized proteins distinct from MOR. Consequently, although genetic data have indicated biological effects caused by defects in specific MOR mRNA variants (43), data have not been published from critical experiments that validate the biological legitimacy of predicted, alternatively spliced MOR cDNA variants, each in a contiguous and mature poly(A)-containing mRNA form. For mouse KOR, on the other hand, at least six mature mRNA variants, generated from the same gene, have been validated by Northern blot analysis. These KOR mRNA variants are generated through the use of two alternative promoters and two alternative polyadenylation sites, in addition to the inclusion of one upstream noncoding exon where alternative splicing occurs to produce different 5′ UTRs (35, 37, 41). Recently, the structure of the gene that encodes human KOR has been shown to also include this upstream noncoding exon (44), suggesting that there has been natural selection for these alternative 5′ UTRs, possibly in the regulation of KOR under certain physiological contexts. Recent studies have presented evidence that the UTRs of KOR mRNA mediate growth factor regulation of KOR protein synthesis and that this is required for neurite extension (33, 45, 46). Finally, the mouse DOR gene has been shown to initiate transcription from two adjacent sites (14), but there has been no report of its alternative splicing.
The similar genomic structures of, and the high degree of sequence conservation among, the three ORs strongly support the idea that the three genes have evolved from a common ancestral gene that spanned at least the three coding exons. Variations have since evolved around such an ancestral gene’s amino terminus for divergent ligand binding abilities and around its carboxyl terminus for divergent intracellular signaling. Further evolution may have taken place to allow alternative splicing in the OR genes’ UTRs for regulation at the level of mRNA stability, mobilization, and/or translation (30, 31, 37). Alternative splicing to change the coding sequences, such as those in MOR, probably evolved to expand the regulatory reservoir for modulation of the OR system.
2.2. Gene Mapping and Promoter Characteristics
The mouse gene that encodes MOR, Oprm, is located in chromosome 10 (47) and spans a distance of 250 kb, with its cDNA estimated to be approximately 11.5–12 kb in length (42). It can utilize two closely positioned promoters: the distal promoter (DP), which drives one major transcript initiated at the −784 position (relative to the translation initiation codon), and the proximal promoter (PP), which drives multiple transcripts initiated from a cluster of transcription initiation sites between the −291 and −268 positions (Figure 2). Both promoters belong to the TATA-less type, are GC-rich, and contain numerous regulatory elements (see Section 3). In animals, PP is preferentially used in most tissues; in cell cultures, PP activity also accounts for the vast majority (>95%) of this gene’s activity (36). In addition, the mouse gene can use a TATA-containing promoter (named E11) located more than 10 kb upstream (48). The rat gene structure is similar to that of the mouse (49). The human gene for MOR, OPRM, as determined by multiple groups (29, 50, 51), has been found to be highly homologous to the structure of the mouse and rat genes, including its multiple TATA-less promoters and complex TF-binding sequences.
Figure 2.

The regulatory DNA elements and corresponding transcription factors (TFs) of the promoters of mouse Oprm (top) and human OPRM (bottom) genes. Thick colored lines show experimentally validated promoter regions. Solid arrows mark validated transcription initiation sites from proximal promoters (PPs) and distal promoters (DPs), and the dashed arrow marks the predicted transcription initiation site from the human gene. +1 indicates the beginning of the coding region. Nucleotide numbers above each map refer either to the ends of validated promoter regions or regulatory sequences, or to the 5′ ends of the reported corresponding TF-binding sites (blue ovals), in relation to the initiation codon +1. Certain TFs’ exact positions have not been defined and therefore are not shown here. Activating TFs are shown as dark blue ovals, and repressing TFs are shown as light blue ovals. Abbreviations: AP1, activator protein 1; AP2, activator protein 2; CREB, cyclic adenosine monophosphate (cAMP) response element binding protein; NF-κB, nuclear factor κB; NFAT, nuclear factor of activated T cells; NRSE, neurorestrictive silencer element; Oct-1, octamer-1; PARP1, poly(ADP-ribose) polymerase 1; PCBP, poly C binding protein; PU.1, PU box binding; REST, repressor element-1 silencing transcription factor; Sox, Sry-like high-mobility group box gene; Sp1, specificity protein 1; Sp3, specificity protein 3; STAT, signal transducers and activators of transcription; YY1, ying yang-1.
The gene that codes for DOR in the mouse, the Oprd gene, has been mapped to chromosome 4 (52), and its structure has been determined (14). Two major transcription initiation sites have been identified, located between the −324 and −142 positions with respect to the ATG initiation codon (Figure 3). This promoter is also TATA-less and GC-rich, and the gene seems to span only the three coding exons. The human gene that codes for DOR has also been isolated (53) and mapped to chromosome 1 (52).
Figure 3.
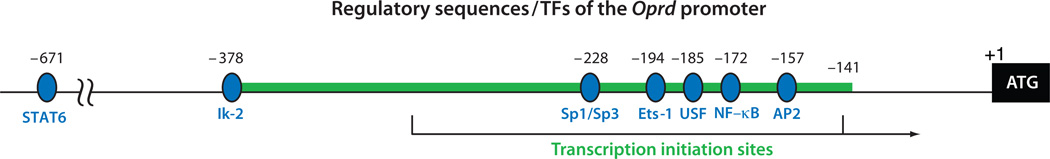
The regulatory DNA elements and the corresponding transcription factors (TFs) of the promoter of Oprd. The heavy green line shows the experimentally validated promoter region. The black arrow shows transcription initiation. +1 indicates the beginning of the coding region. Nucleotide numbers above the map refer either to the ends of validated promoter region or to the 5′ ends of the corresponding TF-binding sites (blue ovals) in relation to the initiation codon +1. All the reported TFs for this gene are activating. Abbreviations: AP2, activator protein 2; Ets, E-twenty six; Ik, Ikaros; NF-κB, nuclear factor κB; Sp1, specificity protein 1; Sp3, specificity protein 3; STAT, signal transducers and activators of transcription; USF, upstream stimulatory factor.
The gene for KOR has been examined for the mouse (12, 15), rat (16), and human (44, 54). It is mapped to human chromosome 8 (54, 55) and mouse chromosome 1 (15, 56, 57). Both the human and mouse genes contain a noncoding exon upstream of the initiating codon (35, 44, 58), and two functional promoters named promoter 1 (P1) and promoter 2 (P2) have been validated for the mouse gene, Oprk (Figure 4). P1 initiates transcription from a cluster of residues between the −1098 and −719 positions with respect to the ATG initiation codon. P2 is located within intron 1 and initiates transcription from a fixed residue at the −93 position (35). Furthermore, alternative splicing can take place in intron 1 of the mouse gene. Therefore, P1 can drive the expression of KOR mRNAs with either one of the two types of 5′ UTR, and the mRNAs span four exons. P2, on the other hand, drives the expression of KOR mRNA with a single type of 5′ UTR containing merely 93 nucleotides, and the mRNA spans only the three coding exons. P1 is constitutively active in cultured cells and is also the major promoter used in most cell lines and animal tissues (58). P2 is active only in certain brain areas, such as the brain stem, and in later stages of in vitro neuronal differentiation (59). Both promoters are TATA-less and GC-rich. The mouse Oprk gene, extending from promoter 1 to its 3′-most of two alternative polyadenylation signals, spans roughly 16 kb, whereas the human gene spans roughly 25 kb.
Figure 4.
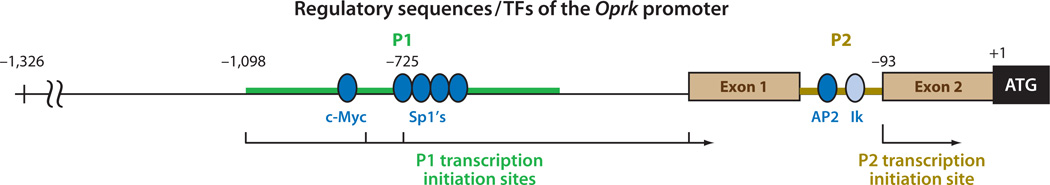
The regulatory DNA elements and corresponding transcription factors (TFs) of the promoter of Oprk. Two brown boxes show the two exons flanking intron 1 where promoter 2 (P2) resides. Both promoter 1 (P1) and P2 have been experimentally validated and are shown in thick colored lines. The arrows indicate transcription initiation sites; the sites of initiation are numbered above the map in relation to the initiation codon +1. Activating TFs are shown as dark blue ovals, and one repressing TF is shown as a light blue oval. Abbreviations: AP2, activator protein 2; Ik, Ikaros; Sp1, specificity protein 1.
In summary, the three OR genes share a conserved genomic structure in the 7-transmembrane domain that is encoded by three similarly arranged exons, but their amino and carboxyl terminal regions are very different. Their promoters are TATA-less and GC- rich except for the upstream E11 promoter of the Oprm gene. The genes encoding MOR and KOR undergo alternative splicing in their 5′ UTRs and 3′ UTRs, and both genes use alternative promoters. In addition, the Oprk gene uses alternative polyadenylation sites. Alternative promoter/polyadenylation/splicing does not affect the main coding sequence of KOR mRNA but could alter the efficiency of KOR protein production through posttranscriptional control (26). For MOR mRNA variants, alternative splicing would drastically alter the protein’s amino and carboxyl termini, making variant proteins predicted to be pharmacologically distinct from MOR (27, 29).
