

Abstract
Recent work has identified novel point mutations in isocitrate dehydrogenase 1 (IDH1) in the majority of the World Health Organization grades II and III infiltrative gliomas and secondary grade IV glioblastomas. Gangliogliomas consist of neoplastic ganglion and glial cells and, in contrast to infiltrative gliomas, are generally indolent. Yet distinguishing between a ganglioglioma and an infiltrative glioma with admixed gray matter can be difficult, perhaps accounting for some “gangliogliomas” that ultimately show aggressive behavior. In this multi‐institutional study, 98 cases originally diagnosed as ganglioglioma were analyzed for IDH1 mutations, 86 of which had follow‐up data available. Eight cases (8.2%) were positive for R132H IDH1 mutations; six had silent IDH2 mutations and two had nonsense IDH2 mutations. The presence of mutant IDH1 in gangliogliomas correlated with a greater risk of recurrence (P = 0.0007) and malignant transformation and/or death (P < 0.0001) compared with tumors that were IDH1 wild type. Furthermore, the age of patients with IDH1‐mutant gangliogliomas was higher than those without mutations (25.5 vs. 46.1 years, P = 0.0033). IDH1/2 testing of tumors suspected of being gangliogliomas may therefore be advisable, particularly in the adult population.
Keywords: ganglioglioma, glioneuronal, isocitrate dehydrogenase
INTRODUCTION
Recent whole genome analysis of glioblastomas identified a novel point mutation in isocitrate dehydrogenase type 1 (IDH1) and, to a lesser extent, in IDH2 2, 33, 46. The mutation, restricted to specific codons encoding one of the arginine residues responsible for binding to the isocitrate substrate (R132 in IDH1 and R172 in IDH2), is present in the majority of the World Health Organization (WHO) grade II and III astrocytomas and oligodendrogliomas, as well as grade IV glioblastomas arising from lower grade tumors (so‐called “secondary” glioblastomas). The majority of IDH1/2‐mutant gliomas occur in adults, although they can be seen in adolescents (36). This mutation nearly always exists as a heterozygote, with one wild‐type allele; similarly, concomitant mutation of both IDH1 and IDH2 is exceedingly rare (13). Consistent with these findings, other studies have shown that mutant IDH1 and IDH2 produce gain‐of‐function mutant enzymes that, instead of converting isocitrate to alpha‐ketoglutarate, convert alpha‐ketoglutarate to R‐2‐hydroxyglutarate 9, 12, 40. While the exact effects of this gain‐of‐function are still unclear, mutant IDH1/2 is tightly correlated with a CpG island hypermethylator profile, which has been shown to have a favorable prognostic effect in many gliomas 31, 39. It is also associated with 1p/19q codeletion in oligodendrogliomas and with TP53 mutations in astrocytomas, suggesting that IDH1/2 mutation is one of the earliest steps in gliomagenesis 20, 23, 33, 41, 42. Furthermore, the presence of mutant IDH1/2 has been consistently shown to be a favorable prognostic marker in all grades of infiltrative gliomas 30, 33, 37, 42, 43, 46.
IDH1/2 mutations are relatively specific to gliomas 3, 32, 46, with only rare reports of a few mutants (often not in the R132 codon) being detected in prostate carcinomas and paragangliomas, among others 11, 21, 29. One notable exception to this is acute myeloid leukemia, in which IDH1/2 mutations are seen in 20%–30% of cases with normal karyotype 1, 27, 28, 38, 40. Even within the glioma family, IDH1/2 mutations are mostly restricted to diffusely infiltrative gliomas. For example, the combination of IDH1/2 mutation and BRAF rearrangement assays may distinguish grade I pilocytic astrocytomas from higher‐grade infiltrative gliomas (22).
Like pilocytic astrocytoma, gangliogliomas are a low‐grade glioma that have a generally favorable prognosis; over 90% of patients have a recurrence‐free survival rate exceeding 7 years (25). Unfortunately, the diagnosis of ganglioglioma can be difficult, as it requires the neuropathologist to distinguish a true mixed glioneuronal neoplasm from an infiltrative glioma that contains admixed non‐neoplastic neurons. Radiology is not always helpful in differentiating between the two, as gangliogliomas do not always show a classic cyst with a contrast‐enhancing mural nodule, and enhancement is also seen in higher‐grade infiltrative gliomas. As these neoplasms are relatively uncommon, comprising approximately 1% of all brain tumors (24), data on molecular biomarkers with outcome is rare. One recent study detected IDH1 mutations in three of eight gangliogliomas (37), but impact on outcome was not available. Our prior work suggested that the presence of IDH1 mutation was an adverse prognostic marker in gangliogliomas (17), but a larger sample size was needed to draw definitive conclusions.
Herein we describe results from a multi‐institutional cohort of 98 gangliogliomas, including data on IDH1/2 mutation status and its association with outcome.
MATERIALS AND METHODS
Cohort
A retrospective cohort of 98 tumors originally diagnosed as “ganglioglioma” were obtained from the University of Kentucky, University of Pittsburgh, University of Rochester, Massachusetts General Hospital and the University of Pennsylvania (Table 1). This cohort included 12 cases previously described (17). Each case was individually reviewed by a respective neuropathologist at each institution, and then confirmed centrally by a single neuropathologist (J. Kofler). All unique identifiers were removed as per local Institutional Review Board specifications. Age and gender and, when available, tumor location, surgical procedure, adjuvant therapy and outcome were included. However, only 37 of the 98 cases had any data on surgical procedure, extent of resection and adjuvant therapy, rendering statistical analysis of such parameters unreliable. Any recurrence, progression, high‐grade transformation or death were considered “adverse outcomes,” analogous to criteria previously described 16, 18. For the purposes of WHO correlation in this study, any case wherein the original diagnosis conveyed the impression of “ganglioglioma with atypical features” was considered higher than WHO grade I. All cases were collected at the University of Pittsburgh, where J. Kofler scored mitoses per 10 high power fields via light microscopy.
Table 1.
