

Abstract
Familial hypercholesterolemia (FH) is a genetic disorder of lipoprotein metabolism characterized by high plasma concentrations of low-density lipoprotein cholesterol (LDLc), tendon xanthomas, and increased risk of premature coronary heart disease. FH is one of the most common inherited disorders; there are 10,000,000 people with FH worldwide, mainly heterozygotes. The most common FH cause is mutations along the entire gene that encode for LDL receptor (LDLR) protein, but it has been also described that mutations in apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 genes produce this phenotype. About 17%–33% of patients with a clinical diagnosis of monogenic hypercholesterolemia do not harbor any genetic cause in the known loci. Because FH has been considered as a public health problem, it is very important for an early diagnosis and treatment. Recent studies have demonstrated the influence of the LDLR mutation type in the FH phenotype, associating a more severe clinical phenotype and worse advanced carotid artherosclerosis in patients with null than those with receptor-defective mutations. Since 2004, a molecular FH diagnosis based on a genetic diagnostic platform (Lipochip®; Progenika-Biopharma, Derio, Spain) has been developed. This analysis completes the adequate clinical diagnosis made by physicians. Our group has recently proposed new FH guidelines with the intention to facilitate the FH diagnosis. The treatment for this disease is based on the benefit of lowering LDLc and a healthy lifestyle. Actually, drug therapy is focused on using statins and combined therapy with ezetimibe and statins. This review highlights the recent progress made in genetics, diagnosis, and treatment for FH.
Keywords: LDLR, APOB, PCSK9, LDL cholesterol
Introduction
The object of this review is to update the status of familial hypercholesterolemia (FH, MIMN#143890), with special consideration on the genetics and diagnosis. Heterozygous FH (heFH) is presented with high prevalence around the entire world, thus supposing elevated costs in health care.1 Although this disease has been exhaustively studied, new locus and mutations associated with FH are described each year. Taking all things together, it is important to research about the FH causes, and the effectiveness in the diagnosis and treatment.
FH is an autosomal codominant inherited disorder of lipoprotein metabolism characterized by very high plasma concentrations of low-density lipopropotein cholesterol (LDLc), tendon xanthomas (TX), and increased risk of premature coronary heart disease (CHD).1 The penetrance of FH is almost 100%, which means that half of the offspring of an affected parent have a severely increased plasma cholesterol level from birth onwards, being both males and females equally affected.
FH was first described by Müller in 1938.2 Initially, the FH defect was long thought to be caused by cholesterol oversynthesis.3 In the middle 1970s, Brown and Goldstein4,5 found that FH defect was due to the absence of a high affinity receptor for uptake of serum low-density lipoprotein (LDL). These investigators characterized the LDL receptor (LDLr) pathway with its implications in other pathways and identifying the genetic defect that caused malfunction of the LDLr.6 Nowadays, FH has become one of the best known genetic diseases.
Although the vast majority of FH cases are caused by mutations in the LDL receptor gene (LDLR) gene, there are other causative genes such as apolipoprotein B (APOB) that codifies for the natural ligand of the LDLr protein7 And a third gene, proprotein convertase subtilisin/kexin type 9 (PCSK9) has been more recently identified as a cause of FH;8,9 however, mutations in this latter gene seems to be rare in the populations studied so far (Table 1).10
Table 1.
Frequency of different types of primary hypercholesterolemia
| Monogenic hypercholesterolemia (1:500) | ||
| Autosomal dominant | ||
| FH | LDLR | 60%–80% |
| FDB-100 | APOE | 1%–5% |
| FH type 3 | PCSK9 | 0%–3% |
| Unknown | Unknown | 20%–40% |
| Autosomal recessive (1:1,000,000) | ||
| ARH | ARH | |
| Phytosterolemia | ABCG5/G8 | |
| Cholesterol 7 | CYP7A1 | |
| α-hydroxylase deficiency | ||
| Complex hypercholesterolemia (1:50) | ||
| FCH | Unknown | |
| Polygenic hypercholesterolemia (1:25) | APOB, APOE, LDLR, unknown | |
Abbreviations: FH, familial hypercholesterolemia; FDB, familial defective Apo B gene; FCH, familial combined hyperlipidemia; LDLR, low-density lipoprotein receptor; APOE, apolipoprotein E; PCSK9, proprotein convertase subtilisin/kexin type 9; APOE, apolipoprotein B gene; ARH, autosomal recessive hypercholesterolemia.
Epidemiology
FH is one of the most common inherited disorders with frequencies of heterozygotes and homozygotes estimated to be 1:500 and 1:1,000,000, respectively. In certain populations, a small number of mutations predominate due to founder effects and therefore, there is a high heFH frequency in these populations including French Canadians, 11 Christian Lebanese,12 Druze,13 Finns,14 South African Afrikaner,15 and Ashkenazi Jews of Lithuanian descent.16
FH heterozygous patients display a twofold increase in plasma cholesterol (generally above the 95th percentile value for population). In patients with FH, the age–sex standardized mortality ratios are 4–5 times higher than in the general populations.17
Due to the high incidence of premature (<55 years in men and <65 years in women) cardiovascular disease (CVD) and reduction in the life expectancy in many families with this disease, FH has been considered as a worldwide public health problem.18 Approximately, 85% of males and 50% of females will suffer a coronary event before the age of 65 years if they are not treated.19 It is noteworthy that up to 9% of the total premature CHD in eastern Finland and Germany is associated with FH.20–22
Long-term follow-up studies have shown that the main cause of death in FH patients is CHD.23,24 With adequate long-term pharmacological treatment, many FH patients could achieve substantial reductions in LDLc, and probably increase their life expectancy by 10–30 years.25
Intervention studies in FH
Scientific evidences, coming from large clinical trials, have demonstrated the benefit of LDLc reduction in the prevention of CVD in a broad spectrum of populations, especially in subjects with symptomatic CHD or with absolute high risk.26,27 As mentioned earlier, FH patients should be considered high risk subjects due to the prevalence of CHD, and they should benefit as a group at least as much as other high risk groups. According to the presence of major risk factors and/or clinical or subclinical atherosclerosis, three categories of risk for heFH are suggested (Table 2): (1) low 10-year risk, with no major risk factors; (2) moderate 10-year risk, with one major risk factor; and (3) high 10-year risk: (a) with ≥2 major risk factors, (b) subclinial atherosclerosis, or (c) clinical CVD.
Table 2.
Major CVD risk factors in heterozygous FH
| 1 Age |
| Men: ≥30 yo |
| Women: ≥45 yo or postmenopausal |
| 2 Cigarette smoking: active smokers |
| 3 Family history of premature CHD |
| 4 Male first-degree relative <55 yo |
| 5 Female first degree <65 yo |
| 6 Very high LDLc: >330 mg/dL (8.5 mmol/L) |
| 7 Diabetes mellitus |
| 8 Lp(a): >60 mg/dL |
Abbreviations: CHD, coronary heart disease; LDLc, low-density lipoprotein cholesterol; yo, years old.
In the last 15 years, different studies have used well-established surrogates of CVD to study the effects of aggressive LDLc reduction in FH. These studies demonstrate that coronary lesions measured by coronary angiography in SCOR,28 LARS,29 L-CAPS,30 LAARS and FHRS,31,32 or by intracoronary ultrasonography in LACMART;33 aortic lesions evaluated by transesophageal echocardiography;34 carotid intima-media thickness measured by quantitative B-mode ultrasound in ASAP;35 endothelial dysfunction measured by flow mediated dilatation and E-selectin;36 myocardial ischemia detected by exercise test in LAARS;31 and myocardial perfusion abnormalities assessed by digital angiography in LAARS,37 all improve with aggressive LDLc reduction obtained with LDL apheresis and/or lipid lowering drugs.