3. TRANSCRIPTION FACTORS AND REGULATORY SIGNALS
3.1. Transcription Factors and Regulatory Signals of the Gene That Encodes μ Opioid Receptor
Transcriptional regulation of MOR expression has been examined mostly for the mouse and the human genes. A number of TFs for the mouse gene, Oprm, have been experimentally evaluated as positive (activating) or negative (repressing). Positive TFs of this gene include, from the 5′ to the 3′ ends, Sry-like high-mobility group box gene (Sox) (60, 61), activating protein 2 (AP2)/specificity protein 1 (Sp1) (62), Sp1 on an inverted GA motif (iGA) (63), poly C binding protein (PCBP) on a sequence adjacent to iGA (64), and cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) in the 5′ UTR (65). Negative TFs include, from the 5′ to the 3′ ends, octamer-1 (Oct-1) (66), PU box binding (PU.1) on a 34-bp silencer region (67, 68), poly(ADP-ribose) polymerase 1 (PARP1) on a double-stranded poly-C sequence (69), and two TFs binding to the 5′ UTR—specificity protein 3 (Sp3) (70) and repressor element-1 silencing transcription factor (REST); the latter binds the neurorestrictive silencer element (NRSE) overlapping the initiation codon ATG (71, 72) (Figure 2, top). These upstream regulatory sequences can be grouped into three clusters, each responsible for binding by multiple TFs. These include the cluster from −775 to −687 (downstream of DP) consisting of several Sox proteins and PU.1; a cluster flanking the iGA sequence consisting of PARP1, AP2, Sp1, and PCBP; and a cluster within the relatively short 5′ UTR consisting of Sp3, CREB, and REST. Because the identification of these clustered TFs has been conducted in different cellular backgrounds, it remains to be verified whether these TFs act in concert or if they are enriched in different types of cells to differentially regulate this gene.
Positive TFs for the human gene, OPRM, include, from the 5′ to the 3′ ends, activator protein 1 (AP1) (73), nuclear factor κB (NF-κB) (74), signal transducers and activators of transcription (STAT) (75, 76), GATA-binding protein (GATA) (77), STAT6 (78), NF-κB (74), nuclear factor of activated T cells (NFAT)/AP1 (79), Sp1/3 (50), ying yang-1 (YY1) (80),NF-κB (74), and PARP (81). Negative TFs include one that binds to a 35-bp silencer region similar to the 34-bp silencer found in the mouse gene (82) and another that binds to a NRSE also similar to that found in the mouse gene (83) (Figure 2, bottom).
In general, transcriptional regulation of the mouse Oprm gene can be elicited by stimuli such as endocrine vitamin A or its active ingredient retinoic acid (RA) (84), cytokines of the interleukin family (78, 85), interferon-γ (46), insulin-like growth factor-1 (76), phorbol ester (73), and so on. These studies have been conducted through the use of the mouse embryonal carcinoma cell line P19; neuroblastoma cell lines such as SH-SY5Y; immune cells such as primary T cells, B cells, and mononuclear cells; and established immune cell lines such as Raji and U-937. In P19 cells, RA induces chromatin remodeling on the Oprm gene locus during its induction of neuronal differentiation (see Section 4). In other cell types that are already differentiated, most signals directly activate TFs. This is consistent with the finding that the Oprm gene is usually heavily methylated and silenced in undifferentiated cells and that it requires activation (86). However, the caveat is the artificial nature of gene reporter constructs used in most of the earlier studies. This raises a concern about the interpretation of these data. More recent studies of the endogenous Oprm gene, i.e., studies examining its chromatin state and remodeling process, have in fact revealed that it has varied epigenetic states in different cells. This would affect the actions of these TFs on the endogenous gene locus (see Section 4). Therefore, the physiological relevance of most of these TFs to transcriptional activation of this gene remains to be validated.
3.2. Transcription Factors and Regulatory Signals of the Gene That Encodes δ Opioid Receptor
The Oprd gene utilizes a single promoter but can initiate transcription from two major sites between the −324 and −142 positions with respect to the initiation codon (14). Studies of its gene transcriptional regulation have focused on activating factors, which include, from the 5′ to the 3′ ends, STAT6 (74), Ikaros (Ik) (87, 88), Sp1/Sp3 (89), E-twenty six 1 (Ets-1) (90), upstream stimulatory factor (USF) (88, 91), NF-κB (74), and AP2 (92). All these studies have been conducted using reporter constructs in a heterologous cellular background; therefore, the physiological relevance of these regulatory signals again remains to be validated. In this regard, an examination of DNA methylation of the endogenous Oprd locus has established that it is epigenetically regulated (93, 94) (see Section 4).
In terms of signals stimulating Oprd transcription, it is known that activation of phosphatidylinositol 3-kinase (PI3K) elevates DOR mRNA levels (95). In the P19 system, the Oprd gene is constitutively active in predifferentiated cells but is repressed during neuronal differentiation (96).
3.3. Transcription Factors and Regulatory Signals of the Gene That Encodes κ Opioid Receptor
Transcriptional regulation of the gene that encodes KOR has primarily been examined for the mouse gene, Oprk. This gene is regulated by multiple TFs including, from the 5′ to the 3′ ends, three positive TFs [c-Myc (97, 98), Sp1 (99), and AP2 (100)] and a negative TF, Ik, that binds to intron 1 (101) (Figure 4). The Oprk gene promoter has been functionally validated in a transgenic animal model (58). In this model, a role for vitamin A (or its active ingredient RA) in transcriptional regulation of this gene was first revealed (59), and it has been validated in a series of studies examining the Oprk gene’s chromatin remodeling and epigenetic regulation (100, 102) (see Section 4). In addition to RA, nitric oxide (NO) also appears to play a role in transcriptional regulation of this gene—by inactivating NF-κB (97), an activator of c-Myc that binds to and activates Oprk’s P1 promoter (98). The physiological relevance of NO to KOR expression in animals remains to be validated, but it might involve feedback regulation for ischemic neuroprotection (103).
3.4. Transcriptional Control in Relation to Distinct Opioid Receptor Expression Patterns
The different expression patterns of MOR, DOR, and KOR mRNAs have been documented in animal studies (25) and in studies of the P19 neuronal differentiation model (96). Although it remains unclear how the three genes are differentially controlled in different brain areas, clues have emerged—mostly from the study of P19 neuronal differentiation—to shed light on the mechanistic basis of the genes’ differential expression. In undifferentiated P19 cells, the Oprk gene is highly expressed, the Oprd gene is weakly expressed, and the Oprm gene is entirely silenced.
As cells undergo RA-triggered neuronal differentiation, the initially active expression of KOR gradually subsides then later reactivates, with concurrent elevation of previously silenced expression of MOR, in the more differentiated cultures (which mostly contain neurons). The initially weak expression of DOR quickly decreases further during early differentiation but then is elevated again in later differentiated cells (96). Comparing the chromatin states of the Oprm and Oprk gene loci from the stem cell stage to the differentiated stages makes it clear that these two gene loci exist in different states of chromatin conformation and that each is remodeled differently during the differentiation process (see Section 4). Importantly, the fundamentally different remodeling mechanisms of the Oprm and Oprk genes, assessed in the neuronal differentiation process, are consistent with the long-observed early expression of KOR and late induction of MOR in this culture model (96). The finding is also in line with previous observations made in animal studies that reveal earliest expression of KOR mRNA in animals even before neurons are born (58) and relatively late expression of MOR mRNA only after mature neurons begin to appear (L.-N. Wei & H.H. Loh, unpublished data). The chromatin state of the Oprd gene remains to be examined.
In summary, the three genes use multiple common TFs such as the Sp family members, AP1 family members, Ik, NF-κB, and the STAT family members. Their promoters are mostly TATA-less and can initiate transcription from multiple sites. They also use similar TFs to act on the 5′ UTR. All these shared features suggest that a fundamentally similar mechanism underlies transcriptional regulation of the three OR genes. This common mechanism is probably exploited only in cells where the chromatin of these gene loci has been remodeled and made accessible, i.e., in an open configuration. Therefore, transcriptional regulation of the three OR genes likely is initiated by differential chromatin remodeling processes during cellular differentiation. Only in cells where the gene loci have been remodeled can the different TFs act to facilitate transcription or silencing under various conditions or stimuli. The commonly expressed basal TFs could be responsible for the overlapped expression patterns of the three OR genes in certain fully differentiated cell types, such as neurons and immune cells. Divergence in their TFs presumably facilitates responses to signal inputs such as cytokines and growth factors, which may be specific to different cell types. This is consistent with observations in animals showing that the three genes, although all generally active in the brain, are differentially expressed in different brain regions and that they also exhibit different temporal patterns of expression during development. Importantly, no validated “cell-specific” TF has been identified for any of these genes. Consequently, the spatial and temporal specification of each gene’s activity may be imposed through the use of different combinations of multiple TFs and, as discussed next, epigenetic regulation.
4. EPIGENETIC REGULATION
Epigenetics refers to reversible and heritable changes in gene activities (phenotypes) without changes in the DNA sequences. The term was initially used to describe specific events occurring during development (104–106) and later extended to also describe changes in gene activity in adults that correlate with, or are caused by, alteration in chromatin (DNA and histone) covalent modifications but not in DNA sequences. This phenomenon has been widely detected in complex diseases such as cancers (107) and disorders of the nervous systems (108); in learning and memory formation (109); in neuronal plasticity (110); and in neurogenesis (111). Extensive studies in the past decade have shown that epigenetic regulation can involve alteration not only in chromatin modification but also in higher-order chromatin structure and nuclear architecture (112); however, most studies of epigenetic regulation examine only changes in chromatin modification rather than changes in conformation or higher-order structures.
