

Abstract
Familial hypercholesterolemia (FH) is a common inherited disorder that results in premature atherosclerosis. Diagnosis of FH is suspected on the basis of clinical criteria, but confirmation requires genetic testing. In the era of statins, early diagnosis and initiation of treatment can modify disease progression and outcomes. Therefore, cascade screening with a combination of lipid concentration measurements and DNA testing should be used to identify relatives of index cases with a clinical diagnosis of FH. Autosomal dominant FH is related to mutations in the low-density lipoprotein receptor (LDLR), apolipoprotein B-100 (APOB), or proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. Genetic screening of the LDLR gene is challenging to achieve at a feasible cost, especially in people who do not have a founder effect. Nucleotide sequencing of all exons and flanking splicing regions in combination with multiplex ligation probe amplification to detect large insertions or deletions is considered the gold-standard approach to screen for LDLR mutations. Alternatively, the cDNA can be sequenced; however, this procedure is not suitable for use in large populations, because of the need of RNA extraction. Multiplex analysis can be appropriate for population with founder effects or a low number of different mutations. Finally, there are many techniques for a mutation scanning approach, which have some benefits over sequencing, and also with the potential for detecting known and novel mutations. Familial defective Apo B is amenable to genetic diagnosis by screening for a few mutations. Recently, gain-of-function mutations in PCSK9 gene have been demonstrated to cause FH phenotype. Strategies for population screening, cost-effectiveness of genetic screening, ethical aspects, and insurance policies are discussed and need implementation worldwide.
Keywords: familial hypercholesterolemia, familial defective Apo B, cholesterol, mutations, genetic screening
Historical perspective
Familial hypercholesterolemia (FH) is the most commonly inherited disorder of lipid metabolism, characterized by an increase in total- and low-density lipoprotein cholesterol (LDL-C), and with a high incidence of premature coronary heart disease (CHD).1 The first observations of the disease came from the pathologist Harbitz, who reported in the 18th century several cases of sudden death and xanthomata, before it was described by Müller as a clinical entity in 1938.2,3 The latter observed that the coincidence of dyslipidemia, xanthomatosis, and CHD was very common in families and was inherited as a dominant trait. Only 50 years later, Goldstein and Brown,4–6 studying patients and cultured cells, unraveled the cholesterol synthetic pathway and identified a defect in the internalization of receptor-bound low-density lipoprotein (LDL-R), which won them the Nobel Prize in 1985. These authors related the disease to a defect in this protein. In 1983, the DNA for this gene was finally cloned7 and the gene mapped to the short arm of chromossome 19, referred to as the low-density lipoprotein receptor (LDLR) gene in 1989.8 Mutations in the LDLR gene reduce the number or impair function of such receptors at the hepatocyte surface, leading to decreased LDL catabolism, thus resulting in marked LDL-C elevation and cholesterol deposition in tissues.
In the majority of cases, the genetic basis of FH can be defined as a monogenic disorder, and the mode of inheritance can be autosomal dominant (ADH)1 or recessive (ARH);9,10 however, not all dominant disorders have a gene–dose effect. In the ADH, Khachadurian differentiated the heterozygous from the homozygous form in affected Lebanese families.1
The two best-known genes implicated in such disorders are the LDLR gene and the apolipoprotein B-100 (APOB) gene.11 Mutations in these genes give rise to two types of diseases, known as the classical FH (FH, OMIM# 143890) and familial defective Apo B-100 (FDB, OMIM #144010), respectively. The phenotype is almost indistinguishable; however, FH caused by LDLR gene defects more often presents clinical signs and features a more severe hypercholesterolemia than when related to FDB. Recently, a newly discovered protein, neural apoptosis-regulated convertase 1, the product of the gene proprotein convertase subtilisin/kexin type 9 (PCSK9), has been referred to as autosomal dominant ‘FH3’ (FH3, OMIM #603776). The so-called gainof- function (GOF) mutations in this gene accelerate LDL-R catabolism featuring the ADH phenotype.12
Hypercholesterolemia is inherited in an autosomal recessive manner as a result of mutations in adenosine-binding cassette transporter (ABC) proteins G5 and G8, in a disease called sitosterolemia or phytosterolemia,13,14 when mutations that disrupt the function of an adaptor protein are present,8,9 or due to mutations in 27-hydroxylase (CYP27) gene that cause cerebrotendinous xanthomatosis.15 Although sitosterolemia can have significant hypercholesterolemia, it seldom exceeds the heterozygous FH range. Cerebrotendinous xanthomatosis is not associated with severe hypercholesterolemia.