Consistent with these findings, the use of lipid lowering drugs, especially methylglutaryl coenzyme A (HMGCoA) reductase inhibitors, has been shown to be associated with improved cardiovascular prognosis without any change in noncardiovascular mortality in FH subjects on the Simon Broome Register in the United Kingdom.18
The LDL receptor
The LDLr is synthesized as a 120 KDa precursor protein. The glycosylated mature receptor reaches the cell surface and is directed towards clathrin-coated pits where it binds to Apo B-enriched and apolipoprotein E (Apo E)-enriched lipoproteins via its extracellular domain (Figure 1).38 The complex is endocytosed and migrates to the endosomes. Upon acidification of the endosomal pH, the LDL particle is released and later degraded in lysosomes. The LDLr returns to the membrane and enters in a new cycle. There is extensive evidence that plasma PCSK9 raises LDLc levels by binding to cell surface LDLr and targeting the receptor to lysosomes for degradation.39,40
Figure 1.

The LDL receptor pathway.
Notes: The low-density lipoprotein receptor (LDLr) is synthesized as a 120 KDa precursor protein and processed in the golgi apparatus (GOLGI) producing the glycosylated mature receptor that is transported to the cell surface and is directed towards clathrin-coated pits through interactions involving LDL particle, enriched with apolipoprotein B (APOB) where it binds to LDL particle. The complex is transported to endosomes where the acidic pH causes a dissociation of the receptor–ligand complex, releasing the LDLr to its recycling so the LDL is degraded in the lysosomal compartment. The proprotein convertase subtilisin/kexin type 9 (PCSK9) and Idol protein participate in a decreasing of receptor recycling and increasing the LDLr degradation.
The LDLR is mapped to 19p13.1–13.3, spans 45,000 base pairs (bp), and codifies for an ubiquitous transmembrane glycoprotein of 839 amino acids that mediates the transport of LDL into cells via endocytosis.41 It contains 18 exons and 17 introns encoding the six functional domains of the mature protein: signal peptide, ligand-binding domain, epidermal growth factor precursor (EGFP) like, O-linked sugar, transmembrane, and cytoplasmic domain (Figure 2).42 The prediction of the presence of different domains in the protein was possible because of the gene sequencing, determining that each domain was encoded by separate exons or group of them and suggesting that the LDLr might have evolved through shuffling of exons from other genes, because it has parts similar to unrelated proteins.43,44 After Goldstein and Brown6 identified LDLr dysfunction as cause of FH, multiple mutations were associated with this disease.43,45,46
Figure 2.
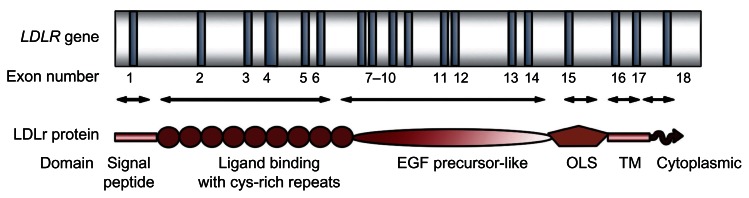
The LDLR gene.
Notes: Exons are shown as dark bars numbered underneath. Arrows indicate exons encoding the different domains of the low-density lipoprotein receptor (LDLR) protein: the signal peptide (exon 1), ligand-binding domain (exons 2–6), EGF precursor-like domain (exons 7–14), the domain named as OLS, O-linked carbohydrate chains (exon 15), transmembrane (TM) domain (exons 16 and 5′ part of exon 17), and the cytoplasmic domain (3′ region of exon 17 and 5′ region of exon 18).
The LDLr production is tightly regulated by a sophisticated feedback mechanism that controls the transcription of the LDLR in response to variations in the intracellular sterol concentration and the cellular demand for cholesterol.47 DNA motifs are essential for the transcriptional regulation of the LDLR and are located within 280 bp of the proximal promoter (Figure 3). This region contains all the cis-acting elements for basal expression and sterol regulation and includes three imperfect direct repeats of 16 bp each, repeats 1–3. Repeats 1 and 3 contain binding sites for Sp1 transcription factor, and contribute to the basal expression of the gene requiring the contribution of the repeat 2 for a strong expression.48 Repeat 2 contains a sterol regulatory element (SRE) that enhances transcription when the intracellular sterol concentration is low through interaction with a transcriptional factor designated as sterol regulatory element binding protein-1.49 Other two regions, named as FP1 and FP2 and located between −281 to −269, contain important cis-acting elements that have been described as essential for maximal induction of transcription.50 To date, several naturally occurring mutations have been mapped to the transcriptional regulatory elements of the LDLR.51–59
Figure 3.
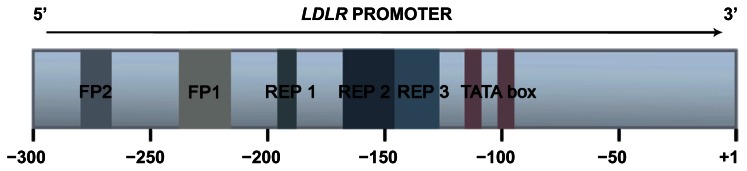
The LDLR promoter regulation.
Notes: Low-density lipoprotein receptor gene (LDLR) 5′ promoter region of 300 bp is represented, numbered the A of the ATG codon as +1. The major regulatory regions are indicated with different colors: FP2 (from −280 to −268); FP1 (−238 to −217); repeat (REP) 1 (−196 to −181); REP 2 (−161 to −146); REP 3 (−145 to −128); and TATA box (from −116 to −110 and −107 to −101). Interaction between cis-element and trans-element at the proximal promoter drives high levels transcription when sterol level becomes deficient. The sterol regulatory element (SRE) binding protein transcription factor interacts with the SRE-1 of REP 2, whereas SP1 transcription factors interact with REP 1 and REP 3 to promoter high levels of LDLR gene transcription in response to low intracellular sterol concentrations. SP1 also involved with constitutive, basal-level expression of the LDLR gene. The TATA boxes recruit and direct the assembly of general transcription factors at the promoter.
Exon 1 encodes the signal peptide, a hydrophobic sequence of 21 amino acids. This peptide is cleaved from the protein during the translocation into the endoplasmic reticulum. Currently, 79 (4.6% of total variants described) frameshift, missense, and nonsense sequence variants have been described in this exon (see http;//www.ucl.ac.uk/fh, http://www.umd.necker.fr).
Exons 2–6 encode the ligand-binding domain, which consists of seven tandem repeats of 40 amino acids each. There is a cluster of negatively charged amino acids, Asp- X-Ser-Asp-Glu in each repeat and six cysteine residues that form three disulfide bonds.37 Binding of lipoproteins to the LDLr appears to be mediated by an interaction between acidic residues in the LDLr-binding domain and basic residues of Apo E and Apo B-100.60,61 Deletion of individuals repeats R3–R7 results in a loss of LDL binding (Apo B-100-mediated), but a LDLr fragment consisting of R4 and R5 is sufficient to bind to Apo E-phospholipids vesicles.62 Recently, a new mechanism for the release of LDL particles in the endosome has been proposed. It is based on the instability of R5 at endosomal low pH and low Ca2+. Under this kind of condition, R5 is unable to bind Ca2+ and appears in an unfolded conformation not expected to bind LDL particles.63 To date, in this region, 693 allelic variants (40.7%) have been found.