Principal forms of chromatin modifications include DNA (particularly cytosine) methylation and various histone-protein covalent modifications such as acetylation, methylation, phosphorylation, ubiquitination, sumoylation, and adenosine diphosphate (ADP) ribosylation (112–114). DNA methylation, particularly of the CG islands within genes’ promoters or upstream regions, is thought to cause gene silencing. The so-called histone code refers to specific histone modifications detected on activated or repressed gene loci; it was proposed on the basis of studies of selected model genes, especially in fission yeast (115, 116). However, as more genes were examined in various organisms, ideas evolved as some changes were found to be transient (117). Nevertheless, certain seemingly more general codes have been adopted from the study of yeast and widely applied in studies of higher organisms, including mammals.
Several general, currently accepted rules in epigenetic regulation are the following:
A promoter sequence rich in methylated cytosine is usually suppressed or silenced.
A gene whose regulatory region’s Histone 3 (H3) and/or Histone 4 (H4) are hyperacetylated is usually considered to be in an activated state but to be repressed when its histones recruit histone deacetylases (HDACs).
A gene positive for H3’s lysine 4 or 79 methylation (di- or trimethylation) is usually considered active, in contrast with the suppression that occurs if a gene is methylated on H3 lysine 9 or H3 27.
A gene enriched in heterochromatin protein 1 (HP1) likely exists in a heterochromatin state and therefore is likely to be silenced (112, 113).
According to these rules, all three OR genes would be expected to be under epigenetic regulation because (a) all three genes are rich in islands and can be heavily methylated; (b) their promoters exhibit various types of modifications in different cellular states or culture conditions; and (c) chromatin remodeling occurs on their promoters with corresponding changes in their patterns of expression in an in vitro neuronal differentiation model.
The enzymatic machinery for, and the mechanisms underlying, epigenetic regulation are extensive; the enzymes include histone acetylases, deacetylases, lysine methyltransferases, arginine methyltransferases, and DNA methyltransferases (118). DNA methylation is usually detected through direct sequencing to identify methylated cytosine residues; therefore, changes in DNA methylation can be detected in a more specific manner. Most studies of histone modification are conducted through the use of chromatin immunoprecipitation (ChIP), which lacks the resolution power to precisely locate the specific types of marks on the chromatin. It also seems true that the most important forms of histone modifications occur on lysine residues in the amino-terminal unstructured domain of histone H3. Other forms of modifications cannot be generalized to predict gene activity. Furthermore, the term hyper or hypo to describe chromatin modification is used in a relative, but not quantitative, manner. It cannot differentiate the number of modification types; rather, it merely indicates that changes have occurred in certain modifications on a chromatin segment. Recently, researchers have developed new ChIP-derived techniques to address the resolution issue (112). MNase-ChIP, for example, can detect the marks on individual nucleosomes by coupling micrococcal nuclease (MNase) digestion with ChIP. The term ChIP-chip denotes the use of microarray chips to determine the targets of ChIP, narrowing down the range of a modified chromatin segment in the genome (119). In addition, ChIP-seq, by sequencing the targets of ChIP, can even more confidently assign these marks to precise DNA sequences of the genome (120).
Despite these technological improvements, several important issues remain to be addressed. First, because all these methods are used to analyze histone mixtures in a pool of cells, it is not possible to determine a target gene’s specific chromatin changes within a particular cell. Second, defining spatial changes in chromatin conformation, which can be determined only through challenging methodologies, is still rarely conducted. Third, plasticity in epigenetic regulation is increasingly being recognized; therefore, the phenomenon of plasticity and reversibility should be addressed, but it has rarely been followed. Understanding epigenetic regulation is important because its mechanisms and effects are pertinent to the context of various physiological or disease conditions. Most epigenetic findings reported for the mammalian systems are loosely defined, merely describing histone modifications through the use of limited, commercially available antibodies in ChIPs. It is of concern that many of these types of tools have not been validated in terms of their appropriate use to interpret activities or changes in genes and gene expression. Fortunately, the chromatin of Oprm and Oprk gene loci in the stem cell–neuronal differentiation process has been examined through more rigorous biochemical methods, which have helped validate the analysis of their epigenetic regulation. However, for the sake of this review, we use a loose definition for epigenetic regulation and include biochemical studies of OR genes’ chromatin remodeling and results that have been obtained almost solely through the use of ChIP technology.
4.1. Epigenetic Regulation of the Gene That Encodes μ Opioid Receptor
The Oprm gene promoter is heavily methylated in the undifferentiated P19 cells where the MOR gene is silenced (86, 121) (Figure 5). The silenced Oprm gene can be activated by decreasing the expression of methyl-CpG-binding protein 2 (MeCP2), an important player in gene silencing (86), or by the addition of a pan-histone acetylation inducer such as trichostatin A (TSA) (122). Furthermore, DNA methylation on the Oprm promoter can be reduced by the addition of an artificial demethylation agent, 5-Aza-2′-dexoycytidine (5-Aza-C) (121). For the human gene OPRM, several older studies reported DNA methylation on its promoter (123, 124). Recent studies have reported increased DNA methylation on the OPRM locus in heroin addicts (125) and on the Oprm locus in animals under ischemic insults (126). Another recent study (84) provided solid evidence for higher-order chromatin conformational remodeling of the Oprm gene promoter during P19 neuronal differentiation. Consistent with the constitutive silencing of MOR in predifferentiated cultures, the promoter region is initially organized into an ordered nucleosome array in the undifferentiated precursor cells. However, as neuronal differentiation proceeds, nucleosomes of the Oprm promoter region spanning P1 and P2 change their positions; concurrently, recruitment of specific chromatin remodelers (including subunits Brg1/SNF2b and BAF155) remodel this promoter and activate its gene. These studies conclude that the Oprm gene undergoes epigenetic regulation with chromatin remodeling that activates its transcription in RA-induced differentiating neurons.
Figure 5.

Epigenetic regulation and chromatin remodeling of the Oprm gene in P19 neuronal differentiation. (Top) In undifferentiated P19 cells, as well as cerebellar cells, where μ opioid receptor (MOR) is silenced, the gene promoter exists in a condensed chromatin conformation with a linear array of nucleosomes that are heavily methylated; these nucleosomes can recruit methyl-CpG-binding protein 2 (MeCP2). (Bottom) In cells treated with retinoic acid (RA), 5-Aza-2′-dexoycytidine (5-Aza-C), or trichostatin A (TSA), chromatin remodeling occurs so that methylation is reduced, with concurrent hyperacetylation of the chromatin and gene activation. The chromatin conformation in this activated state has not been determined, but a chromatin remodeler component, Brg1, has been found on the activated promoter. Transcription initiation from the proximal promoter and distal promoter is indicated with arrows under the chromatin. Abbreviations: Ac, acetylated histone; meC, methyl C.
4.2. Epigenetic Regulation of the Gene That Encodes δ Opioid Receptor
The implication of epigenetic regulation of the Oprd gene came from a report showing heavy DNA methylation on its promoter in Neuro2A cells, where DOR is not expressed, and demethylation on this promoter in NS20Y cells, where DOR is highly expressed (93). In Neuro2A cells, adding 5-Aza-C to the culture elevates DOR expression. Furthermore, in addition to playing a role in regulating MOR expression, MeCP2 contributes to repression of DOR expression, which can be relieved through the addition of TSA to the culture (94). In addition, a nerve growth factor (NGF)/PI3K signal can reduce H3 lysine 9 trimethylation, a repressive chromatin mark, and increase H3 acetylation, an activating chromatin mark (127), consistent with an earlier finding that PI3K elevates DOR mRNA levels (95). Whereas there have been no data for the chromatin state of this gene, results indicate that it is epigenetically regulated, although it is unclear what stimuli initiate this regulation.
4.3. Epigenetic Regulation of the Gene That Encodes κ Opioid Receptor
Specific chromatin remodeling during cell differentiation to elicit epigenetic regulation of OR genes was first described for the Oprk gene (Figure 6). Expression of this gene in mouse brain is altered, at the transcriptional level, by feeding animals a vitamin A–depleted diet (59). This finding implicated epigenetic regulation triggered by diet, or a specific nutrient. Because whole-brain animal studies precluded a molecular investigation that required a pure cell population, the P19 neuronal stem cell model was used to determine the process of transcriptional regulation of KOR during development or in the cell differentiation process. In this model, the KOR gene is constitutively and highly expressed in proliferating cells (59, 97), which would predict the presence of active chromatin marks in these undifferentiated cells. As cells undergo RA-induced neuronal differentiation, KOR expression is gradually lowered, which would predict its acquisition of repressive chromatin marks.
Figure 6.