Ganglioglioma cohort characteristics, according to IDH mutation status. Abbreviations: NA = not applicable; WHO = World Health Organization.
| Variable | IDH1 wild type | IDH1‐mutant | P |
|---|---|---|---|
| # Cases | 90 | 8 | NA |
| Age <20 years | 43 (47.8%) | 0 (0%) | 0.0087 |
| Age ≥20 years | 47 (52.2%) | 8 (100%) | |
| Male | 48 (53.3%) | 6 (75%) | 0.29 |
| Female | 42 (46.7%) | 2 (25%) | |
| Temporal lobe location* | 45/81 (55.6%) | 1/7 (14.3%) | 0.051 |
| Extratemporal location* | 36/81 (44.4%) | 6/7 (85.7%) | |
| WHO grade I | 79 (87.8%) | 3 (37.5%) | 0.0026 |
| “Atypical” features | 6 (6.7%) | 4 (50%) | |
| WHO grade III | 5 (5.6%) | 1 (12.5%) | |
| Follow‐up information available | 79 (87.8%) | 8 (100%) | NA |
Ninety‐eight gangliogliomas were collected. The median patient age was 21.5 years (range 1–77 years). Eighty‐six cases had follow‐up information available with a mean interval of 4.9 years (median 4.0 years). “Atypical” cases denote those that did not quite meet the criteria for a WHO III anaplastic ganglioglioma, but originally appeared more worrisome than WHO I tumors (see Results). P values were calculated using Fisher's exact test; in the case of WHO grade, comparison was made between WHO grade I and combining atypical and WHO grade III cases together. *The original tumor location was not retrievable in nine IDH1 wild‐type tumors and one IDH1‐mutant tumor.
Also included for R132H IDH1 immunohistochemical analysis (see further discussion) is a separate cohort of five oligodendrogliomas with ganglioglioma‐like foci (GGLF) that had been previously described (35) (Table 6). These cases, however, were not originally diagnosed as gangliogliomas and are not part of the patient outcome data analysis.
Table 6.
Mutant IDH1 is present in infiltrative gliomas with ganglioglioma‐like foci (GGLF).
| Case | Age | Sex | Location | Grade | 1p/19q status | Follow‐up | IDH1 status |
|---|---|---|---|---|---|---|---|
| 1 | 44 | Male | Right frontal | III | Codeletion | Radiologic tumor progression 2.5 years after surgery, still alive 2.7 years after surgery | Positive |
| 2 | 40 | Male | Right frontal‐parietal | III | Codeletion | Alive 4.9 years after surgery | Positive |
| 3 | 29 | Female | Left frontal | III | Codeletion | Recurred 2.2 years later, still alive 3.5 years later | Positive |
| 4 | 55 | Male | Right frontal | II | 1p‐deleted 19q normal | Alive 11 months after surgery | Negative |
| 5 | 42 | Male | Right frontal | III | 1 and 19 polysomy | Not applicable | Positive |
A separate cohort of cases previously described as infiltrative oligodendrogliomas with striking ganglioid morphology (35) were analyzed for R132H IDH1 mutation via immunohistochemistry. All four cases that were R132H IDH1‐positive showed expression in both the glial and ganglioid components (Figure 6).
IDH1/2 mutation analysis
IDH1/2 mutation analysis on FFPE tissues was performed using real‐time polymerase chain reaction (PCR) and post‐PCR fluorescence melting curve analysis and Sanger sequencing method as described previously 17, 19. Cases identified with R132H IDH1 were further subjected to immunohistochemical analysis using an R132H IDH1‐specific antibody 6, 7.
Interobserver histologic concordance analysis
To assess the interobserver reliability of hematoxylin and eosin (H&E) diagnosis of gangliogliomas, 18 cases were selected for analysis. Two of the 18 (cases 10 and 17, Table 5) were histologically classic gangliogliomas selected to ensure consistent interobserver evaluations, eight had R132H IDH1 mutations and another eight were selected based on their histologic ambiguity. All cases were independently reviewed by seven board‐certified neuropathologists, including two full professors, two associate professors and three assistant professors. Reviewers were blinded to age, tumor location, radiologic appearance, IDH1 status and patient outcome. Each reviewer rendered their opinion on whether the H&E material was most consistent with a ganglioglioma, an infiltrative glioma or whether the diagnosis was “uncertain/other.”“LM consensus” (Table 5) denotes what the most frequent diagnosis was in each case via light microscopy.
Table 5.
Interobserver variability in histologically difficult ganglioglioma‐vs.‐infiltrative glioma tumors. Abbreviations: Inf G = infiltrative glioma; GG = ganglioglioma; NA = not available; LM = light microscopy.
| Case | Reviewer 1 | Reviewer 2 | Reviewer 3 | Reviewer 4 | Reviewer 5 | Reviewer 6 | Reviewer 7 | LM consensus | IDH1 | Biological behavior thus far |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Inf G | GG | GG | GG | Inf G | Inf G | Inf G | Inf G | R132H | Inf G |
| 2 | Uncertain/other | Inf G | GG | GG | Inf G | Uncertain/other | Inf G | Inf G | Wild type | GG |
| 3 | Uncertain/other | Uncertain/other | Uncertain/other | GG | GG | Uncertain/other | Inf G | Uncertain/other | Wild type | NA |
| 4 | GG | GG | GG | GG | GG | Inf G | Uncertain/other | GG | R132H | Inf G |
| 5 | GG | GG | GG | GG | GG | Inf G | GG | GG | R132H | Inf G |
| 6 | Inf G | GG | GG | GG | GG | Inf G | GG | GG | Wild type | GG |
| 7 | GG | GG | GG | GG | Inf G | Uncertain/other | Inf G | GG | Wild type | NA |
| 8 | GG | GG | GG | Inf G | Inf G | Inf G | Inf G | Inf G | Wild type | GG |
| 9 | Inf G | Inf G | Inf G | GG | GG | Inf G | Inf G | Inf G | R132H | Inf G |
| 10 | GG | GG | GG | GG | GG | GG | GG | GG | Wild type | GG |
| 11 | GG | Uncertain/other | Inf G | GG | Uncertain/other | Inf G | Inf G | Inf G | R132H | Inf G |
| 12 | GG | GG | GG | Uncertain/other | Inf G | Inf G | Uncertain/other | GG | Wild type | Inf G |
| 13 | GG | Inf G | GG | Inf G | GG | Inf G | GG | GG | R132H | Inf G |
| 14 | GG | Inf G | GG | GG | GG | Inf G | GG | GG | R132H | GG |
| 15 | GG | GG | GG | GG | GG | GG | GG | GG | R132H | Inf G |
| 16 | Inf G | Inf G | Inf G | Inf G | Inf G | Inf G | Inf G | Inf G | Wild type | Inf G |
| 17 | GG | GG | GG | GG | GG | GG | GG | GG | Wild type | GG |
| 18 | Inf G | Inf G | Inf G | Inf G | GG | Uncertain/other | Inf G | Inf G | Wild type | Inf G |
Of the 98 cases originally diagnosed as “ganglioglioma,” 18 were selected for independent review, including all IDH1‐mutant tumors, by seven board‐certified neuropathologists (see Methods). Reviewers were blinded as to patient age, tumor location, IDH1 status and outcome; cases were chosen based on their histologic difficulty, with two classic pediatric temporal lobe gangliogliomas (cases 10 and 17) included as “controls.” There was a high rate of interobserver variability (percent of overall agreement = 55%; kappa = 0.33). Regarding recurrence, progression in grade and tumor‐related death as behaviors more consistent with an infiltrative glioma than a ganglioglioma, IDH mutation analysis correlated with known biological behavior in 12 of 16 cases, compared with 8 of 16 via light microscopic consensus.