The second domain of the human LDLr consists of a 411 amino acid sequence, encoded by exons 7–14. This sequence shows a 33% of homology of the human EGFP. Like the ligand-binding domain, this region also contains three repeats of 40–50 amino acids with cysteine-rich sequences. The EGFP-like domain is required for the acid-dependent dissociation of the LDL particles from the LDLr and clathrin-coated pits that takes place in the endosome during receptor recycling. When the EGFP domain is deleted from the LDLr, the receptor can no longer bind LDL but it still binds lipoproteins that contain Apo E.64 Zhang et al65 showed that PCSK9 bind to EGFP-A repeat (the first one) of LDLr, decreasing receptor recycling and increasing degradation. From all mutations described to date, approximately 788 (46%) have been associated with this domain (see http://www.ucl.ac.uk/fh, http://www.umd.necker.fr).
The third domain of the LDLr is a region of 58 amino acids rich in threonine and serine residues that is encoded by exon 15. The function of this domain is unknown, but has been observed that this region serve as attachment sites for O-linked carbohydrate chains.1,66 This region shows minimal sequence conservation among six species analyzed and can be deleted without adverse effects on receptor function in cultured fibroblasts.66 It is thought that this domain plays a role in the stabilization of the receptor.1 Actually, 41 allelic variants within exon 15 are registered in LDLR databases.
The transmembrane domain that contains 22 hydrophobic amino acids is coded by exon 16 and the 5′ end of exon 17. This domain is essential for anchor the LDLr to the cell membrane. The cytoplasmic domain of the LDLr, that compromises 50 amino acid residues, is encoded by the remainder 3′ region of the exon 17 and the 5′ end of the exon 18.1 This domain contains two sequence signals for targeting the LDLr to the surface and for localizing the receptor in coated pits.67 This domain is the most conserved region of the LDLr, which is more than 86% identical among six species.1 Only a few allelic variants, 5.9% of total, have been identified within these domains.
Nowadays, over 1,000 mutations in LDLR have been described in FH patients along many populations (see http://www.ucl.ac.uk/fh, http://www.umd.necker.fr). The naturally occurring LDLR can produce defects in transcription, posttranscription processes, translation, and posttranslation processes.59 FH mutations have been classified into five classes depending on the phenotypic behavior of mutant protein.43
Class 1 mutations are known as “null alleles.” These kinds of mutations are due to LDLR promoter deletion, by frameshift, nonsense, splicing mutations, or rearrangements in a way that messenger RNA (mRNA) is not produced; it generates an abnormal mRNA or normal in size but in a reduced concentration.68
Class 2 mutations are transport defective alleles which encode for proteins that do not have an adequate three- dimensional structure after being synthesized and keep blocked, complete or partially (2A and 2B, respectively) in transport process between endoplasmic reticulum and golgi apparatus. This defect is caused, normally, by missense mutations or small deletions in LDLR avoiding partial or completely the folding protein. These mutations are located within exons that encode ligand-binding domain and EGFP-like domain.69
Class 3 mutations are binding defective alleles which encode for LDLr that are synthesized and transported to cell surface but are not able to bind LDL particles. This is a heterogeneous group, because LDL binding activity goes from 2% to 30% of normal. This defect is due to rearrangements in repeat cysteine residues in binding ligand domain or repeat deletions in EGFP-like domain.70
Class 4 mutations are known as internalization-defective alleles which produce proteins that are not able to group into clathrin-coated pits; therefore, LDLr are not internalized (4A: only cytoplasmic domain is affected and 4B: also affected transmembrane domain).66
Finally, recycling-defective alleles are also named as class 5 mutations, such as missense mutations in EGFP-like domain, which encode for LDLr that are not able to release LDL particles in endosomes avoiding receptor return to cell surface.59
The heterogeneity observed in FH patients in relation to plasma LDLc levels and CHD has been suggested due to differences in the nature of the mutation in the LDLR and several studies have been published in support of this.39,40,71–73 Even HMGCoA reductase inhibitors may depend on the nature of the mutation in the LDLR gene.74,75 Recently, a study carried out by our group, with 436 Spanish FH patients with known LDLR mutations classified as null alleles or defective alleles, has demonstrated that patients with a molecular diagnosis of FH characterized by null allele mutations of LDLR show a more severe clinical phenotype and worse advanced carotid artherosclerosis than those with receptor-defective mutations, independently of age, gender, lipid, and nonlipid risk factor.76
Other genes associated with FH
Apolipoprotein B
In 1986, Vega and Grundy77 showed that some patients (5 of 15 studied) with hypercholesterolemia have reduced clearance of LDL not because of decreased activity of LDLr but because of a defect in structure or composition of LDL that reduces its affinity for receptors. Innerarity et al78 found that moderate hypercholesterolemia, presented in the five subjects previously studied, could be attributed to a defective receptor binding of a genetically altered Apo B-100 to the LDLr.77,78 The inherited nature of this disease was indicated by the findings of the same defect in proband’s first-degree relatives. These findings resulted in referring this disease as Familial Defective Apo B-100 (FDB).78
The first mutation found as a FDB cause was demonstrated by Soria et al79 who sequenced the two alleles of APOB from patients of three families. They observed the mutation R3500Q.79 Two new mutations were described in 1995 as cause of FDB: R3500W and R3531C.80,81 For the classical mutation, R3500Q, frequency was estimated in 1:500–1:700 in several Caucasian populations in North America and Europe (Table 1).82 On the other hand, R3500W and R3531C have been found in a minor frequency.
Recently, a novel mutation H3543Y in APOB associated with FDB has been described with a high prevalence (4 times R3500Q) in a German population.83
Recent data reveal that compared with FH patients with LDLR mutations, FDB patients have lower LDLc levels by 20%–25% (Table 3),84 respond better to statins and have lower risk of CHD.85 This difference could be due to normal clearance of very low-density lipoprotein remnants through Apo E-mediated uptake in FDB.86
Table 3.
Common hypercholesterolemia and hypertriglyceridemia
| Molecular mechanism | Clinical features | |
|---|---|---|
| Dominant inheritance | ||
| Familial hypercholesterolemia | LDLr defect | TX, arcus cornealis, premature CHD, TC: >400 mg/dL (>10.3 mmol/L) or TC: 190–400 mg/dL (4.9–10.3 mmol/L) in heFH |
| Familial defective APOB-100 | APOB defect | Xanthomas, arcus cornealis, premature CHD, and TC: 250–350 mg/dL (7–13 mmol/L) |
| Familial hypercholesterolemia type 3 | PCSK9 | Premature CHD TC: 250–500 mg/dL (6.5–9 mmol/L) |
| Familial hypertriglyceridemia | Possible multiple unknown defects | No symptoms TG: 200–500 mg/dL (2.3–5.7 mmol/L) |
| Familial combined hyperlipidemia | Possible multiple unknown defects | Premature CHD, Apo B elevated, TC: 250–500 mg/dL (6.5–13 mmol/L) TG: 250–750 mg/dL (2.8–8.5 mmol/L) |
| Recessive inheritance | ||
| Autosomal recessive hypercholesterolemia | ARH | Xanthomas, arcus cornealis, xanthelasmas, premature CHD. TC: >350 mg/dL (>9 mmol/L) |
| LPL deficiency | Endothelial LPL defect | Failure to thrive, xanthomas, hepatosplenomegaly, pancreatitis TG: >750 mg/dL (8.5 mmol/L) |
| Apo C-II deficiency | Apo C-II defect | Pancreatitis and metabolic syndrome. TG: >750 mg/dL (8.5 mmol/L) |
| Hepatic lipase deficiency | Hepatic lipase | Premature CHD TC: 250–1,500 mg/dL TG: 395–8,200 mg/dL |
| Cerebrotendinous xanthomatosis | Hepatic mitochondrial 27-hydroxylase defect | Cataracts, premature CHD, neuropathy, ataxia |
| Sitosterolemia | ABCG5/G8 | Tendon xanthomas, premature CHD |
| Variable inheritance | ||
| Familial dysbetalipoproteinemia | APOE (usually e2/e2 homozygotes) | Palmar xanthomas, yellow palmar creases, premature CHD. TC: 250–500 mg/dL (6.5–13 mmol/L) TG: 250–500 mg/dL (2.8–5.6 mmol/L) |
| Polygenic hypercholesterolemia | Possibly multiple unknown defects | Premature CHD TC: 250–350 mg/dL (6.5–9 mmol/L) |
Abbreviations: CHD, coronary heart disease; LDLr, low-density lipoprotein receptor protein; LPL, lipoprotein lipase; TC, total cholesterol; TG, triglycerides; TX, tendon xanthomas.