Other causes of hypercholesterolemia8 are polygenic which can be sporadic or due to APOE gene polymorphisms (ɛ4) alleles.
Usually, the clinical pattern consists of severe hypercholesterolemia due to the accumulation of LDL in the plasma, cholesterol deposition in tendons and occasionally in skin, and with a high risk of premature atherosclerosis, manifested mainly as CHD.1
The frequency of ‘classical’ FH, attributed to LDLR gene defects, is estimated to be 1:500 for heterozygotes in most populations (European, North American, and Japanese) and is therefore thought to be one of the two most common human diseases caused by mutations in a single gene; however, homozygotes are very rare (1:1,000,000).16 FH is more prevalent in certain populations: 1:100 Afrikaners, 1:170 Christian Lebanese, and 1:270 French Canadians, where a founder effect can be found.17 Heterozygous subjects have half the number of functioning receptors compared to normal individuals, suggesting a codominant effect.
Familial defective apolipoprotein B-100 was first described in 1989 with the identification of a mutation at codon 3500 of the APOB gene resulting in the substitution of the amino acid arginine by glutamine (R3500Q).11 Mutations around the 3500 codon affect the recognition and binding of Apo B-100 to LDL receptor, causing increase in the plasma values of LDL-C. FDB phenotype is similar to heterozygous FH and occurs in 1:300–1:700 individuals in central Europe.18,19 The prevalence of FH secondary to GOF mutations in PCSK9 gene is not established. Autosomal recessive FH is very rare; sitosterolemia, although very rare, is relatively common in Sardinia. Table 1 presents the major characteristics of genetic causes of hypercholesterolemia. For the purposes of this review, we will focus on ADH.
Table 1.
Major characteristics of familial hypercholesterolemia
| Lipid phenotype | Genetic alteration | Mutation | Mode of inheritance | Population frequency | Presentation in infancy | Clinical features | Premature CHD | Ref # |
|---|---|---|---|---|---|---|---|---|
| LDL-C ↑↑↑ (TC ↑↑↑) | Classical FH | LDL-R | Codominant | 1:500 (heterozygous) | + | +++ | 1,8,16,17 | |
| 1:1,000,000 (homozygous) | + | Tendon xanthomas, Xanthelasmas, Corneal arcus | ++++ | |||||
| Defective Apo-B (FDB) | APOB Arg 3500 Gln | Dominant | 1:700 | + | ++ | 11,18,19 | ||
| Autosomal dominant 3 (HF3) | PCSK9 GOF mutations (1p32) | Dominant | 0.2%–34% | + | ++ | 12 | ||
| Autosomal recessive | Adaptor protein | Recessive | rare | + | +++ | 8,9 | ||
| Sitosterolemia | ARH | Recessive | rare | + | +++ | 13,14 | ||
| Sterol 27-hydroxylase gene |
ABC G5/G8 CYP27 |
Recessive | rare | + | +++ | 15 | ||
| TC ↑ or ↑↑ LDL-C ↑ or ↑↑ |
Polygenic hypercholesterolemia | ? | Multiple genes | 1:100–5:100 | + | – | + | 8 |
| TC ↑ or ↑↑ LDL-C ↑ or ↑↑ |
– | APOE4 | Codominant | ~5% of TC variation | Variable | – | + if LDL-C ↑↑ | 8 |
Diagnosis criteria of FH
Diagnosis of FH is mainly based on lipid levels, clinical characteristics of the proband, family history of dyslipidemia/premature CHD, and genetic analysis. There are not absolute clinical criteria for the diagnosis, and therefore, arbitrary criterion should be used. Initially, a clinical diagnosis of heterozygous FH should be considered in individuals with LDL-C concentrations greater than 190 mg/dL (4.9 mmol/L), and they should be referred to a specialist center.20,21 Secondary causes of hypercholesterolemia should be excluded before a diagnosis of FH is made. Diagnosis of FH needs to be confirmed by at least two measurements of elevated LDL-C concentrations, due to biological and analytical variability of the assay. In addition, absence of clinical signs (corneal arcus and tendon xanthomata) in adults and children does not exclude the diagnosis of FH. A family history should be obtained from an individual being investigated for FH to determine the pattern of inheritance. A pedigree analysis should include at least the third generation of the proband with relatives’ age of onset of CHD and lipid concentrations. For deceased relatives, the age and cause of death should be reported.21
There are three different diagnosis criteria developed for FH. The USA Make Early Diagnosis to Prevent Early Deaths (USA MEDPED) Program diagnostic criteria has focused mainly on LDL-C levels in the individual and in family history of hypercholesterolemia with evidence for a dominant transmission. If there are children with hypercholesterolemia, the probability of the diagnosis increases.20 In addition to lipid values, clinical examination showing corneal arcus before age 45 years and tendon xanthomata increases the diagnostic accuracy.