Statistics
Fisher's exact test was used to compare relative risk (RR) between two groups. Multivariate analysis was performed using multiple regressions with outcome as the key variable. All statistical analyses were performed using InStat software version 3.06 (GraphPad, La Jolla, CA, USA). Free‐range kappa coefficient was calculated using a method described previously (4) (http://justusrandolph.net/kappa/).
RESULTS
Cohort characteristics
Ninety‐eight gangliogliomas were collected (Table 1). The median patient age was 21.5 years (range 1–77 years). Eighty‐six cases had follow‐up information available with a mean interval of 4.9 years (median 4.0 years). Ten cases were considered histologically “atypical,” with increased cellularity and/or nuclear atypia. Another six cases were diagnosed as anaplastic gangliogliomas; the remaining 82 cases were WHO grade I tumors. Of the 88 cases in which tumor location was known, 46 (52.3%) were located in the temporal lobes.
Eight cases (8.2%) were positive for the R132H IDH1 mutation. Eight unrelated cases had IDH2 mutations, but six were silent, including five G > C transversions at codon L143 (CTG > CTC) and one G > A transition at codon R172 (AGG > AGA). Of the six tumors with silent IDH2 mutations, two had patient‐matched non‐neoplastic tissue available for analysis. In both cases, the same CTG > CTC mutation at codon 143 was identified in the non‐neoplastic tissue (Figure 1A,B). The seventh and eighth IDH2‐mutant tumors were stop codon mutations: one at W164 (TGG > TGA) in a grade I ganglioglioma in the right temporal lobe of a 4‐year‐old female, with no evidence of tumor recurrence as of 6 years postsurgery; nearby non‐neoplastic cortical tissue was negative for that mutation (Figure 1C,D). The other nonsense IDH2 mutation occurred at L132 (TGG > TGA) in a grade I ganglioglioma in the right parietal lobe of a 9‐year‐old male whose tumor recurred 3 years after initial resection, but has had no further events in the past 10 years. No non‐neoplastic tissue was available from that patient.
Figure 1.
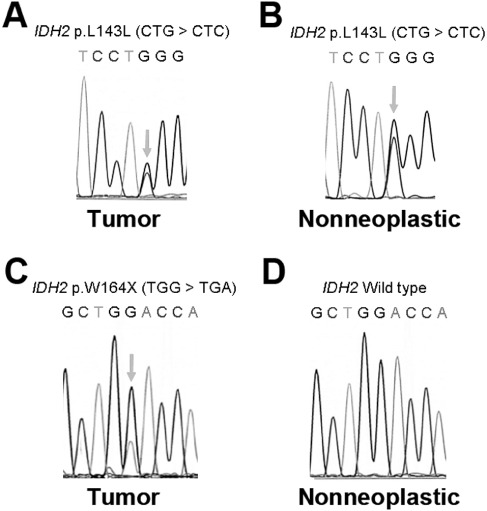
Silent and nonsense IDH2 mutations in gangliogliomas. A left frontal ganglioglioma in a 7‐year‐old male showed a silent L143L mutation in IDH2 (A), as did a separate patient‐matched scar tissue specimen (B). Identical findings in tumor and nontumor brain tissues were seen in a 2‐year‐old female with a left temporal ganglioglioma (not shown). In contrast, the nonsense W164X IDH2 mutation in a right temporal ganglioglioma from a 4‐year‐old female (C) was not seen in nearby non‐neoplastic cortical tissue (D).
Of note, one of the IDH1/2 wild‐type cases, a 52‐year‐old male with a cerebellar tumor, was suspected to have Lhermitte Duclos. His tumor had originally been excised at an outside hospital six years prior to the recurrent specimen. He presented for a re‐resection, which was the material included in this cohort, yet the histology was equivocal for Lhermitte Duclos (not shown).
Correlation of IDH1 status with clinical parameters, histology and outcome
The mean patient age of IDH1 wild‐type gangliogliomas was 25.5 years [95% confidence interval (CI) 21.7–29.4] vs. 46.1 years (95% CI 33.5–58.7) for IDH1‐mutated tumors (P = 0.0033). The youngest patient in this cohort with an IDH1 mutation was 20 years old; none of the 43 patients under 20 years had a mutation. Thirty‐six of 81 IDH1 wild‐type gangliogliomas (44.4%) were in a non‐temporal lobe area, whereas six of seven (85.7%) IDH1‐mutated gangliogliomas were located outside the temporal lobes (location was unknown in one case), trending towards significance (P = 0.051). While 48/90 (53.3%) IDH1 wild‐type patients were male, 6/8 (75%) of the IDH1‐mutated gangliogliomas were in males, though this was not significant (P = 0.29).
Concerning histology and WHO grade, of the 90 gangliogliomas wild type for IDH1, 79 were WHO grade I (Figure 2), six were considered “atypical,” and five others met the criteria for anaplastic ganglioglioma. In comparison, four of eight IDH1‐mutated gangliogliomas were considered atypical but not assigned a grade III, while one was grade III at the time of original diagnosis (P = 0.0026) (Table 2). A histologic grade higher than WHO I (ie, “atypical” or anaplastic grade III) also correlated with a higher risk of adverse outcome (P = 0.0046) and malignant transformation/death (P = 0074) (Table 3). Furthermore, 13.3% of IDH1 wild‐type tumors had identifiable mitoses in biopsy material, compared with 50.0% of IDH1 mutant tumors (P = 0.029). Cases with even just a single mitosis trended toward a higher risk of any adverse outcome (P = 0.060) or high‐grade progression/death (P = 0.061).
Figure 2.