Epigenetic regulation and reversible chromatin remodeling of the Oprk gene promoter in P19 neuronal differentiation. (Top) In P19 undifferentiated cells, the Oprk gene promoter 1 (P1) exists in an open chromatin configuration with no nucleosomes detected, allowing the E box (c-Myc binding), GC boxes (Sp1 binding), and transcription initiation sites to be exposed for transcription initiation from P1 (black arrow). Intron 1, where promoter 2 (P2) resides, is organized in a condensed conformation (gray ovals); therefore, P2 is inactive in the predifferentiated cells (as indicated with a black X). (Middle) In retinoic acid (RA)-induced differentiating cells, c-Myc/Max is replaced with Mad/Max on the E box, which recruits histone deacetylases (HDACs) and other repressive remodeling components such as BRG and BAF155. The transcription factor Ikaros (Ik) also recruits HDACs, together with the Mad/Max complex, to remodel this piece of chromatin into a tightly packed conformation with a regular nucleosome array covering both P1 and P2, which are decorated with repressive chromatin marks such as K9-me2. Transcription is shut down in the differentiating cells (as indicated with Xs). (Bottom) Upon further neuronal differentiation, cultures begin to express growth factor receptors such as nerve growth factor (NGF) receptor. After NGF stimulation, the NGF receptor is activated, triggering phosphatidylinositol 3-kinase (PI3K) signaling to activate transcription factor activator protein 2 (AP2) that binds a target site on P2. Together with remodeling machineries yet to be described, this initiates chromatin remodeling that opens up P2, which becomes decorated by active chromatin marks. It remains to be determined if the reactivated chromatin conformation at P2 spreads more distally to also permit transcription initiation from P1 (as indicated with a question mark). Abbreviation: Ac, acetylated histone; KOR, κ opioid receptor.
To biochemically confirm the prediction that chromatin remodeling underlies the developmental changes in the expression of KOR, the chromatin conformation of the endogenous Oprk gene locus was determined in response to RA-induced neuronal differentiation (102). This study revealed an entirely open chromatin conformation (with the absence of nucleosomes) on the gene locus’s P1 and a closed chromatin conformation on its P2 in proliferating, undifferentiated P19 cells, where this gene is constitutively active through P1 activity. However, in differentiating P19 cells, where KOR expression is reduced, this gene locus is remodeled to exhibit an ordered chromatin conformation because nucleosomes are detected on both P1 and P2 and because this chromatin segment becomes compact in the differentiating cells. This finding was based on results from two experiments: MNase mapping, which revealed a nucleosome ladder on the organized chromatin; and a restriction accessibility assay, which assessed the open/accessible chromatin regions of this promoter. In conjunction with a third experiment, ligation-mediated polymerase chain reaction (LM-PCR), which defined the nucleosome borders on the nucleosome array, these experiments clearly established that, during RA-induced neuronal differentiation, nucleosomes are gradually assembled and deposited onto the specific regions of this promoter, shutting down its transcription (102). This provides the first solid biochemical evidence for the process of chromatin remodeling that occurs on the Oprk gene’s P1, which results in changes in this gene activity during the differentiation process. The fact that the chromatin of this gene promoter is changed from an open (disorganized) state to a closed (organized) state in the early neuronal differentiation process provides unambiguous evidence for epigenetic regulation of the Oprk gene.
There is an interesting biphasic pattern of transcriptional activation of the Oprk gene. Following suppression of the gene in early differentiation stages, Oprk transcription becomes active again as the culture progresses toward its fully differentiated stage. How is closed chromatin on this gene locus reactivated in later stages of differentiation? It appears that, in more differentiated cells, the Oprk gene’s transcription can be reactivated not at P1 but at the more proximal P2—and reactivated by NGF, because the now-differentiated neuronal cells express NGF receptors to transmit signals and activate TF AP2 that binds to P2. Concurrently, chromatin modification is changed from H3 lysine 9 methylation (repression) to H3 lysine 4 methylation (activation) on P2 (100). Presumably, the NGF-stimulated reactivation phase of this gene involves rearrangement of the nucleosomal positions, or their disassembly, on this promoter so that AP2 can access its binding site on P2; however, this remains to be determined. Nevertheless, changes in the chromatin structure and modification have been conclusively demonstrated for the Oprk gene, and these changes alter its activity during the process of stem cell neuronal differentiation. More interestingly, for this gene, epigenetic regulation operates in two directions—for both gene silencing and activation— which may provide the crucial control for the presumed plasticity in epigenetic regulation. This type of reversibility is rarely reported for mammalian genes. Figure 6 illustrates these changes in chromatin conformation, modification, and nucleosome arrangement in P1 and P2 of the Oprk gene during RA-induced P19 neuronal differentiation.
In summarizing epigenetic regulation of OR genes, it can be concluded that all three OR genes show epigenetic regulation. DNA methylation is biochemically confirmed for Oprm and Oprd, whereas extensive chromatin remodeling and altered epigenetic marks are biochemically confirmed for Oprm and Oprk. Although the three gene promoters are similar, they exploit different forms of epigenetic regulation and exhibit different patterns of expression during P19 neuronal differentiation. This suggests that epigenetic regulation may contribute to the “cell specificity” of the activation of these genes. So far, only Oprk has been validated to undergo reversible epigenetic changes in a relevant model system, i.e., RA-induced neuron cell differentiation. For Oprm, RA stimulates its chromatin remodeling to activate this gene. For Oprd, the physiological signal for its chromatin remodeling remains unclear.
5. FUTURE PERSPECTIVES
5.1. Tools to Study Transcription in a Relevant Biological Context
To deduce the roles of TFs in regulating OR gene expression, it is important to address several unresolved issues. First, transcriptional regulation should be examined in relevant cellular contexts, and transcriptional activation should be measured by rigorous biochemical assays that determine transcription per se, not merely the steady-state accumulation of total RNA. Many studies have used cells (such as COS and CV-1) with dubious relevance to differentiated cells (such as neurons) and have rarely detected transcription because of technical challenges. Moreover, assays of reporter genes that detect steady-state mRNA levels can be complicated by posttranscriptional regulation. The nature and arrangement of the reporter, e.g., whether it is in-frame with the coding region or its position relative to the transcription initiation site, is crucial. An in-frame reporter would more faithfully report activities of the gene, but it could be compromised by translational control that is usually mediated by the 5′ UTR. A reporter containing only a regulatory element without its transcription initiation site may introduce artifacts resulting from the fusion of the short element to an irrelevant initiation site of transcription. Therefore, data generated from such artificial constructs and heterologous systems should be interpreted cautiously. Although this review has described TFs that have been reported to transcriptionally regulate the three OR genes, readers should be aware that most published data describe only the level of accumulated mRNA rather than transcription and that many studies have utilized only reporter genes and not the endogenous target genes of interest.
Second, whereas a relatively large number of TFs have been reported to be involved in the regulation of each OR gene, it is more likely that not all these TFs act in concert and in all the different cell types. Instead, most of these TFs likely function in a context-dependent or stimulus-dependent manner. Third, few studies have examined upstream signals in the context of whole animals, which is the ultimate goal but one that may not be easily attained because of technical challenges. Fourth, it is highly likely that some TFs that have been studied are involved in epigenetic regulation. Those responding to cytokines or growth factors, in particular, could serve to transmit environmental signals into the cell and trigger epigenetic changes. This is the most important direction in future transcriptional studies of these genes.
5.2. Future Studies of Epigenetic Regulation of Opioid Receptor Genes
Determination of TFs that can regulate a specific gene’s expression will identify only the pool of TFs that might play a regulatory role in the expression of that gene in various cellular backgrounds. An even more important factor in the gene’s activity is the state of chromatin configuration of that gene locus in a specific cellular context. Without such information about the chromatin state of a specific gene locus in such a context, it is not possible to determine whether specific TFs can act on the target gene. As such, future studies will be required to examine the contextual chromatin state of endogenous gene loci. Although ChIP-based detection may be informative, reliable information would still depend on biochemical assays of chromatin.
ChIP-based studies will correlate chromatin marks, before and after epigenetic changes, with activities of the genes of interest. In conducting ChIP experiments, investigators must be cautious in their use of the required reagents, such as antibodies, because many such reagents do not differentiate between multiple forms of histone modifications such as mono-, di-, or trimethylation. Furthermore, a general description of chromatin modification as hyper or hypo must be replaced by more specific documentation of the extent of specific modification relevant to gene activation or repression. To this end, more tools and techniques to determine chromatin marks in selective cell types must be developed. Different cells in a heterogeneous context (such as in a tissue or brain region) can have distinct developmental or maturation states. A difficult technical challenge in addressing this issue is to develop ChIP assays that can detect specific types of chromatin marks in a single cell.
It will be very important to define the mechanisms underlying epigenetic regulation. For this, the field awaits the improvement of existing methodologies such as MNase, restriction accessibility, and LM-PCR, and the development of new methods that are more user friendly.
For the three OR genes, it is critical to validate whether the ChIP-based description of epigenetic changes on the Oprm, Oprd, and Oprk loci indeed reflect changes in their activity under physiological and pharmacological conditions. Extended from this, work in this field should seek to define how and why a cell is guided to alter its specific epigenetic marks on these genes in particular conditions and settings. Results of such studies would potentially provide insight into the extensive networks of interaction between genes and environment, which may underlie problems associated with the therapeutic use and abuse of these drugs. In addition, it will be important to analyze multiple interacting factors related to this gene family in association studies of a complex disorder such as drug addiction. For this type of complex disease, useful insights may derive from studies that define whether and how the epigenetic marks on multiple gene targets can be erased or altered—individually or in any combination—by various factors from the environment, physiological processes or activities, nutrients, and therapeutic agents.