Proprotein convertase subtilisin/kexin type 9 gene
In 1999, Varret et al87 identify a new autosomal dominant hypercholesterolemia (ADH) locus in 1q34.1-p32 chromosome (Tables 1 and 2). PCSK9 was first identified as a member of proprotein convertase family with hepatic, intestine, and kidney expression.88 Mutations in PCSK9 gene (S127R, P216L, and D374Y y N157K) that produce gain of function were associated with a decrease in LDLr number and ADH.89 Initially, it was thought the hypothesis of a PCSK9 role in LDLr degradation in the cell surface.89 Nowadays, as we have described earlier, there are enough evidence to think that PCSK9 participates in LDLr lysosomal degradation (Figure 1).39,40,90 Recently, it has been proposed that PCSK9 may induce internalization and degradation of LDLr by leading receptor to ubiquitination by Idol.91
PCSK9 mutations have been also classified into five classes, including “null alleles”, mutations that affect autocatalytic scission avoiding the protein transport through endoplasmic reticulum or from the endoplasmic reticulum to cell surface, alleles that affects PCSK9 stability and finally mutations that produce gain of function because of gene overexpression.92–94 Some mutations in PCSK9 (Y142X, C679X, and R46L) produce a loss of function and are associated with low LDLc.95,96
About 17%–33% of patients with a clinical diagnosis of monogenic hypercholesterolemia based on Simon Broome Register Group (SBRG) criteria do not harbor any genetic cause in the known loci suggesting a possibility of additional hypercholesterolemia loci (Table 1).9,10
FH diagnosis
Clinical criteria used to identify patients with FH include high plasma levels of total and LDLc (>250 mg/dL or >7 mmol/L), family history of hypercholesterolemia especially in children, deposition of cholesterol in extravascular tissues such as TX or corneal arcus, and personal and family history of premature CVD.1 Heterozygous FH (heFH) patients have LDLc levels approximately twice those of the normal population, ranging from 190 to 400 mg/dL (4.9–10.3 mmol/L). Triglycerides (TG) levels are usually in the normal range. However, some patients with FH have increased TG levels, explained in part by the interaction with other genes (ie, E2/E2 genotype) or environmental factors (ie, alcohol, overweight, and diabetes mellitus).
TX are pathognomonic of FH; however, their identification is not always easy and they are considered insensitive diagnostic markers. A high variability of xanthoma presence in FH patients has been reported.97 Xanthelasmas occur commonly in heterozygotes, but are rare in homozygotes. Xanthelasmas are not specific for FH and can appear in subjects with normal lipid levels.1 TX may appear in patients with cerebrotendinous xanthomatosis and are indistinguishable from those FH. This kind of xanthomas also occur in subjects affected by FDB, dysbetalipoproteinemia, and sitosterolemia (Table 3).1
Variability in the frequency observed in different studies depends in part on the clinical criteria used for FH (some of them included the presence of xanthomas), as well as the methods used for the identification of xanthomas.15
There are no absolutely predictive clinical criteria for the diagnosis of FH, and arbitrary criteria must be used. Several criteria have been proposed (Table 4) by SBRG,18 the USA Make Early Diagnosis to Prevent Early Death (MEDPED) Program,98 and the Dutch MEDPED Program.99 Our group demonstrated that MEDPED programs resulted in high sensitivities and specificities being more accurate as more complex is the scoring system.100 The best approach in most populations is to determine LDLc in all first degree of a FH proband and it is recommended that all second-degree family members are also screened.101
Table 4.
Familial hypercholesterolemia diagnostic criteria
| SBRG | Definitive | TC >290 mg/dL or LDLc >190 mg/dL + familial history of TX presence | |
| Possible | TC >290 mg/dL or LDLc >190 mg/dL + familial history of myocardial infarction or family history of hypercholesterolemia | ||
| USA MEDPED | Family with clinical suspicions of FH | Age <20 yo, TC >270 mg/dL | |
| Age 20–29 yo, TC >290 mg/dL | |||
| Age 30–39 yo, TC >340 mg/dL | |||
| Age ≥40 yo, TC >360 mg/dL | |||
| DLCN MEDPED | Familial history | Hypercholesterolemia | 1 (score) |
| Premature vascular disease | 1 | ||
| TX and/or arcus cornealis | 2 | ||
| Children <18 yo with LDLc >95th percentile | 2 | ||
| Personal history | Premature vascular disease | 1–2 | |
| Physical exam | TX presence | 6 | |
| Arcus cornealis (<45 yo) | 4 | ||
| LDLc levels | ≥330 mg/dL | 8 | |
| 250–329 mg/dL | 5 | ||
| 190–249 mg/dL | 3 | ||
| 155–189 mg/dL | 1 | ||
| Civeira et ala | Familal history of TX + LDLc ≥190 mg/dL | ||
| Nonfamily history of TX | Age <30 yo with LDLc >220 mg/dL | ||
| Age 30–39 yo with LDLc >225 mg/dL | |||
| Age >40 yo with LDLc >235 mg/dL | |||
Notes:
Clinical criteria proposed for genetic testing.100
Abbreviations: SBRG, Simon Broom Register Group from United Kingdom;18 MEDPED, Make Early Diagnosis to Prevent Early Death program in the United States;98 DLCN MEDPED: Dutch Lipid Clinic Network MEDPED group criteria scoring system for the diagnosis of heterozygous FH patients (“Definitive” diagnosis >7 points, “Probable” 5–7 points).99
Although clinical diagnosis criteria have been extensively used for FH,18 the genetic testing is the preferred method for FH because it provides an unequivocal diagnosis.1,10,19 Since 2004, a genetic diagnostic platform for FH called Lipochip® (Progenika-Biopharma, Derio, Spain) has been developed, which includes a microarray for the detection of common point mutations and small deletions in the LDLR and APOB genes, the diagnosis of large rearrangements, and a full LDLR coding sequence analysis when the former are negative.102 By providing either a positive (presence of LDLR or APOB mutations) or negative (absence of defects in these genes) diagnosis, this platform has allowed the genetic characterization of >5,000 Spanish patients.100,103 Even though the diagnosis of FH based on the detection of a functional mutation on a causative gene is the recommended procedure in most suspicious cases but it cannot be recommended for all cases of hypercholesterolemia because the genetic testing is still complex, expensive, and require specific families for microarray analysis. For those reasons, clinical diagnosis is still very important and in the FH diagnostic guidelines published by our group, we recommend genetic analysis because these populations use to present a few LDLR mutations that are responsible for most FH cases; the most frequent causative mutations are known; or in subjects from families with known mutations with an uncertain clinical diagnosis (Table 4).19 We have recently published a study that proves the molecular diagnosis usefulness to distinguish familial combined hyperlipidemia (FCH) of FH patients, when clinical presentation can produce a misclassification, which can be solved by finding LDLR mutations that cause the disease.104 At this work, 28 carriers of LDLR mutations were found in 143 unrelated FCH subjects. Our group has recently shown that presence of TX is highly specific of FH when primary hypercholesterolemia, family history of hypercholesterolemia, and premature coronary disease are presented. Moreover, a sonographic evaluation of Achilles tendons for the diagnosis of FH has been performed.105 Thus, taking all these findings together, a new set criteria to maximize the likelihood of genetic confirmation in subjects with clinical suspicions of FH based on age, TX presence, and LDLc levels have been proposed (Table 4).100