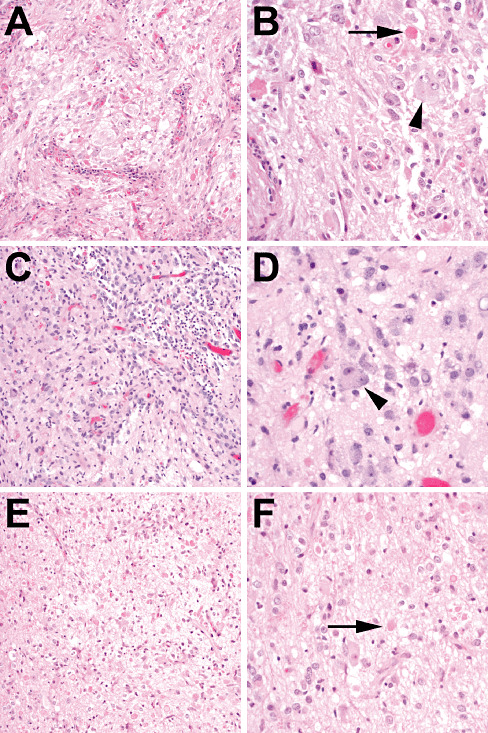
IDH1 wild‐type grade I gangliogliomas. (A,B) A 17‐year‐old male with a tumor in the right parietal lobe underwent resection 13 years ago, which revealed a ganglioglioma with binucleate neurons (arrowhead) and eosinophilic granular bodies (arrow). No further recurrences were reported. (C,D) A right temporal lobe mass from a 10‐year‐old male showed ganglion cells with binucleation (arrowhead) admixed with bland‐appearing glial elements. Even after gross total resection the tumor recurred 2 years later (E,F) but was still recognizable as a grade I ganglioglioma, with abundant ganglion cells and scattered eosinophilic granular bodies (arrow). Follow‐up since then (3 years after first recurrence) has shown no evidence of additional recurrences.
Table 2.
IDH1‐mutant tumors originally diagnosed as ganglioglioma. Abbreviations: DOD = died of disease; WHO = World Health Organization.
| Case | Age | Sex | WHO grade | Mitoses/hpf | Location | Outcome |
|---|---|---|---|---|---|---|
| 1 | 74 | Male | III | 4/10 | Right frontal | DOD 2.5 years later |
| 2 | 51 | Female | “Atypical features” | 0/10 | Left parietal | Recurrence 4 years later with malignant transformation to GBM; DOD 1 year after recurrence |
| 3 | 45 | Male | “Atypical features” | 1/10 | Right temporal | Recurrence 6 years later diagnosed as grade II‐III astrocytoma, treated with temozolomide and radiation |
| 4 | 45 | Male | “Atypical features” | 0/10 | Left frontal | Postsurgical radiation, recurrence 5 years later, progression to grade III oligodendroglioma two years after initial recurrence; DOD 13 years after original diagnosis |
| 5 | 52 | Male | I | 0/10 | Frontal | Radiation after surgery, recurrence 15 years after initial diagnosis; no follow‐up information since then |
| 6 | 20 | Male | I | 2/10 | Left posterior frontal lobe | Recurrence 5 years after diagnosis, treated with radiation |
| 7 | 44 | Male | I | 2/10 | Not specified | Alive 8 years after surgery, no additional information available |
| 8 | 38 | Female | “Atypical features” | 0/10 | Right parietal | Residual/recurrent tumor within 1 year of surgery, treated with radiation |
Eight of the 98 cases studied had IDH1 mutations; all were wild type for IDH2. Common features include advanced age and trends toward extratemporal location and mitoses. Most have already shown aggressive behavior.
Table 3.
Univariate analysis of relative risk of specific variables on outcome in gangliogliomas.
| All adverse outcomes | High‐grade progression/death | ||||
|---|---|---|---|---|---|
| Variable | RR (95% CI) | P | Variable | RR | P |
| IDH1‐mutant | 1.38 (1.05–1.81) | 0.0007 | IDH1‐mutant | 2.89 (0.93–8.96) | 0.0002 |
| WHO grade >I | 1.47 (1.07–2.01) | 0.0046 | WHO grade >I | 4.09 (0.71–23.7) | 0.0074 |
| Any mitoses | 1.61 (0.92–2.01) | 0.060 | Age ≥20 years | ∞ | 0.0055 |
| Non‐temporal | 1.37 (0.80–2.35) | 0.31 | Any mitoses | 1.20 (0.93–1.55) | 0.061 |
| Male gender | 1.17 (0.64–2.15) | 0.62 | Male gender | 1.31 (0.41–4.18) | 0.70 |
| Age ≥20 years | 1.07 (0.64–1.77) | 1.00 | Non‐temporal | 1.10 (0.48–2.51) | 1.00 |
Outcomes of 86 gangliogliomas were scored for either all adverse outcome or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance of relative risk (RR; via Fisher's exact test) in correlating with each type of outcome. (∞ = no case under 20 years old showed high‐grade progression or death.) Of note, the World Health Organization (WHO) grade >I group includes all gangliogliomas with “atypical” features as well as grade III anaplastic gangliogliomas (Table 1).
The presence of mutant IDH1 in gangliogliomas correlated with a greater risk of any adverse outcome (P = 0.0007), including high‐grade transformation and/or death (P = 0.0002) compared with tumors that were IDH1 wild type (Table 3) (Figure 3). On Kaplan–Meier analysis of survival over time, IDH1 mutant gangliogliomas showed shorter recurrence‐free survival, higher rate of malignant transformation and shorter overall survival (Figure 4). On multivariate analysis, only IDH1 mutation (P = 0.001) was an independent risk factor for any adverse outcome (Table 4). Considering only high‐grade transformation and/or death on multivariate analysis, IDH1 status was the most powerful risk factor (P < 0.0001) followed by age (P = 0.025) (Table 4). Neither mitoses, gender, tumor location nor WHO grade emerged as a independent risk factor for any kind of adverse outcome or specifically for high‐grade transformation and/or death.
Figure 3.