Studies of OR gene regulation by transcription factors in the past two decades have revealed little information that is pharmacologically relevant, but the data convincingly show distinct, and sometimes opposite, patterns of epigenetic regulation that deserve serious investigation, particularly into why these three genes have so extensively diverged in their regulatory regions. These genes interact with environment, nutrients, and drugs in ways that are increasingly recognized to be complicated by intertwined biological processes, including temporal and spatial controls that must underlie the activity of any drug receptor in the whole organism. Consequently, future studies of OR genes must go beyond the action of transcription factors to include factors affecting their epigenetic states, physiological contexts, and posttranscriptional control. Only through such a comprehensive approach can novel insights be deduced to shed light on issues of pharmacological and physiological relevance. For example, results showing that Oprk gene regulation is dramatically distinct from that of Oprd and Oprm, that Oprk possesses regulatory plasticity in development, and that its regulated expression is crucial to neurite extension would suggest that Oprk functions as more than just a pharmacologic cousin of Oprm and Oprd. Discovering and validating the complete biological roles of these three genes will require extensive studies of each in multiple physiological and pharmacological contexts and, in particular, studies that define with more precision their transcriptional and posttranscriptional regulation in those contexts.
SUMMARY POINTS.
The three OR genes are highly conserved in their genomic structures and DNA sequences, particularly in the three coding exons, thus strongly suggesting that they probably evolved from a common ancestral gene spanning these exons. Variations have evolved around the genes’ amino and carboxyl termini for divergent ligand binding and intracellular signaling characteristic of each OR. KOR mRNA variants are further generated by alternative splicing, and they harbor varied UTRs for regulation at the level of mRNA stability, mobilization, and translation. MOR mRNA variants are also generated by alternative splicing and probably produce hitherto uncharacterized proteins distinct from those of MOR.
All three OR genes utilize TATA-less promoters, except for the upstream E11 promoter of the Oprm gene. Additionally, the Oprk gene uses alternative polyadenylation sites. All three genes are regulated by both common (such as the Sp family members, AP1 family members, Ik, NF-κB, and the STAT family members) and varied TFs specific to each gene and are distinctly sensitive to epigenetic regulation. The specificity of spatial and temporal regulation of each gene’s activity may be imposed by the use of different combinations of multiple TFs and epigenetic regulation.
DNA methylation occurs for the Oprm and Oprd gene promoters, whereas extensive chromatin remodeling is biochemically confirmed for the Oprm and Oprk gene loci. The three genes exploit different forms of epigenetic regulation, suggesting that epigenetic regulation may provide the principal, or fundamental, guidance for their distinct “cell specificity.” The Oprm gene chromatin is remodeled to activate its activity in the RA-induced neuron cell differentiation model, whereas the Oprk gene can undergo reversible epigenetic changes to both activate and silence its activity in this model.
Past studies of transcriptional regulation of OR genes revealed little of pharmacological relevance, but recent studies of their epigenetic regulation suggest their potential interactions with certain physiological, environmental, and nutritional states. Significant divergence of the Oprk gene’s regulatory mechanisms and its newly recognized physiological function suggest that KOR has evolved further away from other members of the OR family and is not just a pharmacologic cousin of MOR and DOR.
FUTURE ISSUES.
What are the relevant cellular and physiological contexts for studying OR gene regulation? What rigorous, quantitative biochemical assays are appropriate for studying transcription and posttranscriptional regulation of OR genes?
What are the roles of TFs in epigenetic regulation of OR genes? How do cytokines, growth factors, drug exposure, and environmental signals trigger epigenetic changes of OR genes? Are these regulatory pathways relevant to important pharmacological problems of opioids, such as induction of tolerance?
Can single-cell ChIP methodology be developed to examine epigenetic marks in situ? What is the contextual chromatin state of the endogenous OR gene locus in different physiological or pharmacological settings?
What are the posttranscriptional regulatory mechanisms that control temporal and spatial specificity of expression of each OR protein? What is the relationship of transcriptional regulation to posttranscriptional regulation of each OR gene?
ACKNOWLEDGEMENTS
This work was supported by NIH grants DA11190, DA11806, DK54733, DK60521, and K02-DA13926 in addition to grants from Philip Morris USA Inc. and Philip Morris International to L.-N. Wei, and by NIH grants DA00564, DA01583, DA11806, and K05-DA70554 to H.H. Loh, as well as grants from the F. and A. Stark Fund of the Minnesota Medical Foundation (H.H. Loh) and the Distinguished McKnight University Professorship (L.-N. Wei). The authors appreciate valuable comments and input from Dr. F. Burton during the preparation of this review.
Glossary
- OR
opioid receptor
- MOR
μ opioid receptor
- DOR
δ opioid receptor
- KOR
κ opioid receptor
- cDNA
complementary DNA (reverse-transcribed from processed mRNA)
- GPCR
G protein–coupled receptor
- mRNA
messenger RNA
- UTR
untranslated region
- TF
transcription factor
- RT-PCR
reverse transcription–polymerase chain reaction
- DP
distal promoter
- PP
proximal promoter
- P1
promoter 1
- P2
promoter 2
- Sp1
specificity protein 1
- iGA
inverted GA motif
- CREB
cAMP response element binding protein
- PARP1
poly(ADP-ribose) polymerase 1
- Sp3
specificity protein 3
- REST
repressor element-1 silencing transcription factor
- NRSE
neurorestrictive silencer element
- NF-κB
nuclear factor κB
- RA
retinoic acid
- Ik
Ikaros
- PI3K
phosphatidylinositol 3-kinase
- NO
nitric oxide
- H3
Histone 3
- H4
Histone 4
- HDAC
histone deacetylase
- HP1
heterochromatin protein 1
- ChIP
chromatin immunoprecipitation
- MNase
micrococcal nuclease
- MeCP2
methyl-CpG-binding protein 2
- TSA
trichostatin A
- 5-Aza-C
5-Aza-2′-dexoycytidine
- NGF
nerve growth factor
- LM-PCR
ligation-mediated polymerase chain reaction
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Pert CB, Snyder SH. Properties of opiate-receptor binding in rat brain. Proc. Natl. Acad. Sci. USA. 1973;70:2243–2247. doi: 10.1073/pnas.70.8.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic [3H]Etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA. 1973;70:1947–1949. doi: 10.1073/pnas.70.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terenius L. Characteristics of the “receptor” for narcotic analgesics in synaptic plasma membrane fraction from rat brain. Acta Pharmacol. Toxicol. 1973;33:377–384. doi: 10.1111/j.1600-0773.1973.tb01539.x. [DOI] [PubMed] [Google Scholar]
- 4.Chang KJ, Cuatrecasas P. Multiple opiate receptors: enkephalins and morphine bind to receptors of different specificity. J. Biol. Chem. 1979;254:2610–2618. [PubMed] [Google Scholar]
- 5.Chang KJ, Cooper BR, Hazum E, Cuatrecasas P. Multiple opiate receptors: different regional distribution in the brain and differential binding of opiates and opioid peptides. Mol. Pharmacol. 1979;16:91–104. [PubMed] [Google Scholar]
- 6.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol. Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 9.Yasuda K, Espinosa R, III, Takeda J, Le Beau MM, Bell GI. Localization of the κ opioid receptor gene to human chromosome band 8q11.2. Genomics. 1994;19:596–597. doi: 10.1006/geno.1994.1117. [DOI] [PubMed] [Google Scholar]
- 10.Thompson RC, Mansour A, Akil H, Watson SJ. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- 11.Min BH, Augustin LB, Felsheim RF, Fuchs JA, Loh HH. Genomic structure analysis of promoter sequence of a mouse μ opioid receptor gene. Proc. Natl. Acad. Sci. USA. 1994;91:9081–9085. doi: 10.1073/pnas.91.19.9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu HC, Lu S, Augustin LB, Felsheim RF, Chen HC, et al. Cloning and promoter mapping of mouse κ opioid receptor gene. Biochem. Biophys. Res. Commun. 1995;209:639–647. doi: 10.1006/bbrc.1995.1547. [DOI] [PubMed] [Google Scholar]