Treatment
The standard of care for patients with homozygous FH has been LDL apheresis.106,107 The LDLc reduction observed with LDL apheresis is over 60%, and similar values were observed in Lp(a). LDL apheresis is an invasive and expensive but safe procedure. The inconvenience of this method is that it has to be performed at 1-weekly or 2-weekly intervals.108
A statin is the drug of first choice in the majority of cases. The safety and efficacy of statins as LDLc lowering drugs and their demonstrated performance in preventing CVD morbidity and mortality in primary and secondary prevention trials have been amply demonstrated. In addition, statins can be safely combined with either resins or ezetimibe. Furthermore, the LDLc-lowering effect of these drugs is not modified by the concomitant use of plant sterols/stanols that can be recommended also to these patients.109
Considering that <100 mg/dL (2.6 mmol/L) is the optimal LDLc concentration defined by ATPIII guidelines,26 it would be necessary to achieve mean reductions between 50% and 75% to reach that goal. Based on published data from both heFH and other high risk populations, three different LDLc goals can be recommended for heFH (Table 5). An important aspect of FH treatment with many benefits beyond LDLc lowering is a healthy lifestyle, which includes a healthy diet, ideal body weight, no smoking, and moderate physical activity. Although LDLc is the main CVD risk factor in FH patients, other risk factors such as smoking habit or low HDLc concentration are great modifiers of the CVD development.110
Table 5.
LDLc treatment goals according to categories of risk
| Categories | Optimal goala | |
|---|---|---|
|
|
||
| mg/dL | mmol/L | |
| Low 10-year risk | 160 | 4.1 |
| Moderate 10-year risk | 130 | 3.4 |
| High 10-year risk | 100 | 2.6 |
Notes:
If these optimal goals are not reached, the minimum reductions in LDLc to be achieved are: 40, 50, and 60%, respectively.
Abbreviation: LDLc, low-density lipoprotein cholesterol.
Several intervention studies have clearly demonstrated that a healthy diet reduce cardiovascular risk factors independently of classic risk factors (Table 2). Moreover, this kind of diet can increase the LDLc-lowering power of drugs.19 The beneficial effect is probably mediated by a variety of mechanisms including improved carbohydrate metabolism, lower blood pressure, greater antioxidant protection, and regulating inflammatory and thrombogenic processes.
Summary
In this manuscript, we have reviewed the recently genetic and molecular mechanisms described as target for hypercholesterolemia and updated FH diagnosis and treatment.
FH has been studied widely since its first description in 1938.2 Despite this knowledge, new mutations and genes implicated in cholesterol pathway, as PCSK9 or Idol,88,91 have been recently associated with hypercholesterolemia. In fact, over 17% of ADH is caused by unknown mechanisms, because of that an exhaustive research in this way has to be done.9
Moreover, an accurate FH clinical diagnosis combined with genetic testing allows physicians to discriminate FH from other dyslipemias.104 The treatment goal in FH for decreasing CHD risk is LDLc levels but not denying the benefits obtained with lifestyle changes that reduce the environmental risk factors.
Acknowledgments
This work has been partially funded by grants FIS PS09/00665, PI06/1238, and PSE 010000-2008-5.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, AB, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001. pp. 2863–2913. [Google Scholar]
- 2.Müller C. Xanthomata, hypercholesterolemia, angina pectoris. Acta Med Scand. 1938;(89):75–84. [Google Scholar]
- 3.Langer T, Strober W, Levy RI. The metabolism of low density lipoprotein in familial type II hyperlipoproteinemia. J Clin Invest. 1972;51:1528–1536. doi: 10.1172/JCI106949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci U S A. 1973;70:2084–2088. doi: 10.1073/pnas.70.10.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 1974;71:788–792. doi: 10.1073/pnas.71.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 7.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damgaard D, Jensen JM, Larse ML, et al. NO genetic linkage or molecular evidence for involvement of the PCSK9, ARH or CYP7A1 genes in the familial hypercholesterolemia phenotype in a sample of Danish families without pathogenic mutations in the LDL receptor and APOB genes. Atherosclerosis. 2004;177:415–422. doi: 10.1016/j.atherosclerosis.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 9.Graham CA, McIlhatton BP, Kirk CW, et al. Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate. Atherosclerosis. 2005;182:331–340. doi: 10.1016/j.atherosclerosis.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 11.Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common low-density lipoprotein receptor mutations in the French Canadian population. J Clin Invest. 1990;85:1014–1023. doi: 10.1172/JCI114531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehrman MA, Russell DW, Goldstein JL, Brown MS. Alu- Alu recombination deletes splice acceptor sites and produces secreted low density lipoprotein receptor in a subject with familial hypercholesterolemia. J Biol Chem. 1987;262:3354–3361. [PubMed] [Google Scholar]
- 13.Landsberger D, Meiner V, Reshef A, et al. A nonsense mutation in the LDL receptor gene leads to familial hypercholesterolemia in the Druze sect. Am J Hum Genet. 1992;50:427–433. [PMC free article] [PubMed] [Google Scholar]
- 14.Koivisto UM, Turtola H, Aalto-Setala K, et al. The familial hypercholesterolemia (FH)-North Karelia mutation of the low density lipoprotein receptor gene deletes seven nucleotides of exon 6 and is a common cause of FH in Finland. J Clin Invest. 1992;90:219–228. doi: 10.1172/JCI115839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotze MJ, Langenhoven E, Warnich L, du Plessis L, Retief AE. The molecular basis and diagnosis of familial hypercholesterolaemia in South African Afrikaners. Ann Hum Genet. 1991;55:115–121. doi: 10.1111/j.1469-1809.1991.tb00404.x. [DOI] [PubMed] [Google Scholar]
- 16.Meiner V, Landsberger D, Berkman N, et al. A common Lithuanian mutation causing familial hypercholesterolemia in Ashkenazi Jews. Am J Hum Genet. 1991;49:443–449. [PMC free article] [PubMed] [Google Scholar]