The Dutch MedPed developed a scoring system that includes personal and familial LDL-C levels, history of cardiovascular disease (coronary, carotid, and peripheral arteries), and the presence of corneal arcus before the age of 45 years, xanthomas, and the detection of functional mutations in LDLR gene.20 The relative weight of each of these clinical signs, alone or in combination with others, receives a score that has been constructed for the population of The Netherlands. The third scoring system for the diagnosis of FH is the Simon Broome21 that includes a combination of family history, clinical examination (arcus and tendon xanthomata), lipid profile, and presence of mutations in the LDLR gene. This score was developed in the UK.
These criteria are easy to use in clinical practice, and some centers prefer a combination of them for the diagnosis of FH. However, the diagnosis is not always unequivocal. A study that compared the sensitivity and specificity of different clinical criteria for diagnosing FH showed that the Simon Broome criteria performed at least as well as the Dutch criteria for individuals with definite FH, and both Simon Broome and the Dutch criteria demonstrated better performance than the USA MEDPED.22
In adults and children, the effectiveness of the following tests to diagnose heterozygous FH in individuals with a family history of early heart disease and/or hypercholesterolemia has been discussed. The use of corneal arcus for case finding was studied in a UK population, in which a graded prevalence of corneal arcus with age is found.23 Some degree of arcus affected 50% of individuals with FH by age 31–35 years and 50% of healthy individuals by age 41–45 years; complete full ring arcus affected 50% of the FH group by the age of 50 years, with only 5% similarly affected in the healthy group. In addition, arcus grade was not related to the presence of coronary disease. Even in patients with mutations in the LDLR gene, many heterozygous individuals do not present evident xanthomas. The accuracy in the detection of xanthomas can be improved by using ultrasonography of the Achilles tendon; Achilles tendon’s thickness is higher in FH than in non-FH hypercholesterolemia. Thickened tendons were more prevalent (63%) in FH when compared to non-FH individuals (4%) as well as low or mixed echogenicity of tendons (90% in FH versus 6% in non-FH hypercholesterolemia).24 In addition, xanthomatosis was detected in 68% of FH with mutations and in none of the mutation-negative individuals. Ultrasonography of the Achilles tendon shows a good negative predictive value; however, guidelines do not recommend the use of ultrasonography of the Achilles tendon in the diagnosis of FH.25
Two years ago, a full recommendation had been issued in the UK, the NICE Clinical Guideline on FH, which accomplishes major points in the management of the disease.21
Genetic screening appears as the only certainty criterion in the assessment of individuals with FH and enables us to screen relatives in order to manage dyslipidemia and prevent premature cardiovascular disease. However, in many suspected FH subjects, no mutation is found.