- 13.Simonin F, Befort K, Gaveriaux-Ruff C, Matthes H, Nappey V, et al. The human δ-opioid receptor: genomic organization, cDNA cloning, functional expression, and distribution in human brain. Mol. Pharmacol. 1994;46:1015–1021. [PubMed] [Google Scholar]
- 14.Augustin LB, Felsheim RF, Min BH, Fuchs SM, Fuchs JA, Loh HH. Genomic structure of the mouse δ opioid receptor gene. Biochem. Biophys. Res. Commun. 1995;207:111–119. doi: 10.1006/bbrc.1995.1160. [DOI] [PubMed] [Google Scholar]
- 15.Nishi M, Takeshima H, Mori M, Nakagawara K, Takeuchi T. Structure and chromosomal mapping of genes for the mouse κ-opioid receptor and an opioid receptor homologue (MOR-C) Biochem. Biophys. Res. Commun. 1994;205:1353–1357. doi: 10.1006/bbrc.1994.2814. [DOI] [PubMed] [Google Scholar]
- 16.Yakovlev AG, Krueger KE, Faden AI. Structure and expression of a rat κ opioid receptor gene. J. Biol. Chem. 1995;270:6421–6424. doi: 10.1074/jbc.270.12.6421. [DOI] [PubMed] [Google Scholar]
- 17.Law PY, Loh HH, Wei LN. Insights into the receptor transcription and signaling: implications in opioid tolerance and dependence. Neuropharmacology. 2004;47(Suppl. 1):300–311. doi: 10.1016/j.neuropharm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Offermanns S, Simon MI. Organization of transmembrane signaling by heterotrimeric G proteins. Cancer Surv. 1996;27:177–198. [PubMed] [Google Scholar]
- 19.Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 20.Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, Lambert DG. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br. J. Anaesth. 2009;103:38–49. doi: 10.1093/bja/aep129. [DOI] [PubMed] [Google Scholar]
- 21.Gentilucci L, Squassabia F, Artali R. Re-discussion of the importance of ionic interactions in stabilizing ligand-opioid receptor complex and in activating signal transduction. Curr. Drug Targets. 2007;8:185–196. doi: 10.2174/138945007779315704. [DOI] [PubMed] [Google Scholar]
- 22.van Rijn RM, Whistler JL. The δ1 opioid receptor is a heterodimer that opposes the actions of the δ2 receptor on alcohol intake. Biol. Psychiatry. 2009;66:777–784. doi: 10.1016/j.biopsych.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decaillot FM, Rozenfeld R, Gupta A, Devi LA. Cell surface targeting of μ-δ opioid receptor heterodimers by RTP4. Proc. Natl. Acad. Sci. USA. 2008;105:16045–16050. doi: 10.1073/pnas.0804106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei LN, Loh HH. Regulation of opioid receptor expression. Curr. Opin. Pharmacol. 2002;2:69–75. doi: 10.1016/s1471-4892(01)00123-0. [DOI] [PubMed] [Google Scholar]
- 25.Elde R, Arvidsson U, Riedl M, Vulchanova L, Lee JH, et al. Distribution of neuropeptide receptors: new views of peptidergic neurotransmission made possible by antibodies to opioid receptors. Ann. N.Y. Acad. Sci. 1995;757:390–404. doi: 10.1111/j.1749-6632.1995.tb17497.x. [DOI] [PubMed] [Google Scholar]
- 26.Wei LN, Law PY, Loh HH. Post-transcriptional regulation of opioid receptors in the nervous system. Front. Biosci. 2004;9:1665–1679. doi: 10.2741/1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan YX. Diversity and complexity of the μ opioid receptor gene: alternative pre-mRNA splicing and promoters. DNA Cell Biol. 2005;24:736–750. doi: 10.1089/dna.2005.24.736. [DOI] [PubMed] [Google Scholar]
- 28.Choi HS, Kim CS, Hwang CK, Song KY, Wang W, et al. The opioid ligand binding of human μ-opioid receptor is modulated by novel splice variants of the receptor. Biochem. Biophys. Res. Commun. 2006;343:1132–1140. doi: 10.1016/j.bbrc.2006.03.084. [DOI] [PubMed] [Google Scholar]
- 29.Shabalina SA, Zaykin DV, Gris P, Ogurtsov AY, Gauthier J, et al. Expansion of the human μ-opioid receptor gene architecture: novel functional variants. Hum. Mol. Genet. 2009;18:1037–51. doi: 10.1093/hmg/ddn439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bi J, Tsai NP, Lin YP, Loh HH, Wei LN. Axonal mRNA transport and localized translational regulation of κ-opioid receptor in primary neurons of dorsal root ganglia. Proc. Natl. Acad. Sci. USA. 2006;103:19919–19924. doi: 10.1073/pnas.0607394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bi J, Tsai NP, Lu HY, Loh HH, Wei LN. Copb1-facilitated axonal transport and translation of κ opioid-receptor mRNA. Proc. Natl. Acad. Sci. USA. 2007;104:13810–13815. doi: 10.1073/pnas.0703805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai NP, Bi J, Wei LN. The adaptor Grb7 links netrin-1 signaling to regulation of mRNA translation. EMBO J. 2007;26:1522–1531. doi: 10.1038/sj.emboj.7601598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai NP, Tsui YC, Pintar JE, Loh HH, Wei LN. κ opioid receptor contributes to EGF-stimulated neurite extension in development. Proc. Natl. Acad. Sci. USA. 2010;107:3216–3221. doi: 10.1073/pnas.0912367107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law PY, Erickson LJ, El-Kouhen R, Dicker L, Solberg J, et al. Receptor density and recycling affect the rate of agonist-induced desensitization of μ-opioid receptor. Mol. Pharmacol. 2000;58:388–398. doi: 10.1124/mol.58.2.388. [DOI] [PubMed] [Google Scholar]
- 35.Lu S, Loh HH, Wei LN. Studies of dual promoters of mouse κ-opioid receptor gene. Mol. Pharmacol. 1997;52:415–420. doi: 10.1124/mol.52.3.415. [DOI] [PubMed] [Google Scholar]
- 36.Ko JL, Minnerath SR, Loh HH. Dual promoters of mouse μ-opioid receptor gene. Biochem. Biophys. Res. Commun. 1997;234:351–357. doi: 10.1006/bbrc.1997.6640. [DOI] [PubMed] [Google Scholar]
- 37.Wei LN, Hu X, Bi J, Loh H. Post-transcriptional regulation of mouse κ-opioid receptor expression. Mol. Pharmacol. 2000;57:401–408. [PubMed] [Google Scholar]
- 38.Song KY, Hwang CK, Kim CS, Choi HS, Law PY, et al. Translational repression of mouse μ opioid receptor expression via leaky scanning. Nucleic Acids Res. 2007;35:1501–1513. doi: 10.1093/nar/gkm034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim CS, Hwang CK, Song KY, Choi HS, Kim DK, et al. Novel function of neuron-restrictive silencer factor (NRSF) for posttranscriptional regulation. Biochim. Biophys. Acta. 2008;1783:1835–1846. doi: 10.1016/j.bbamcr.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 40.Song KY, Choi HS, Hwang CK, Kim CS, Law PY, et al. Differential use of an in-frame translation initiation codon regulates human μ opioid receptor (OPRM1) Cell. Mol. Life Sci. 2009;66:2933–2942. doi: 10.1007/s00018-009-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu X, Bi J, Loh HH, Wei LN. Regulation of mouse κ opioid receptor gene expression by different 3′-untranslated regions and the effect of retinoic acid. Mol. Pharmacol. 2002;62:881–887. doi: 10.1124/mol.62.4.881. [DOI] [PubMed] [Google Scholar]
- 42.Wu Q, Hwang CK, Yao S, Law PY, Loh HH, Wei LN. Amajor species of mouse μ-opioid receptor mRNA and its promoter-dependent functional polyadenylation signal. Mol. Pharmacol. 2005;68:279–285. doi: 10.1124/mol.105.012567. [DOI] [PubMed] [Google Scholar]
- 43.Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW. Involvement of exon 11-associated variants of the μ opioid receptor MOR-1 in heroin, but not morphine, actions. Proc. Natl. Acad. Sci. USA. 2009;106:4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuferov V, Fussell D, LaForge KS, Nielsen DA, Gordon D, et al. Redefinition of the human κ opioid receptor gene (OPRK1) structure and association of haplotypes with opiate addiction. Pharmacogenetics. 2004;14:793–804. doi: 10.1097/00008571-200412000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai NP, Bi J, Loh HH, Wei LN. Netrin-1 signaling regulates de novo protein synthesis of κ opioid receptor by facilitating polysomal partition of its mRNA. J. Neurosci. 2006;26:9743–9749. doi: 10.1523/JNEUROSCI.3014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai NP, Lin YL, Tsui YC, Wei LN. Dual action of epidermal growth factor: extracellular signal-stimulated nuclear–cytoplasmic export and coordinated translation of selected messenger RNA. J. Cell Biol. 2010;188:325–333. doi: 10.1083/jcb.200910083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaufman DL, Keith DE, Jr, Anton B, Tian J, Magendzo K, et al. Characterization of the murine μ opioid receptor gene. J. Biol. Chem. 1995;270:15877–15883. doi: 10.1074/jbc.270.26.15877. [DOI] [PubMed] [Google Scholar]
- 48.Xu J, Xu M, Pan YX. Characterizing exons 11 and 1 promoters of the μ opioid receptor (Oprm) gene in transgenic mice. BMC Mol. Biol. 2006;7:41–56. doi: 10.1186/1471-2199-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraus J, Borner C, Lendeckel U, Hollt V. Interferon-γ down-regulates transcription of the μ-opioid receptor gene in neuronal and immune cells. J. Neuroimmunol. 2006;181:13–18. doi: 10.1016/j.jneuroim.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 50.Wendel B, Hoehe MR. The human μ opioid receptor gene: 5′ regulatory and intronic sequences. J. Mol. Med. 1998;76:525–532. doi: 10.1007/s001090050246. [DOI] [PubMed] [Google Scholar]