- 17.Scientific Steering Committee on behalf of the Simon Broome Register Group. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Atherosclerosis. 1999;142:105–112. [PubMed] [Google Scholar]
- 18.Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ. 1991;303(6807):893–896. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68. doi: 10.1016/j.atherosclerosis.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Koivisto UM, Hamalainen L, Taskinen MR, Kettunen K, Kontula K. Prevalence of familial hypercholesterolemia among young north Karelian patients with coronary heart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res. 1993;34:269–277. [PubMed] [Google Scholar]
- 21.Baron H, Fung S, Aydin A, Bahring S, Luft FC, Schuster H. Oligonucleotide ligation assay (OLA) for the diagnosis of familial hypercholesterolemia. Nat Biotechnol. 1996;14:1279–1282. doi: 10.1038/nbt1096-1279. [DOI] [PubMed] [Google Scholar]
- 22.Schuster H. High risk/high priority: familial hypercholesterolemia – a paradigm for molecular medicine. Atheroscler Suppl. 2002;2:27–30. doi: 10.1016/s1567-5688(01)00019-8. [DOI] [PubMed] [Google Scholar]
- 23.Mabuchi H, Koizumi J, Shimizu M, Takeda R. Development of coronary heart disease in familial hypercholesterolemia. Circulation. 1989;79:225–232. doi: 10.1161/01.cir.79.2.225. [DOI] [PubMed] [Google Scholar]
- 24.Miettinen TA, Gylling H. Mortality and cholesterol metabolism in familial hypercholesterolemia. Long-term follow-up of 96 patients. Arteriosclerosis. 1988;8:163–167. doi: 10.1161/01.atv.8.2.163. [DOI] [PubMed] [Google Scholar]
- 25.WHO. Familial Hypercholesterolemia, report of a WHO consultation. Paris: 1997. Oct, Human Genetic Program. WHO/HGN/FH/CONS/98.7. [Google Scholar]
- 26.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol Adults (Adult Treatment Panel III): final report. Circulation. 2002;106:3143–3221. [PubMed] [Google Scholar]
- 27.Gotto AM., Jr Management of dyslipidemia. Am J Med. 2002;112(Suppl 8A):10S–18S. doi: 10.1016/s0002-9343(02)01085-9. [DOI] [PubMed] [Google Scholar]
- 28.Kane JP, Mallow MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ. Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimens. JAMA. 1990;264:3007–3012. [PubMed] [Google Scholar]
- 29.Tatami R, Inoue N, Itoh H, et al. Regression of coronary atherosclerosis by combined LDL-apheresis and lipid-lowering drug therapy in patients with familial hypercholesterolemia: a multicenter study. The LARS Investigators. Atherosclerosis. 1992;95:1–13. doi: 10.1016/0021-9150(92)90170-l. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura S, Sekiguchi M, Kano T, et al. Effects of intensive lipid lowering by low-density lipoprotein apheresis on regression of coronary atherosclerosis in patients with familial hypercholesterolemia: Japan Low-density Lipoprotein Apheresis Coronary Atherosclerosis Prospective Study (L-CAPS) Atherosclerosis. 1999;144:409–417. doi: 10.1016/s0021-9150(98)00328-1. [DOI] [PubMed] [Google Scholar]
- 31.Kroon AA, Aengevaeren WR, van der Werf T, et al. LDL-Apheresis Atherosclerosis Regression Study (LAARS). Effect of aggressive versus conventional lipid lowering treatment on coronary atherosclerosis. Circulation. 1996;93:1826–1835. doi: 10.1161/01.cir.93.10.1826. [DOI] [PubMed] [Google Scholar]
- 32.Thompson GR, Maher VM, Matthews S, et al. Familial Hypercholesterolaemia Regression Study: a randomised trial of low-density-lipoprotein apheresis. Lancet. 1995;345:811–816. doi: 10.1016/s0140-6736(95)92961-4. [DOI] [PubMed] [Google Scholar]
- 33.Matsuzaki M, Hiramori K, Imaizumi T, et al. Intravascular ultrasound evaluation of coronary plaque regression by low density lipoprotein-apheresis in familial hypercholesterolemia: the Low Density Lipoprotein- Apheresis Coronary Morphology and Reserve Trial (LACMART) J Am Coll Cardiol. 2002;40:220–227. doi: 10.1016/s0735-1097(02)01955-1. [DOI] [PubMed] [Google Scholar]
- 34.Pitsavos CE, Aggeli KI, Barbetseas JD, et al. Effects of pravastatin on thoracic aortic atherosclerosis in patients with heterozygous familial hypercholesterolemia. Am J Cardiol. 1998;82:1484–1488. doi: 10.1016/s0002-9149(98)00691-2. [DOI] [PubMed] [Google Scholar]
- 35.Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 2001;357:577–581. doi: 10.1016/s0140-6736(00)04053-8. [DOI] [PubMed] [Google Scholar]
- 36.Alonso R, Mata P, De Andres R, Villacastin BP, Martinez-Gonzalez J, Badimon L. Sustained long-term improvement of arterial endothelial function in heterozygous familial hypercholesterolemia patients treated with simvastatin. Atherosclerosis. 2001;157:423–429. doi: 10.1016/s0021-9150(00)00733-4. [DOI] [PubMed] [Google Scholar]
- 37.Aengevaeren WR, Kroon AA, Stalenhoef AF, Uijen GJ, van der Werf T. Low density lipoprotein apheresis improves regional myocardial perfusion in patients with hypercholesterolemia and extensive coronary artery disease. LDL-Apheresis Atherosclerosis Regression Study (LAARS) J Am Coll Cardiol. 1996;28:1696–1704. doi: 10.1016/s0735-1097(96)00388-9. [DOI] [PubMed] [Google Scholar]
- 38.Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105:13045–13050. doi: 10.1073/pnas.0806312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koivisto PV, Koivisto UM, Kovanen PT, Gylling H, Miettinen TA, Kontula K. Deletion of exon 15 of the LDL receptor gene is associated with a mild form of familial hypercholesterolemia. FH-Espoo. Arterioscler Thromb. 1993;13:1680–1688. doi: 10.1161/01.atv.13.11.1680. [DOI] [PubMed] [Google Scholar]
- 40.Gudnason V, Day IN, Humphries SE. Effect on plasma lipid levels of different classes of mutations in the low-density lipoprotein receptor gene in patients with familial hypercholesterolemia. Arterioscler Thromb. 1994;14:1717–1722. doi: 10.1161/01.atv.14.11.1717. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:5153–5162. [PubMed] [Google Scholar]
- 42.Sudhof TC, Van der Westhuyzen DR, Goldstein JL, Brown MS, Russell DW. Three direct repeats and a TATA-like sequence are required for regulated expression of the human low density lipoprotein receptor gene. J Biol Chem. 1987;5(262):10773–10779. [PubMed] [Google Scholar]
- 43.Tolleshaug H, Goldstein JL, Schneider WJ, Brown MS. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell. 1982;30:715–724. doi: 10.1016/0092-8674(82)90276-8. [DOI] [PubMed] [Google Scholar]
- 44.Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133–170. doi: 10.1146/annurev.ge.24.120190.001025. [DOI] [PubMed] [Google Scholar]
- 45.Schneider WJ, Beisiegel U, Goldstein JL, Brown MS. Purification of the low density lipoprotein receptor, an acidic glycoprotein of 164,000 molecular weight. J Biol Chem. 1982;257:2664–2673. [PubMed] [Google Scholar]
- 46.Beisiegel U, Schneider WJ, Goldstein JL, Anderson RG, Brown MS. Monoclonal antibodies to the low density lipoprotein receptor as probes for study of receptor-mediated endocytosis and the genetics of familial hypercholesterolemia. J Biol Chem. 1981;256:11923–11931. [PubMed] [Google Scholar]