Diagnosing FH in children
Children of parents with known FH should be investigated, but clinical signs are less frequent than in affected adults. In Afrikaners, total and LDL-C concentrations were significantly higher in children with a detected mutation compared to those without a mutation,26 but the sensitivity and specificity are considerably lower at young ages. Clinical signs are far less frequent in affected children. When ultrasonography of the Achilles tendon was assessed, thickness was higher than in controls: one-third presented abnormal tendons before age 10, and 42% between 10 and 18 years.27
Assessment of cardiovascular risk stratification
Familial hypercholesterolemia accounts for 5%–10% of CHD in patients less than 55 years of age.28 Without treatment, 50% of heterozygous males will develop CHD before the age of 50 and 100% by the age of 70 years, and about 12% of heterozygous females will have CHD by the age of 50 years, increasing to 74% by 70 years.29 However, clinical expression of CHD in heterozygous FH patients is highly heterogeneous in terms of the age of onset and severity. CHD tends to cluster with higher frequency in certain families, but marked differences among individuals can occur,30 even among subjects coming from families sharing the same mutations in the LDLR gene, indicating that other genetic or environmental factors play an important role in the development of atherosclerosis in FH.31 Therefore, a number of risk factors have been established for FH, in order to stratify risk. The application of standard risk estimates for cholesterol seriously underestimates CHD risk in FH; therefore, Framingham risk score should not be used in this situation.32,33 Major risk factors for CHD in FH patients are: age (men ≥ 30 years or women ≥ 45 years); active smokers; family history of premature CHD (male first-degree relative < 55 years or female first-degree relative < 65 years); very high LDL-C (> 330 mg/dL or 8.5 mmol/L); low HDL-C (< 40 mg/dL or 1.0 mmol/L); high blood pressure (> 140/90 mm Hg); diabetes mellitus, Lp (a) > 60 mg/dL.20
Asymptomatic individuals with FH should be evaluated for subclinical CHD based on the demonstration of myocardial ischemia through a stress test (exercise electrocardiogram test, stress echocardiography, or radio nuclide test) or demonstration of coronary atherosclerotic plaques through myocardial perfusion imaging, detection of coronary calcium by electron beam computed tomography, or multislice (spiral) CT. Subclinical atherosclerosis in other locations should be assessed by noninvasive ankle-brachial index, carotid intimal medial thickening by sonography, and abdominal sonography for the detection of aortic aneurysms.20
Cascade testing
When an individual is being investigated for FH, a family history should be obtained to determine if a dominant pattern of inheritance is present. In children of an affected parent, LDL-C concentrations should usually be measured by the age of 10 years and repeated after puberty before a diagnosis of FH can be excluded. However, other authors propose that blood samples are obtained at 15 months of age, when attending the vaccination.34 Making a diagnosis as early as possible will ensure preventive and therapeutic strategies. It is recommended to use systematic methods for case identification of FH, and there should be a nationwide family-based follow-up system to enable comprehensive identification of affected individuals. However, these tools are not well standardized worldwide. A diagnosis confirmation by a specialist with expertise in FH should be made with initiation of cascade testing. Cascade testing uses a combination of lipid concentration measurement and DNA testing, and should be used to identify relatives of index cases with a clinical diagnosis of FH. In the absence of a DNA diagnosis, cascade testing using lipid measurements alone should be undertaken. Health care professionals should discuss the implications of cascade testing with individuals. In families in which a mutation has been identified, the mutation should be used to identify affected relatives. During the assessment and communication of familial risk, individuals should receive clear and appropriate educational information about FH and about the process of family testing. A specialist with expertise in FH should provide information to affected individuals on their specific level of risk of CHD, the implications for them and their families, lifestyle advice, and treatment options. Individuals with FH should be encouraged to contact their relatives to inform them about their potential risk thus facilitating cascade testing. Individuals and families with FH should be offered written advice and information about patient support groups.21
In whom should the investigation be performed?
Any individual with a personal or family history of CHD or hypercholesterolemia should be investigated for FH. In addition, as FH is an autosomal dominant disorder, a child with the disease will have an affected parent. Identification of an affected child from 1 to 9 years provides an opportunity to identify affected parents. Therefore, screening in children accomplishes two aims: diagnosing children and their parents.