- 51.Xu Y, Carr LG. Binding of Sp1/Sp3 to the proximal promoter of the hMOR gene is enhanced by DAMGO. Gene. 2001;274:119–128. doi: 10.1016/s0378-1119(01)00624-2. [DOI] [PubMed] [Google Scholar]
- 52.Befort K, Mattei MG, Roeckel N, Kieffer B. Chromosomal localization of the δ opioid receptor gene to human 1p34.3-p36.1 and mouse 4D bands by in situ hybridization. Genomics. 1994;20:143–145. doi: 10.1006/geno.1994.1146. [DOI] [PubMed] [Google Scholar]
- 53.Uhl GR, Childers S, Pasternak G. An opiate-receptor gene family reunion. Trends Neurosci. 1994;17:89–93. doi: 10.1016/0166-2236(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 54.Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, et al. κ-opioid receptor in humans: cDNAand genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc. Natl. Acad. Sci. USA. 1995;92:7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasuda K, Raynor K, Kong H, Breder CD, Takeda J, et al. Cloning and functional comparison of κ and δ opioid receptors from mouse brain. Proc. Natl. Acad. Sci. USA. 1993;90:6736–6740. doi: 10.1073/pnas.90.14.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kozak CA, Filie J, Adamson MC, Chen Y, Yu L. Murine chromosomal location of the μ and κ opioid receptor genes. Genomics. 1994;21:659–661. doi: 10.1006/geno.1994.1331. [DOI] [PubMed] [Google Scholar]
- 57.Giros B, Pohl M, Rochelle JM, Seldin MF. Chromosomal localization of opioid peptide and receptor genes in the mouse. Life Sci. 1995;56:PL369–PL375. doi: 10.1016/0024-3205(95)00119-q. [DOI] [PubMed] [Google Scholar]
- 58.Hu X, Cao S, Loh HH, Wei LN. Promoter activity of mouse κ opioid receptor gene in transgenic mouse. Brain Res. Mol. Brain Res. 1999;69:35–43. doi: 10.1016/s0169-328x(99)00077-7. [DOI] [PubMed] [Google Scholar]
- 59.Bi J, Hu X, Loh HH, Wei LN. Regulation of mouse κ opioid receptor gene expression by retinoids. J. Neurosci. 2001;21:1590–1599. doi: 10.1523/JNEUROSCI.21-05-01590.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Im HJ, Smirnov D, Yuhi T, Raghavan S, Olsson JE, et al. Transcriptional modulation of mouse μ-opioid receptor distal promoter activity by Sox18. Mol. Pharmacol. 2001;59:1486–1496. doi: 10.1124/mol.59.6.1486. [DOI] [PubMed] [Google Scholar]
- 61.Hwang CK, Wu X, Wang G, Kim CS, Loh HH. Mouse μ opioid receptor distal promoter transcriptional regulation by SOX proteins. J. Biol. Chem. 2003;278:3742–3750. doi: 10.1074/jbc.M208780200. [DOI] [PubMed] [Google Scholar]
- 62.Ko JL, Liu HC, Loh HH. Role of an AP-2-like element in transcriptional regulation of mouse μ-opioid receptor gene. Brain Res. Mol. Brain Res. 2003;112:153–162. doi: 10.1016/s0169-328x(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 63.Ko JL, Liu HC, Minnerath SR, Loh HH. Transcriptional regulation of mouse μ-opioid receptor gene. J. Biol. Chem. 1998;273:27678–27685. doi: 10.1074/jbc.273.42.27678. [DOI] [PubMed] [Google Scholar]
- 64.Kim SS, Pandey KK, Choi HS, Kim SY, Law PY, et al. Poly(C) binding protein family is a transcription factor in μ-opioid receptor gene expression. Mol. Pharmacol. 2005;68:729–736. doi: 10.1124/mol.105.012245. [DOI] [PubMed] [Google Scholar]
- 65.Lee PW, Lee YM. Transcriptional regulation of μ opioid receptor gene by cAMP pathway. Mol. Pharmacol. 2003;64:1410–1418. doi: 10.1124/mol.64.6.1410. [DOI] [PubMed] [Google Scholar]
- 66.Liang Y, Carr LG. Identification of an octamer-1 transcription factor binding site in the promoter of the mouse μ-opioid receptor gene. J. Neurochem. 1996;67:1352–1359. doi: 10.1046/j.1471-4159.1996.67041352.x. [DOI] [PubMed] [Google Scholar]
- 67.Choe C, Im HJ, Ko JL, Loh HH. Mouse μ opioid receptor gene expression: a 34-base pair cis-acting element inhibits transcription of the μ opioid receptor gene from the distal promoter. J. Biol. Chem. 1998;273:34926–34932. doi: 10.1074/jbc.273.52.34926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang CK, Kim CS, Choi HS, McKercher SR, Loh HH. Transcriptional regulation of mouse μ opioid receptor gene by PU.1. J. Biol. Chem. 2004;279:19764–19774. doi: 10.1074/jbc.M400755200. [DOI] [PubMed] [Google Scholar]
- 69.Choi HS, Hwang CK, Kim CS, Song KY, Law PY, et al. Transcriptional regulation of mouse μ opioid receptor gene in neuronal cells by poly(ADP-ribose) polymerase-1. J. Cell. Mol. Med. 2008;12:2319–2333. doi: 10.1111/j.1582-4934.2008.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi HS, Hwang CK, Kim CS, Song KY, Law PY, et al. Transcriptional regulation of mouse μ opioid receptor gene: Sp3 isoforms (M1, M2) function as repressors in neuronal cells to regulate the μ opioid receptor gene. Mol. Pharmacol. 2005;67:1674–1683. doi: 10.1124/mol.104.008284. [DOI] [PubMed] [Google Scholar]
- 71.Kim CS, Hwang CK, Choi HS, Song KY, Law PY, et al. Neuron-restrictive silencer factor (NRSF) functions as a repressor in neuronal cells to regulate the μ opioid receptor gene. J. Biol. Chem. 2004;279:46464–46473. doi: 10.1074/jbc.M403633200. [DOI] [PubMed] [Google Scholar]
- 72.Kim CS, Choi HS, Hwang CK, Song KY, Lee BK, et al. Evidence of the neuron-restrictive silencer factor (NRSF) interaction with Sp3 and its synergic repression to the μ opioid receptor (MOR) gene. Nucleic Acids Res. 2006;34:6392–63403. doi: 10.1093/nar/gkl724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Borner C, Hollt V, Kraus J. Involvement of activator protein-1 in transcriptional regulation of the human μ-opioid receptor gene. Mol. Pharmacol. 2002;61:800–805. doi: 10.1124/mol.61.4.800. [DOI] [PubMed] [Google Scholar]
- 74.Kraus J, Borner C, Giannini E, Hollt V. The role of nuclear factor κB in tumor necrosis factor-regulated transcription of the human μ-opioid receptor gene. Mol. Pharmacol. 2003;64:876–884. doi: 10.1124/mol.64.4.876. [DOI] [PubMed] [Google Scholar]
- 75.Borner C, Woltje M, Hollt V, Kraus J. STAT6 transcription factor binding sites with mismatches within the canonical 5′-TTC…GAA-3′ motif involved in regulation of δ- and μ-opioid receptors. J. Neurochem. 2004;91:1493–1500. doi: 10.1111/j.1471-4159.2004.02846.x. [DOI] [PubMed] [Google Scholar]
- 76.Bedini A, Baiula M, Spampinato S. Transcriptional activation of human μ-opioid receptor gene by insulin-like growth factor-I in neuronal cells is modulated by the transcription factor REST. J. Neurochem. 2008;105:2166–2178. doi: 10.1111/j.1471-4159.2008.05303.x. [DOI] [PubMed] [Google Scholar]
- 77.Borner C, Hollt V, Kraus J. Cannabinoid receptor type 2 agonists induce transcription of the μ-opioid receptor gene in Jurkat T cells. Mol. Pharmacol. 2006;69:1486–1491. doi: 10.1124/mol.105.018325. [DOI] [PubMed] [Google Scholar]
- 78.Kraus J, Borner C, Giannini E, Hickfang K, Braun H, et al. Regulation of μ-opioid receptor gene transcription by interleukin-4 and influence of an allelic variation within a STAT6 transcription factor binding site. J. Biol. Chem. 2001;276:43901–43908. doi: 10.1074/jbc.M107543200. [DOI] [PubMed] [Google Scholar]
- 79.Borner C, Kraus J, Bedini A, Schraven B, Hollt V. T-cell receptor/CD28-mediated activation of human T lymphocytes induces expression of functional μ-opioid receptors. Mol. Pharmacol. 2008;74:496–504. doi: 10.1124/mol.108.046029. [DOI] [PubMed] [Google Scholar]
- 80.Liu H, Li H, Guo L, Li M, Li C, et al. Mechanisms involved in phosphatidylinositol 3-kinase pathway mediated up-regulation of the μ opioid receptor in lymphocytes. Biochem. Pharmacol. 2010;79:516–523. doi: 10.1016/j.bcp.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 81.Ono T, Kaneda T, Muto A, Yoshida T. Positive transcriptional regulation of the human μ opioid receptor gene by poly(ADP-ribose) polymerase-1 and increase of its DNA binding affinity based on polymorphism of G−172 → T. J. Biol. Chem. 2009;284:20175–20183. doi: 10.1074/jbc.M109.019414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu Y, Carr LG. Transcriptional regulation of the human μ opioid receptor (hMOR) gene: evidence of positive and negative cis-acting elements in the proximal promoter and presence of a distal promoter. DNA Cell Biol. 2001;20:391–402. doi: 10.1089/104454901750361451. [DOI] [PubMed] [Google Scholar]
- 83.Andria ML, Simon EJ. Identification of a neurorestrictive suppressor element (NRSE) in the human μ-opioid receptor gene. Brain Res. Mol. Brain Res. 2001;91:73–80. doi: 10.1016/s0169-328x(01)00124-3. [DOI] [PubMed] [Google Scholar]