- 47.Dawson PA, Hofmann SL, van der Westhuyzen DR, Sudhof TC, Brown MS, Goldstein JL. Sterol-dependent repression of low density lipoprotein receptor promoter mediated by 16-base pair sequence adjacent to binding site for transcription factor Sp1. J Biol Chem. 1988;263:3372–3379. [PubMed] [Google Scholar]
- 48.Smith JR, Osborne TF, Goldstein JL, Brown MS. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265:2306–2310. [PubMed] [Google Scholar]
- 49.Jeon H, Meng W, Takagi J, Eck MJ, Springer TA, Blacklow SC. Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat Struct Biol. 2001;8:499–504. doi: 10.1038/88556. [DOI] [PubMed] [Google Scholar]
- 50.Mehta KD, Chang R, Underwook J, Wise J, Kumer A. Identification of a novel cis-acting element participating in maximal induction of the human LDL-receptor gene transcription in response to low cellular cholesterol levels. J Biol Chem. 1996;271:33616–33622. doi: 10.1074/jbc.271.52.33616. [DOI] [PubMed] [Google Scholar]
- 51.Dedoussis GV, Genschel J, Bochow B, et al. Molecular characterization of familial hypercholesterolemia in German and Greek patients. Hum Mutat. 2004;23:285–286. doi: 10.1002/humu.9218. [DOI] [PubMed] [Google Scholar]
- 52.Smith AJ, Ahmed F, Nair D, et al. A functional mutation in the LDLR promoter (−139C > G) in a patient with familial hypercholesterolemia. Eur J Hum Genet. 2007:1186–1189. doi: 10.1038/sj.ejhg.5201897. [DOI] [PubMed] [Google Scholar]
- 53.Mozas P, Castillo S, Tejedor D, et al. Molecular characterization of familial hypercholesterolemia in Spain: identification of 39 novel and 77 recurrent mutations in LDLR. Hum Mutat. 2004;24:187. doi: 10.1002/humu.9264. [DOI] [PubMed] [Google Scholar]
- 54.Scholtz CL, Peeters AV, Hoogendijk CF, et al. Mutation −59c→t in repeat 2 of the LDL receptor promoter: reduction in transcriptional activity and possible allelic interaction in a South African family with familial hypercholesterolemia. Hum Mol Genet. 1999;8:2025–2030. doi: 10.1093/hmg/8.11.2025. [DOI] [PubMed] [Google Scholar]
- 55.Francová H, Trbusek M, Zapletalová P, Kuhrová V. New promoter mutations in the low-density lipoprotein receptor gene which induce familial hypercholesterolaemia phenotype: molecular and functional analysis. J Inherit Metab Dis. 2004;27:523–528. doi: 10.1023/B:BOLI.0000037337.93335.c4. [DOI] [PubMed] [Google Scholar]
- 56.Mozas P, Galetto R, Albajar M, Ros E, Pocoví M, Rodríguez-Rey JC. A mutation (−49C > T) in the promoter of the low density lipoprotein receptor gene associated with familial hypercholesterolemia. J Lipid Res. 2002;43:13–18. [PubMed] [Google Scholar]
- 57.Sun XM, Neuwirth C, Wade DP, Knight BL, Soutar AK. A mutation (T-45C) in the promoter region of the low-density lipoprotein (LDL)- receptor gene is associated with a mild clinical phenotype in a patient with heterozygous familial hypercholesterolaemia (FH) Hum Mol Genet. 1995;4:2125–2129. doi: 10.1093/hmg/4.11.2125. [DOI] [PubMed] [Google Scholar]
- 58.Koivisto UM, Palvimo JJ, Jänne OA, Kontula K. A single-base substitution in the proximal Sp1 site of the human low density lipoprotein receptor promoter as a cause of heterozygous familial hypercholesterolemia. Proc Natl Acad Sci U S A. 1994;91:10526–10530. doi: 10.1073/pnas.91.22.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 60.Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem. 1988;263:13282–13290. [PubMed] [Google Scholar]
- 61.Russell DW, Brown MJ, Goldstein JL. Different combinations of cysteine-rich repeats mediated binding of low-density lipoprotein receptor to two different proteins. J Biol Chem. 1989;264:21682–21688. [PubMed] [Google Scholar]
- 62.Fisher C, Abdul-Aziz D, Blacklow SC. A two-modulate region of the low-density lipoprotein receptor sufficient for formation of complexes with apolipoprotein E ligands. Biochemistry. 2004;43:1037–1044. doi: 10.1021/bi035529y. [DOI] [PubMed] [Google Scholar]
- 63.Arias-Moreno X, Velázquez-Campoy A, Rodríguez JC, Pocoví M, Sánchez J. Mechanism of low density lipoprotein (LDL) release in the Endosome. J Biol Chem. 2008;283:22670–22679. doi: 10.1074/jbc.M802153200. [DOI] [PubMed] [Google Scholar]
- 64.Brown MS, Herz J, Goldstein JL. Calcium cages, acid baths and recycling receptors. Nature. 1997;388:629–630. doi: 10.1038/41672. [DOI] [PubMed] [Google Scholar]
- 65.Zhang DW, Lagace TA, Garuti R, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:20502–20512. doi: 10.1074/jbc.M702027200. [DOI] [PubMed] [Google Scholar]
- 66.Davis CG, Elhammer A, Russell DW, et al. Deletion of clustered O-linked carbohydrates does not impair function of low density lipoprotein receptor in transfected fibroblasts. J Biol Chem. 1986;261:2828–2838. [PubMed] [Google Scholar]
- 67.Yokode M, Pathak RK, Hammer RE, Brown MS, Goldstein JL, Anderson RG. Cytoplasmic sequence required for basolateral targeting of LDL receptor in livers of transgenic mice. J Cell Biol. 1992;117:39–46. doi: 10.1083/jcb.117.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hobbs HH, Leitersdorf E, Goldstein JL, Brown MS, Russel DW. Multiple crm-mutations in familial hypercholesterolemia. Evidence for 13 alleles, including four deletions. J Clin Invest. 1988;81:909–917. doi: 10.1172/JCI113402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamamoto T, Davis CG, Brown MS, et al. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 70.Lehrman MA, Schnedier WJ, Südhof TC, Brown MS, Goldstein JL, Russell DW. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science. 1985;227:140–146. doi: 10.1126/science.3155573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gudnason V, King-Underwood L, Seed M, Sun XM, Soutar AK, Humphries SE. Identification of recurrent and novel mutations in exon 4 of the LDL receptor gene in patients with familial hypercholesterolemia in the United Kingdom. Arterioscler Thromb. 1993;13:56–63. doi: 10.1161/01.atv.13.1.56. [DOI] [PubMed] [Google Scholar]
- 72.Kotze MJ, De Villiers WJ, Steyn K, et al. Phenotypic variation among familial hypercholesterolemics heterozygous for either one of two Afrikaner founder LDL receptor mutations. Arterioscler Thromb. 1993;13:1460–1468. doi: 10.1161/01.atv.13.10.1460. [DOI] [PubMed] [Google Scholar]
- 73.Sun XM, Webb JC, Gudnason V, et al. Characterization of deletions in the LDL receptor gene in patients with familial hypercholesterolemia in the United Kingdom. Arterioscler Thromb. 1992;12:762–770. doi: 10.1161/01.atv.12.7.762. [DOI] [PubMed] [Google Scholar]
- 74.Leitersdorf E, Eisenberg S, Eliav O, et al. Genetic determinants of responsiveness to the HMG-CoA reductase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation. 1993;87:III35–III44. [PubMed] [Google Scholar]
- 75.Jeenah M, September W, Graadt van Roggen F, de Villiers W, Seftel H, Marais D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia. Atherosclerosis. 1993;98:51–58. doi: 10.1016/0021-9150(93)90222-g. [DOI] [PubMed] [Google Scholar]