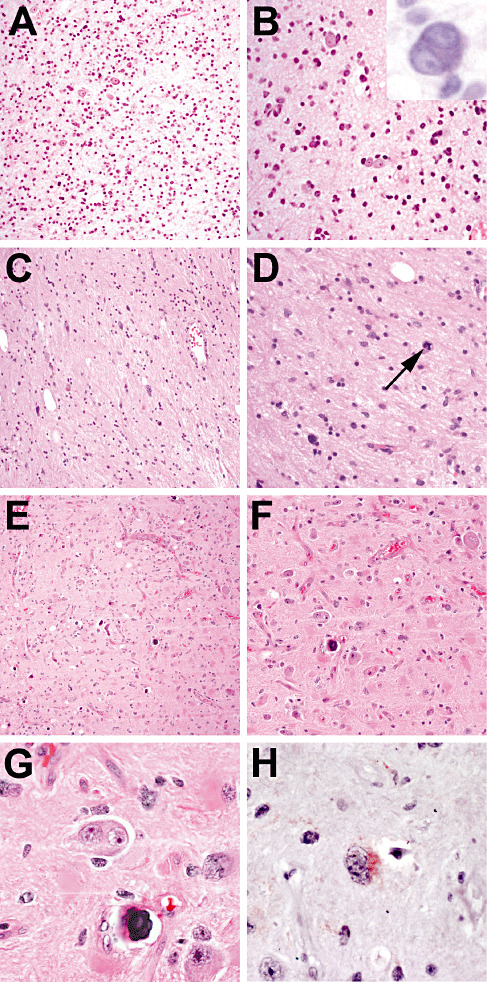
IDH1‐mutated tumors originally diagnosed as gangliogliomas. A 45‐year‐old male underwent incomplete resection of a right temporal mass (A,B) that showed disorganized neurons admixed with atypical glial cells. Occasional binucleated neurons were present (B, inset, cresyl violet stain). The residual tumor was treated with radiation. Six years later, the tumor progressed to a WHO grade III anaplastic astrocytoma (C,D) with mitoses (D, arrow). Both the original and recurrent tumors were positive for an R132H IDH1 mutation via polymerase chain reaction (PCR) (insufficient remaining tissue for immunohistochemical analysis). Another case was of a 38‐year‐old female who had a right parietal tumor incompletely resected (E,F). The tumor consisted mostly of abnormal‐appearing ganglion cells, frequently binucleated (G). Both via PCR and an R132H‐specific anti‐IDH1 antibody (H), this tumor contained an R132H IDH1 mutation. The follow‐up interval has been less than a year, yet the patient has already required adjuvant radiation for residual/recurrent tumor.
Figure 4.
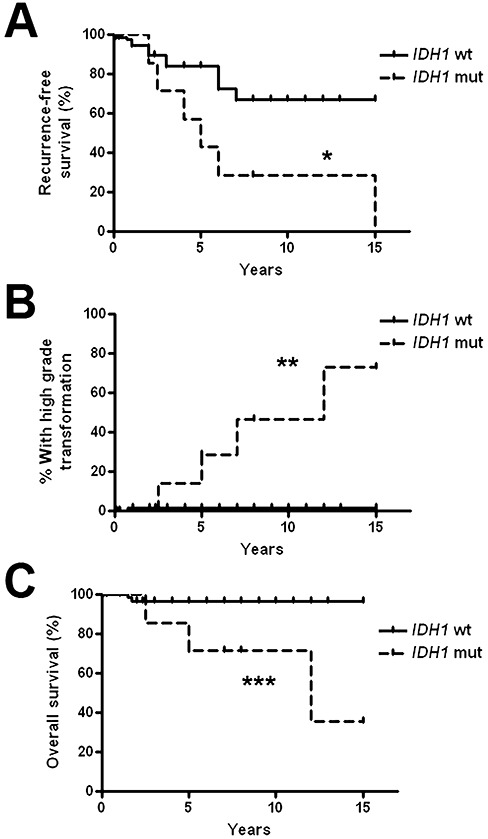
IDH1‐mutant “gangliogliomas” have a more aggressive clinical course than wild‐type gangliogliomas. Eighty‐six gangliogliomas with follow‐up information were assessed for recurrence‐free survival, rate of high‐grade progression and overall survival. As shown by Kaplan–Meier analysis, gangliogliomas with IDH1 mutations showed lower recurrence‐free survival (A), more frequent progression to high‐grade glioma (B) and shorter overall survival (C) compared with wild‐type gangliogliomas. *P = 0.014, **P < 0.0001, ***P = 0.0028 via Kaplan–Meier analysis.
Table 4.
Multivariate analysis of clinical, histologic and IDH mutation status in outcomes of gangliogliomas.
| Variable | All adverse outcomes | High‐grade progression/death |
|---|---|---|
| IDH1 status | P = 0.001 | P < 0.0001 |
| Age | P = 0.86 | P = 0.025 |
| Mitoses | P = 0.21 | P = 0.67 |
| World Health Organization grade | P = 0.47 | P = 0.46 |
| Location | P = 0.55 | P = 0.49 |
| Gender | P = 0.78 | P = 0.94 |
Outcomes of 86 gangliogliomas were scored for either all adverse outcomes or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance in correlating with each type of outcome. IDH1 mutation status was the only independent prognostic variable for all adverse outcomes, and was the most powerful variable for high‐grade progression and/or death.
Focusing on IDH1 wild‐type gangliogliomas, only the presence of mitoses correlated with increased risk of any adverse outcome on univariate analysis (RR 1.41, 95% CI 0.94–2.1, P = 0.024) (Supporting Information Table S1). Yet none of the variables showed significant correlation with malignant transformation and/or death. Neither age, gender, location, mitoses nor WHO grade was an independent prognostic factor for any adverse outcome (P = 0.4605) (Supporting Information Table S2). Age (P = 0.012) was the sole independent prognostic factor for malignant transformation and/or death in IDH1 wild‐type tumors; specifically, only two adult patients (55 and 62 years old) had such outcomes in this cohort.
The majority of recurrent IDH1 wild‐type tumors continued to exhibit low‐grade morphology; specifically, only one of the 14 recurrent IDH1 wild‐type tumors transformed into a high‐grade malignant glial neoplasm, consistent with glioblastoma. In that case, the transformed tumor maintained its wild‐type IDH1 status. In all four R132H IDH1‐mutated cases in which material from the recurrent/progressive tumor was available for testing, the same R132H mutation was detected in the recurrent material (not shown).
Immunohistochemical analysis using an R132H IDH1‐specific antibody allowed for more detailed study of what cells had the mutation. In most instances, the atypical ganglioid‐appearing cells that contributed to the diagnosis of “ganglioglioma” were in fact negative for R132H IDH1 (Figure 5). In only one case did ganglioid‐appearing cells show mutant IDH1 expression (Figure 3E–H).
Figure 5.