- 84.Hwang CK, Kim CS, Kim DK, Law PY, Wei LN, Loh HH. Up-regulation of μ opioid receptor gene is mediated through chromatin remodeling and transcriptional factors in differentiated neuronal cells. Mol. Pharmacol. 2010;78:58–68. doi: 10.1124/mol.110.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Borner C, Kraus J, Schroder H, Ammer H, Hollt V. Transcriptional regulation of the human μ-opioid receptor gene by interleukin-6. Mol. Pharmacol. 2004;66:1719–1726. doi: 10.1124/mol.104.003806. [DOI] [PubMed] [Google Scholar]
- 86.Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, et al. Evidence of endogenous μ opioid receptor regulation by epigenetic control of the promoters. Mol. Cell. Biol. 2007;27:4720–4736. doi: 10.1128/MCB.00073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun P, Loh HH. Transcriptional regulation of mouse δ-opioid receptor gene: role of Ikaros in the stimulated transcription of mouse δ-opioid receptor gene in activated T cells. J. Biol. Chem. 2002;277:12854–12860. doi: 10.1074/jbc.M112268200. [DOI] [PubMed] [Google Scholar]
- 88.Sun P, Loh HH. Transcriptional regulation of mouse δ-opioid receptor gene: Ikaros-2 and upstream stimulatory factor synergize in trans-activating mouse δ-opioid receptor gene in T cells. J. Biol. Chem. 2003;278:2304–2308. doi: 10.1074/jbc.M208162200. [DOI] [PubMed] [Google Scholar]
- 89.Smirnov D, Im HJ, Loh HH. δ-opioid receptor gene: effect of Sp1 factor on transcriptional regulation in vivo. Mol. Pharmacol. 2001;60:331–340. doi: 10.1124/mol.60.2.331. [DOI] [PubMed] [Google Scholar]
- 90.Sun P, Loh HH. Transcriptional regulation of mouse δ-opioid receptor gene: role of Ets-1 in the transcriptional activation of mouse δ-opioid receptor gene. J. Biol. Chem. 2001;276:45462–45469. doi: 10.1074/jbc.M104793200. [DOI] [PubMed] [Google Scholar]
- 91.Liu HC, Shen JT, Augustin LB, Ko JL, Loh HH. Transcriptional regulation of mouse δ-opioid receptor gene. J. Biol. Chem. 1999;274:23617–23626. doi: 10.1074/jbc.274.33.23617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Woltje M, Kraus J, Hollt V. Regulation of mouse δ-opioid receptor gene transcription: involvement of the transcription factors AP-1 and AP-2. J. Neurochem. 2000;74:1355–1362. doi: 10.1046/j.1471-4159.2000.0741355.x. [DOI] [PubMed] [Google Scholar]
- 93.Wang G, Wei LN, Loh HH. Transcriptional regulation of mouse δ-opioid receptor gene by CpG methylation: involvement of Sp3 and a methyl-CpG-binding protein, MBD2, in transcriptional repression of mouse δ-opioid receptor gene in Neuro2A cells. J. Biol. Chem. 2003;278:40550–40556. doi: 10.1074/jbc.M302879200. [DOI] [PubMed] [Google Scholar]
- 94.Wang G, Liu T, Wei LN, Law PY, Loh HH. DNA methylation-related chromatin modification in the regulation of mouse δ-opioid receptor gene. Mol. Pharmacol. 2005;67:2032–2039. doi: 10.1124/mol.105.011056. [DOI] [PubMed] [Google Scholar]
- 95.Chen YL, Law PY, Loh HH. Sustained activation of phosphatidylinositol 3-kinase/Akt/nuclear factor-κB signaling mediates G protein–coupled δ-opioid receptor gene expression. J. Biol. Chem. 2006;281:3067–3074. doi: 10.1074/jbc.M506721200. [DOI] [PubMed] [Google Scholar]
- 96.Chen HC, Wei LN, Loh HH. Expression of μ-, κ- and δ-opioid receptors in P19 mouse embryonal carcinoma cells. Neuroscience. 1999;92:1143–1155. doi: 10.1016/s0306-4522(99)00030-5. [DOI] [PubMed] [Google Scholar]
- 97.Park SW, Li J, Loh HH, Wei LN. A novel signaling pathway of nitric oxide on transcriptional regulation of mouse κ opioid receptor gene. J. Neurosci. 2002;22:7941–7947. doi: 10.1523/JNEUROSCI.22-18-07941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park SW, Wei LN. Regulation of c-myc gene by nitric oxide via inactivating NF-κB complex in P19 mouse embryonal carcinoma cells. J. Biol. Chem. 2003;278:29776–29782. doi: 10.1074/jbc.M303306200. [DOI] [PubMed] [Google Scholar]
- 99.Li J, Park SW, Loh HH, Wei LN. Induction of the mouse κ-opioid receptor gene by retinoic acid in P19 cells. J. Biol. Chem. 2002;277:39967–39972. doi: 10.1074/jbc.M200840200. [DOI] [PubMed] [Google Scholar]
- 100.Park SW, He Y, Ha SG, Loh HH, Wei LN. Epigenetic regulation of κ opioid receptor gene in neuronal differentiation. Neuroscience. 2008;151:1034–1041. doi: 10.1016/j.neuroscience.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu X, Bi J, Loh HH, Wei LN. An intronic Ikaros-binding element mediates retinoic acid suppression of the κ opioid receptor gene, accompanied by histone deacetylation on the promoters. J. Biol. Chem. 2001;276:4597–4603. doi: 10.1074/jbc.M005477200. [DOI] [PubMed] [Google Scholar]
- 102.Park SW, Huq MDM, Loh HH, Wei LN. Retinoic acid–induced chromatin remodeling of mouse κ opioid receptor gene. J. Neurosci. 2005;25:3350–3357. doi: 10.1523/JNEUROSCI.0186-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zeynalov E, Nemoto M, Hurn PD, Koehler RC, Bhardwaj A. Neuroprotective effect of selective κ opioid receptor agonist is gender specific and linked to reduced neuronal nitric oxide. J. Cereb. Blood Flow Metab. 2006;26:414–420. doi: 10.1038/sj.jcbfm.9600196. [DOI] [PubMed] [Google Scholar]
- 104.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 105.Rideout WM, III, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science. 2001;293:1093–1098. doi: 10.1126/science.1063206. [DOI] [PubMed] [Google Scholar]
- 106.Kiefer JC. Epigenetics in development. Dev. Dyn. 2007;236:1144–1156. doi: 10.1002/dvdy.21094. [DOI] [PubMed] [Google Scholar]
- 107.Ross SA, Milner JA. Epigenetic modulation and cancer: effect of metabolic syndrome? Am. J. Clin. Nutr. 2007;86:S872–S877. doi: 10.1093/ajcn/86.3.872S. [DOI] [PubMed] [Google Scholar]
- 108.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 109.Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell. Mol. Life Sci. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Duman RS, Newton SS. Epigenetic marking and neuronal plasticity. Biol. Psychiatry. 2007;62:1–3. doi: 10.1016/j.biopsych.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 111.Hsieh J, Gage FH. Epigenetic control of neural stem cell fate. Curr. Opin. Genet. Dev. 2004;14:461–469. doi: 10.1016/j.gde.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 112.Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim. Biophys. Acta. 2009;1790:863–868. doi: 10.1016/j.bbagen.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 113.Bartova E, Krejci J, Harnicarova A, Galiova G, Kozubek S. Histone modifications and nuclear architecture: a review. J. Histochem. Cytochem. 2008;56:711–721. doi: 10.1369/jhc.2008.951251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Henkels CH, Khorasanizadeh S. Implications of a histone code mimic in epigenetic signaling. Mol. Cell. 2007;27:521–522. doi: 10.1016/j.molcel.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 115.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 116.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 117.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 118.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009;66:596–612. doi: 10.1007/s00018-008-8432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Collas P. The state-of-the-art of chromatin immunoprecipitation. Methods Mol. Biol. 2009;567:561–25. doi: 10.1007/978-1-60327-414-2_1. [DOI] [PubMed] [Google Scholar]
- 120.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, et al. Epigenetic programming of μ-opioid receptor gene in mouse brain is regulated by MeCP2 and Brg1 chromatin remodeling factor. J. Cell. Mol. Med. 2008;13:3591–3615. doi: 10.1111/j.1582-4934.2008.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin YC, Flock KE, Cook RJ, Hunkele AJ, Loh HH, Ko JL. Effects of trichostatin A on neuronal μ-opioid receptor gene expression. Brain Res. 2008;1246:1–10. doi: 10.1016/j.brainres.2008.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Buzas B, Rosenberger J, Cox BM. μ and δ opioid receptor gene expression after chronic treatment with opioid agonist. Neuroreport. 1996;7:1505–1508. doi: 10.1097/00001756-199606170-00013. [DOI] [PubMed] [Google Scholar]
- 124.Andria ML, Simon EJ. Localization of promoter elements in the human μ-opioid receptor gene and regulation by DNA methylation. Brain Res. Mol. Brain Res. 1999;70:54–65. doi: 10.1016/s0169-328x(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 125.Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, et al. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873. doi: 10.1038/npp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MVL, Zukin RS. Ischemic insults promote epigenetic reprogramming of μ opioid receptor expression in hippocampal neurons. Proc. Natl. Acad. Sci. USA. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen YL, Law PY, Loh HH. NGF/PI3K signaling-mediated epigenetic regulation of δ opioid receptor gene expression. Biochem. Biophys. Res. Commun. 2008;368:755–60. doi: 10.1016/j.bbrc.2008.01.164. [DOI] [PMC free article] [PubMed] [Google Scholar]