Treatment to lower cholesterol could be initiated immediately in the parent and judged in affected children. Studies show that early initiation of statin therapy in children with FH might be beneficial in the prevention of atherosclerosis in adolescence.35 The decision to initiate drug therapy for a child or young person should take into account their age, the age of onset of cardiovascular disease within the family, and presence of other cardiovascular risk factors, including LDL-C concentrations greater than 6.0 mmol/L (or 230 mg/dL).20 If the decision to initiate statins has been made in children and young people (aged 10 years upward), physician should choose drugs licensed for use in the appropriate age group.20 Management of lifetime therapy with statins should consider other aspects such as contraception in women, pregnancy, lactation, and genetic counseling, for as many as 50% of the descendants can be affected by the disease.21
Genetic screening
Most people with FH, a highly atherogenic condition, are undiagnosed. Early detection and treatment can reduce morbidity and mortality; however, specialists worldwide agree that clinical diagnostic criteria are lacking. Genetic testing for LDLR and APOB gene defects, and more recently for PCSK9 gene mutations, is the preferable diagnostic method; however, the best phenotype guide when a genetic diagnosis should be performed is not clearly established.
Most geographically based surveys of FH patients show a large number of mutations in a given population, except when a founder effect is present in the population. At the molecular level, FH is most common due to mutations in the LDLR gene (Figure 1),36 in which more than 1100 different allelic variations spanning the entire length of the gene have been detected worldwide.37–39 For PCSK9, there are more than 50 amino acid variants described (most common mutations are presented in Figure 2). These changes are classified as GOF mutations, when associated with high levels of LDL-C, or loss-of-function mutations, when associated with low LDL-C levels.12 There is a limited number of mutations in APOB gene (eg, R3500Q), which disrupts the binding site to the LDL-R and are associated with ADH.17,18 When a founder effect can be found in a population, or when the proband’s genotype is known, investigating mutations can be driven by the previous knowledge of the probable genetic defect. The disease is much more common in some population groups where the majority of FH subjects have few LDLR gene mutations, as a result of a founder effect. These population groups include French Canadians,40,41 Afrikaners,42,43 Lebanes e,44 Finns,45 and Ashkenazi Jews.46,47 In some of these groups, the frequency of the homozygous condition is more than 10-fold higher than in other populations, mainly due to the high prevalence of heterozygosity of FH coupled with a high incidence of consanguinity, which increases the proportion of homozygotes. The already described mutations accomplish missense, nonsense, splice-site mutations, frameshift mutations (insertions and deletions), and in-frame deletions, neutral and silent mutations. All the allelic variants described can be assessed consulting the database at http://www.ucl.ac.uk/ldlr/Current/index.php?select_db=LDLR.
Figure 1.
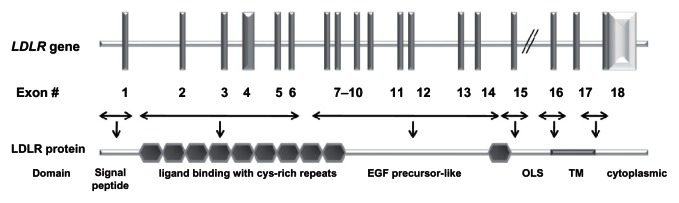
Schematic representation of the LDLR gene and LDLR protein structures.36
Abbreviations: OLS, O-linked sugar domain; TM, transmembrane.
Figure 2.

Schematic representation of the PCSK9 gene structure with gain-of-function and loss-of-function mutations.
Copyright © 2006, American Heart Association. Adapted with permission from Berge KE, Ose L, Leren TP. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol. 2006;26(5):1094–1100.
Abbreviations: NARC1, neural apoptosis-regulated convertase 1; SS, signal sequence; Pro, N-terminal prodomain.
However, if the mutation is unknown then virtually any part of the gene may originate the disease, strategies for the identification of unknown mutations on a gene-wide basis must be employed. Molecular diagnosis of FH can be time-consuming and expensive because of the size of the LDLR, which spans 45 Kb on the short arm of chromosome 19 and consists of 18 exons and 17 introns.48
Type of technique: advantages, disadvantages, and costs
Methods for early detection of FH gene carrier status allow primary prevention strategies in children with FH at a young age without the risk of misclassification of FH status. Because of the lack of mutational hot spots in the LDLR gene, many difficulties in the accurate diagnosis of FH in families can occur using traditional, clinical, and biochemical diagnostic criteria; therefore, there is a need for simple diagnostic tools, such as genetic analysis of the entire coding region of the LDLR gene, that can also be used in populations without founder mutations.