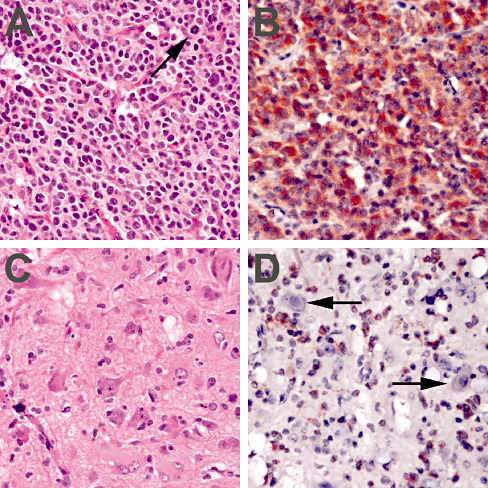
R132H IDH1 immunostain identifies admixed non‐neoplastic neurons in gliomas. A 74‐year‐old male underwent incomplete resection for a right frontal lobe mass that contained obvious glioma with mitoses (A, arrow), but also admixed disorganized neurons (C). Within 2 years the patient died from progressive disease. The original tumor was positive for IDH1 mutation on polymerase chain reaction. Mutant R132H IDH1 localization was seen only in the glial elements (B), not the neurons (D, arrows).
Interobserver reliability vs. IDH testing in difficult ganglioglioma cases
To assess whether H&E or IDH mutation analysis correlates better with clinical outcome in ambiguous cases, 18 of the 98 tumors originally diagnosed as ganglioglioma were selected for multiobserver testing, including all eight IDH1‐mutant tumors (see Methods). The other 10 were IDH1/2 wild‐type tumors, eight of which were considered histologically difficult plus two cases of “classic” gangliogliomas (cases 10 and 17, Table 5). Interobserver reliability in differentiating infiltrative gliomas from gangliogliomas was low, with an overall agreement of 55% and kappa = 0.33. Using clinical outcome as a marker (with the caveat that follow‐up information in some cases was incomplete), IDH mutation analysis matched behavior in 12 of 16 cases, ie, a tumor that was R132H IDH1‐positive showed recurrence, high‐grade transformation and/or tumor‐related death, whereas wild‐type cases were more apt to have no known recurrences postsurgery. In contrast, the consensus light microscopic diagnosis matched behavior in 8 of 16 cases.
IDH1 in gliomas with GGLF
Recent work has shown that oligodendrogliomas with GGLF behave more like infiltrative gliomas than gangliogliomas, and that the GGLF are really neoplastic glial cells with extensive ganglioid differentiation (35). This was done in part by showing that the 1p/19q molecular signature on FISH was the same in the glial and ganglioid cells. To extend these observations, we immunostained five of the original seven GGLF cases with the R132H IDH1 antibody (two had insufficient tissue). Four of the five cases showed immunoreactivity, including all three cases with 1p/19q codeletion (Table 6). In all four cases, both glial and ganglioid cells were positive for R132H IDH1 (Figure 6).
Figure 6.
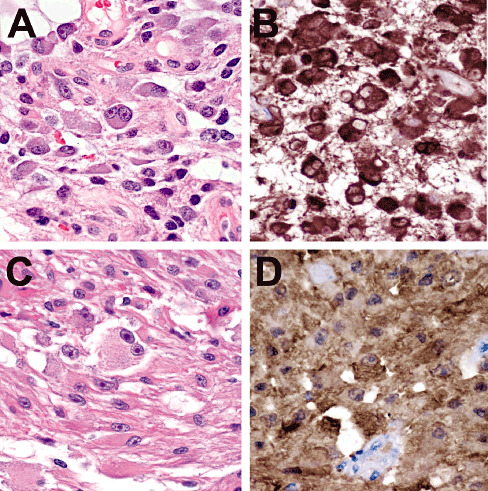
Oligodendrogliomas with ganglioglioma‐like foci (GGLF) show mutant IDH1 expression in both the glial and ganglioid components. A 42‐year‐old male had a right frontal lobe tumor (case 5 in Table 5) that showed ganglioid and glial components, including binucleate neuron‐like cells (A). Both cell types showed strong immunoreactivity for R132H IDH1 (B). Another right frontal tumor in a 44‐year‐old male also showed prominent binucleate ganglioid morphology (C) yet had R132H IDH1 expression (D). In both cases, the ganglioid cells were also positive for GFAP, synaptophysin and chromogranin, but negative for neurofilament protein, NeuN and CD34 (35).
DISCUSSION
We have demonstrated that IDH1 mutations occur in a minority of tumors diagnosed as gangliogliomas, and that such mutations are associated with advanced age, increased atypia and/or WHO grade and worse outcome. Trends were seen toward extratemporal location and increased mitoses. These data suggest that IDH1 mutation screening can refine the diagnosis and management of ganglioglioma, specifically distinguishing between a ganglioglioma, infiltrative glioma in gray matter and infiltrative glioma with focal gangliocytic differentiation.
Diagnosing a ganglioglioma is often challenging as it can show a wide range of appearances histologically. For example, the relative proportions of neuronal and glial elements in a ganglioglioma vary greatly, even within different regions of the same tumor, and thus are subject to sampling variability. “Classic” gangliogliomas have dysplastic neurons with malorientation, but that can be difficult to assess in a surgically manipulated, disoriented biopsy, especially in gray matter that is being overrun by infiltrative glioma. Neuronal binucleation is a helpful sign, but is not present in all cases. Other common histologic features such as eosinophilic granular bodies, Rosenthal fibres, lymphocytic infiltrates, calcifications, microcystic change and a prominent capillary network are also only variably present and of limited specificity. Even the pattern of glial growth varies from case to case, showing features of pilocytic astrocytoma, oligodendroglioma and/or fibrillary astrocytoma 24, 45.
Thus, differentiating a ganglioglioma from gray matter that has been distorted by invading glioma cells is not trivial, a fact underscored by the high interobserver variability seen in our dataset of difficult cases (Table 5). Yet the consequences of misdiagnosis can be significant, as a ganglioglioma is likely to be managed much more conservatively, focused mostly on follow‐up and local radiation if needed, whereas infiltrative gliomas are candidates for temozolomide and more aggressive radiotherapy. Calling an infiltrative glioma a ganglioglioma may therefore result in undertreatment.