- 76.Junyent M, Gilabert R, Jarauta E, et al. Impact of low-density lipoprotein receptor mutational class on carotid atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis. 2010;208:437–441. doi: 10.1016/j.atherosclerosis.2009.07.058. [DOI] [PubMed] [Google Scholar]
- 77.Vega GL, Grundy SM. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia. J Clin Invest. 1986;78:1410–1414. doi: 10.1172/JCI112729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Innerarity TL, Weisgraber KH, Arnold KS, et al. Familial defective apoilpoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A. 1987;84:6919–6923. doi: 10.1073/pnas.84.19.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soria LF, Luewig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apoipoprotein B-100. Proc Natl Acad Sci U S A. 198;86:587–591. doi: 10.1073/pnas.86.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaffney D, Reid JM, Cameron I, et al. Independent mutations at codon 3500 of the Apolipoprotein B gene are associated with hyperlipidemia. Arterioscler Thromb Vasc Biol. 1995;15:1025–1029. doi: 10.1161/01.atv.15.8.1025. [DOI] [PubMed] [Google Scholar]
- 81.Pullinger CR, Hennessy LK, Chatterton JE, et al. Familial ligand defective apolipoprotein B: identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest. 1995;95:1225–1234. doi: 10.1172/JCI117772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rauh G, Keller C, Schuster H, Wolfran G, Zollner N. Familial defective apoilpoprotein B-100: a common cause of primary hypercholesterolemia. Clin Invest. 1992;70:77–84. doi: 10.1007/BF00422946. [DOI] [PubMed] [Google Scholar]
- 83.Soufi M, Sattler AM, Maerz W, et al. A new but frequent mutation of APOB-100-APOB His3543Tyr. Atherosclerosis. 2004;174:11–16. doi: 10.1016/j.atherosclerosis.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 84.Fouchier SW, Kastelein JJ, Defesche JC. Update of the molecular basis of familial hypercholesterolemia in the Netherlands. Hum Mutat. 2005;26:550–556. doi: 10.1002/humu.20256. [DOI] [PubMed] [Google Scholar]
- 85.Fouchier SW, Kastelein JJ, Sijbrands EJ. Familial defective apolipoprotein B versus familial hypercholesterolemia: an assessment of risk. Semin Vasc Med. 2004;4:259–264. doi: 10.1055/s-2004-861493. [DOI] [PubMed] [Google Scholar]
- 86.Schaefer JR, Scharnegl H, Baumstark MW, et al. Homozygous familial defective apolipoprotein B-100. Enhanced removal of apolipoprotein E-containing VLDLs and decreased production of LDLs. Arterio Thromb Vasc Biol. 1997;17:348–353. doi: 10.1161/01.atv.17.2.348. [DOI] [PubMed] [Google Scholar]
- 87.Varret M, Rabès JP, Saint-Jore B, et al. A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet. 199;64:1378–1387. doi: 10.1086/302370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 89.Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069–2074. doi: 10.1073/pnas.0409736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peterson AS, Fong LG, Young SG. Commentary: PCSK9 function and physiology. J Lipid Res. 2008;49:1595–1599. doi: 10.1194/jlr.CX00001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sawamura T. New Idol for cholesterol reduction? Clin Chem. 2009;55:2082–2084. doi: 10.1373/clinchem.2009.134023. [DOI] [PubMed] [Google Scholar]
- 92.Cameron J, Holla ØL, Ranheim T, Kulseth MA, Berge KE, Leren TP. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum Mol Genet. 2006;15:1551–1558. doi: 10.1093/hmg/ddl077. [DOI] [PubMed] [Google Scholar]
- 93.Cunningham D, Danley DE, Geoghegan KF, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14:413–419. doi: 10.1038/nsmb1235. [DOI] [PubMed] [Google Scholar]
- 94.Pandit S, Wisniewski D, Santoro JC, et al. Functional analysis of sites within PCSK9 responsible for hypercholesterolemia. J Lipid Res. 2008;49:1333–1343. doi: 10.1194/jlr.M800049-JLR200. [DOI] [PubMed] [Google Scholar]
- 95.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia C, Hobbs H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Gene. 25;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 96.Cohen JC, boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 97.Descamps OS, Leysen X, Van Leuven F, Heller FR. The use of Achilles tendon ultrasonography for the diagnosis of familial hypercholesterolemia. Atherosclerosis. 2001;157:514–518. doi: 10.1016/s0021-9150(01)00533-0. [DOI] [PubMed] [Google Scholar]
- 98.Williams R, Hunt S, Schumacher C, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–176. doi: 10.1016/0002-9149(93)90155-6. [DOI] [PubMed] [Google Scholar]
- 99.Defesche J. Familial hypercholesterolemia. In: Betteridge J, editor. Lipids and Vascular Disease. Vol. 6. London: Martin Dunitz; 2000. pp. 65–76. [Google Scholar]
- 100.Civeira F, Ros E, Jarauta E, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol. 2008;102:1187–1193. doi: 10.1016/j.amjcard.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 101.Marks D, Wonderling D, Thorogood M, Lambert H, Humphries E, Neil AW. Cost effectiveness analysis of different approaches of screening for familial hypercholesterolemia. BMJ. 2002;324:1303–1308. doi: 10.1136/bmj.324.7349.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tejedor D, Castillo S, Mozas P, et al. Spanish Familial Hypercholesterolemia Group. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem. 2005;51:1137–1144. doi: 10.1373/clinchem.2004.045203. [DOI] [PubMed] [Google Scholar]
- 103.Tejedor D, Castillo S, Mozas P, et al. Comparison of DNA array platform vs DNA sequencing as genetic diagnosis tools for familial hypercholesterolemia. Clin Chem. 2006;52:1971–1972. doi: 10.1373/clinchem.2006.073957. [DOI] [PubMed] [Google Scholar]
- 104.Civeira F, Jarauta E, Cenarro A, et al. Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J Am Coll Cardiol. 2008;52:1546–1553. doi: 10.1016/j.jacc.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 105.Junyent M, Gilabert R, Zambón D. The use of Achilles tendon sonography to distinguish familial hypercholesterolemia from other genetic dyslipidemias. Arterioscler Thromb Vasc Biol. 2005;25:2203–2208. doi: 10.1161/01.ATV.0000183888.48105.d1. [DOI] [PubMed] [Google Scholar]
- 106.Hudgins LC, Gordon BR, Parker TS, Saal SD, Levine DM, Rubin AL. LDL apheresis: an effective and safe treatment for refractory hypercholesterolemia. Cardiovasc Drug Rev. 2002;20:271–280. doi: 10.1111/j.1527-3466.2002.tb00097.x. [DOI] [PubMed] [Google Scholar]
- 107.Thompson GR. LDL apheresis. Atherosclerosis. 2003;167:1–13. doi: 10.1016/s0021-9150(02)00251-4. [DOI] [PubMed] [Google Scholar]
- 108.Thompson GR. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–255. doi: 10.1016/j.atherosclerosis.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 109.Baigent C, Landry M. Study of Heart and Renal Protection (SHARP) Kidney Int Suppl. 2003;63:S07–S10. doi: 10.1046/j.1523-1755.63.s84.4.x. [DOI] [PubMed] [Google Scholar]
- 110.Pearson T, Laurora I, Chu H, Kafonek S. The lipid assessment project. Arch Intern Med. 2000;160:459–467. doi: 10.1001/archinte.160.4.459. [DOI] [PubMed] [Google Scholar]