This assumes, of course, that all cases originally diagnosed as ganglioglioma that ultimately behave aggressively were misinterpretations of pathologic material. Yet it is possible that some infiltrative gliomas simply show focal neuronal/gangliocytic differentiation (34). A set of such cases were recently described, in which otherwise obvious infiltrative gliomas with oligodendroglial and astrocytic features also showed focal ganglioid morphology (35). Thus, there is considerable histologic overlap between a low‐grade, potentially curable tumor that forms glial and neuronal elements and an infiltrative, incurable tumor composed primarily of glial cells that occasionally differentiate. Here, we show that mutant IDH1 helps differentiate between the two entities.
Of interest are the eight gangliogliomas that were wild type for IDH1 but had IDH2 mutations. The most common was a G > C transversion at codon 143, which is not listed as one of the known IDH2 single nucleotide polymorphisms in Entrez SNP yet was detected in non‐neoplastic tissue from both patients in which analysis was possible (Figure 1), suggesting that it is a germline polymorphism. This transversion is a silent CTG > CTC, preserving the lysine at codon 143. Similarly, the G > A transition at codon 172 seen in another ganglioglioma would maintain the arginine residue. However, the nonsense mutations at codons W164 and L132 would produce sharply truncated proteins (normally IDH2 is 452 amino acids long) and likely would be nonfunctional. The W164 mutation was not seen in nearby non‐neoplastic cortical tissue, suggesting it is a somatic mutation exclusive to the tumor. Neither of those gangliogliomas showed any notable atypia, either in histology or biological behavior. Still, to our knowledge, none of these polymorphisms or mutations has been previously identified in any infiltrative gliomas or gangliogliomas. Furthermore, to our knowledge no ganglioglioma has been identified with the neoenzymatic R172 IDH2 mutation described in other infiltrative gliomas. This is not surprising, considering that IDH2 mutations are far less common than IDH1 mutations, and only eight of our gangliogliomas even had an IDH1 mutation. Yet, as R172H IDH2 mutations do exist in diffusely infiltrative gliomas, carry the same gain‐of‐function consequences for the neoenzyme, and appear to carry the same prognostic strength as R132 IDH1 mutations, any “ganglioglioma” that is ultimately found to have an R172 IDH2 mutation may behave more like an infiltrative glioma.
The phenomenon of malignant progression of a ganglioglioma is well known, and is believed to occur in less than 5% of all cases (45). Some clinical and histologic features previously suggested as adverse markers include male gender, age over 40 years, no seizures in the clinical history, non‐temporal location, inability to completely resect and histologic atypia (e.g., nuclear pleomorphism, necrosis, increased mitoses) 10, 25, 26. The histologic criteria in particular have been incorporated into a distinct WHO grade III ganglioglioma, which is associated with worse survival (24). Beyond histology, molecular markers to enhance prognostic accuracy in gangliogliomas are few. Increased p53 immunostaining, which is suggestive of TP53 mutations and is characteristic of WHO grade II and III astrocytic tumors, may correlate with worse outcome in gangliogliomas (14). Array comparative genomic hybridization suggested that CDKN2A/B and DMBT1 deletion or CDK4 amplification were adverse markers (15). Detection of 1p/19q codeletion in “ganglioglioma” has been associated with outcomes more suggestive of WHO grade II or III oligodendroglial tumor containing focal ganglion‐like maturation, as opposed to a true ganglioglioma with oligodendroglial component (35). Our results support the contention that detection of grades II–IV molecular abnormalities in histologic gangliogliomas is an unfavorable prognostic finding. Moreover, as gliomas with true (ie, whole‐arm) 1p/19q codeletion apparently always carry IDH1 or IDH2 mutations (23), and IDH1/2 mutations associate with TP53 mutations in astrocytomas, testing for IDH1 in suspected gangliogliomas would not only detect most lower grade diffusely infiltrative astrocytomas, but also 1p/19q codeleted oligodendrogliomas.
The discovery of point mutation hotspots in IDH1 and IDH2 in gliomas has provided a number of new directions for further study in the field of neuro‐oncology. Not only are these enzymes now targets for research into mechanisms of gliomagenesis and improved therapeutics, but they can also help refine diagnostic and prognostic accuracy. This was demonstrated in our prior work, wherein peripheral biopsies indeterminate for glioma still showed IDH1/2 mutations via molecular analysis (17). Now, a monoclonal antibody is commercially available that detects the most common mutant enzyme, R132H IDH1, in paraffin‐embedded tissues, and can distinguish infiltrative glioma from non‐neoplastic mimickers 5, 6, 7. Another recently published paper showed that the IDH1 antibody could also differentiate oligodendrogliomas from dysembryoplastic neuroepithelial tumor, neurocytoma and clear cell ependymoma (8). While this antibody will not detect less common IDH1 or IDH2 mutations, it nonetheless serves as a good marker for the most common IDH1 variant, and has the additional virtue of specifically identifying which cells in a tumor are producing the mutant enzyme. In our cohort, this enhanced our understanding of just how “dysplastic” otherwise normal neurons can look when they are being overrun by glioma (Figure 5). The R132H IDH1 antibody is therefore fast becoming a staple of the neuropathologist's armamentarium, and can serve as an effective, low‐cost screening tool, reserving PCR analysis for immunonegative or equivocal cases. In the context of ganglioglioma vs. infiltrative glioma, it does appear to be more reliable than neurofilament immunostain, whose utility is unclear (44); in our cohort, neurofilament immunostain did not help in the discrimination between IDH1 wild‐type and mutant tumors (data not shown).
In summary, we present evidence supporting testing for IDH1 mutations in all gangliogliomas of adults, especially those adult tumors in a nontemporal location and/or containing any suspicious histologic features such as mitoses. Such testing is likely to help avoid misdiagnoses and undertreatment of infiltrative gliomas.
Supporting information
Table S1. Univariate analysis of relative risk of specific variables on outcome in IDH1 wild‐type gangliogliomas. Outcomes of 78 gangliogliomas were scored for either all adverse outcomes or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance of relative risk (RR, via Fisher's exact test) in correlating with each type of outcome. (∞ = no case under 20 years old showed high‐grade progression or death.) Of note, the “WHO grade >I group includes all gangliogliomas with “atypical” features as well as grade III anaplastic gangliogliomas.
Table S2. Multivariate analysis of clinical and histologic factors in outcomes of IDH1 wild‐type gangliogliomas. Outcomes of 78 gangliogliomas were scored for either all adverse outcome or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance in correlating with each type of outcome. Age was the only independent prognostic variable for high‐grade progression and/or death; no variable was independently significant for all adverse outcomes.
Supporting info item
ACKNOWLEDGMENTS
The authors thank Lindsey Kelly, Geeta Mantha, Mark Stauffer and Patty Cross for their technical expertise.
REFERENCES
- 1. Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A et al (2010) Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116:2122–2126. [DOI] [PubMed] [Google Scholar]
- 2. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 3. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP et al (2009) IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high‐grade gliomas but not in other solid tumors. Hum Mutat 30:7–11. [DOI] [PubMed] [Google Scholar]
- 4. Brennan RL, Prediger DJ (1981) Coefficient Kappa: some uses, misuses, and alternatives. Educ Psychol Meas 41:687–699. [Google Scholar]
- 5. Camelo‐Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL (2010) Mutant IDH1‐specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol 119:509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2009) Characterization of R132H mutation‐specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601. [DOI] [PubMed] [Google Scholar]
- 8. Capper D, Reuss D, Schittenhelm J, Hartmann C, Bremer J, Sahm F et al (2011) Mutation‐specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma‐like morphology. Acta Neuropathol 121:241–252. [DOI] [PubMed] [Google Scholar]
- 9. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2009) Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 462:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. El Khashab M, Gargan L, Margraf L, Koral K, Nejat F, Swift D et al (2009) Predictors of tumor progression among children with gangliogliomas. Clinical article. J Neurosurg Pediatr 3:461–466. [DOI] [PubMed] [Google Scholar]
- 11. Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF et al (2010) Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 95:1274–1278. [DOI] [PubMed] [Google Scholar]
- 12. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG et al (2010) Cancer‐associated metabolite 2‐hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474. [DOI] [PubMed] [Google Scholar]
- 14. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, VandenBerg SR (1997) Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer 79:989–1003. [PubMed] [Google Scholar]
- 15. Hoischen A, Ehrler M, Fassunke J, Simon M, Baudis M, Landwehr C et al (2008) Comprehensive characterization of genomic aberrations in gangliogliomas by CGH, array‐based CGH and interphase FISH. Brain Pathol 18:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horbinski C, Hamilton RL, Lovell C, Burnham J, Pollack IF (2009) Impact of morphology, MIB‐1, p53 and MGMT on outcome in pilocytic astrocytomas. Brain Pathol 20:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN (2009) Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin‐fixed, paraffin‐embedded glioma tissues. J Neuropathol Exp Neurol 68:1319–1325. [DOI] [PubMed] [Google Scholar]
- 18. Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF (2010) Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol 119:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horbinski C, Kelly L, Nikiforov YE, Durso MB, Nikiforova MN (2010) Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J Mol Diagn 12:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI et al (2009) Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125:353–355. [DOI] [PubMed] [Google Scholar]
- 22. Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H et al (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118:401–405. [DOI] [PubMed] [Google Scholar]
- 23. Labussiere M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C et al (2010) All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 74:1886–1890. [DOI] [PubMed] [Google Scholar]
- 24. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO classification of tumors of the central nervous system. In: World Health Organization Classification of Tumors, Ohgaki H (ed.), pp. 103–105. IARC: Lyon. [Google Scholar]
- 25. Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow‐up of 8 years. Cancer 101:146–155. [DOI] [PubMed] [Google Scholar]
- 26. Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M (2008) Tumor recurrence and malignant progression of gangliogliomas. Cancer 113:3355–3363. [DOI] [PubMed] [Google Scholar]
- 27. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D et al (2010) IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28:2348–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murugan AK, Bojdani E, Xing M (2010) Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun 393:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007. [DOI] [PubMed] [Google Scholar]
- 31. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park SW, Chung NG, Han JY, Eom HS, Lee JY, Yoo NJ, Lee SH (2009) Absence of IDH2 codon 172 mutation in common human cancers. Int J Cancer 125:2485–2486. [DOI] [PubMed] [Google Scholar]
- 33. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955. [DOI] [PubMed] [Google Scholar]
- 35. Perry A, Burton SS, Fuller GN, Robinson CA, Palmer CA, Resch L et al (2010) Oligodendroglial neoplasms with ganglioglioma‐like maturation: a diagnostic pitfall. Acta Neuropathol 120:237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons‐Weiler MA, Laframboise WA et al (2011) IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sonoda Y, Kumabe T, Nakamura T, Saito R, Kanamori M, Yamashita Y et al (2009) Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci 100:1996–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thol F, Damm F, Wagner K, Gohring G, Schlegelberger B, Hoelzer D et al (2010) Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 116:614–616. [DOI] [PubMed] [Google Scholar]
- 39. Toedt G, Barbus S, Wolter M, Felsberg J, Tews B, Blond F et al (2011) Molecular signatures classify astrocytic gliomas by IDH1 mutation status. Int J Cancer 128:1095–1103. [DOI] [PubMed] [Google Scholar]
- 40. Ward PS, Patel J, Wise DR, Abdel‐Wahab O, Bennett BD, Coller HA et al (2010) The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell 17:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J et al (2009) Molecular predictors of progression‐free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750. [DOI] [PubMed] [Google Scholar]
- 43. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F et al (2009) NOA‐04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880. [DOI] [PubMed] [Google Scholar]
- 44. Wierzba‐Bobrowicz T, Schmidt‐Sidor B, Gwiazda E, Bertrand E (1999) The significance of immunocytochemical markers, synaptophysin and neurofilaments in diagnosis of ganglioglioma. Folia Neuropathol 37:157–161. [PubMed] [Google Scholar]
- 45. Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD (1994) Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol 88:166–173. [DOI] [PubMed] [Google Scholar]
- 46. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Univariate analysis of relative risk of specific variables on outcome in IDH1 wild‐type gangliogliomas. Outcomes of 78 gangliogliomas were scored for either all adverse outcomes or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance of relative risk (RR, via Fisher's exact test) in correlating with each type of outcome. (∞ = no case under 20 years old showed high‐grade progression or death.) Of note, the “WHO grade >I group includes all gangliogliomas with “atypical” features as well as grade III anaplastic gangliogliomas.
Table S2. Multivariate analysis of clinical and histologic factors in outcomes of IDH1 wild‐type gangliogliomas. Outcomes of 78 gangliogliomas were scored for either all adverse outcome or strictly for high‐grade progression/death (see Methods). Variables were ranked according to statistical significance in correlating with each type of outcome. Age was the only independent prognostic variable for high‐grade progression and/or death; no variable was independently significant for all adverse outcomes.
Supporting info item