Three basic approaches to LDLR genetic analysis are now available: i) the gold-standard approach of full nucleotide sequencing of the LDLR gene, ii) the use of a selected panel of common LDLR mutations, and iii) a gene scanning approach to analyze the LDLR gene, using denaturing high-performance liquid chromatography (D-HPLC) or high-resolution melting (HRM), as options.
Earlier molecular methods used for detection of LDLR mutations included single-strand conformation polymorphism (SSCP) analysis,49–52 denaturing gradient gel electrophoresis (DGGE),53,54 and denaturing HPLC (DHPLC).55,56 Recent approaches include sequencing of the LDLR cDNA region,57 a SSCP/heteroduplex method followed by capillary electrophoresis,58 and melt-microplate array diagonal gel electrophoresis,59 which are suitable for the detection of new mutations.
SSCP and DGGE are expensive and time-consuming to be practical for population screening. These techniques have disadvantages, such as use of radioactivity, requirement of various hybridizations and electrophoresis conditions, and the need to confirm the exact nature of the alterations by DNA sequencing. In addition, the sensitivity of these scanning methods is low (70%–80%), and they require considerable technical skill. Scanning for mutations had a step forward with the use of DHPLC.55 This approach is automated and has high sensitivity (>96%) if amplicons are well-designed and temperatures are optimized.
If common LDLR mutations are known in a population, a selected panel can be used as first strategy. Tejedor et al60 developed a low-density oligonucleotide microarray to identify 118 DNA sequence variations (117 for the LDLR gene and 1 for the APOB gene). The DNA array uses specific oligonucleotide probes for FH-causing mutations with the more frequent LDLR mutations known to affect certain country and which are regularly updated. They verified specificity and sensitivity by analyzing 1180 previously sequenced DNA samples, and conducted a blind study screening in 407 Spanish patients with a clinical diagnosis of FH. The DNA array confirmed the previous genotyping results in almost all cases and had specificity and sensitivity of 99.7% and 99.9%, respectively. The microarray detected at least 1 mutation in 51% of the patients for whom clinical diagnosis was classified as certain (according to Dutch FH-MEDPED criteria) and in 37% of those with a diagnosis of probable or possible FH, thus giving a definite diagnosis.61 Oliva et al62 used the LIPOchip® platform to screen close relatives of affected individuals followed by treatment, which included three diagnostic procedures, sequentially: an updated version of the previous DNA array; in case DNA array analysis was negative, multiplex quantitative PCR was used to identify significant rearrangements, and if the two previous analyses were negative, the sample was analyzed through complete sequencing of the LDLR gene. They found that genetic screening of close relatives followed by treatment was cost-effective compared with no screening, considering the increment in costs for screening and treatment, and the life years gained. Blesa et al63 proposed a combination of Southern blot technique to detect large rearrangements and a semiquantitative fragment analysis covering the entire LDLR gene. Small sequence variations in genes were detected by amplification of fragments containing individual exons and their splice intron–exon junctions, and sequencing. These authors compared their results with those observed by Tejedor et al60,61 and found that even in a population with the same ethnic background, a large variability can occur, with the full sequencing being cheaper and preferable to microarray, for many mutations could not be detected with the chip. Comparing the two techniques, the throughput of the platform was estimated by the authors to be 5000 analyses/year, with costs estimated in €425 (or U$505.00); however, a laboratory using a midsized DNA sequencer can analyze 1000 sequences, or more than 20,000 fragments/day, which means that 25 patients could have the full LDLR gene sequenced, with large rearrangements detected by semi-quantitative procedures in 300 patients/day, at costs of €250 (or U$297.00).61,63
Laios and Drogari64 tested a multiplex assay to detect common LDLR gene mutations in FH patients from Greece. Basically, they used the microelectronic array technology of the NanoChip® Workstation to develop a multiplex method to analyze three single-nucleotide polymorphisms (SNP) (the Greece II, Genoa, and Afrikaner-2 mutations) accounting for 49% of FH cases in Greece. This assay was compared with three monoplex assays (1 SNP/site) and resulted in agreement in most of the cases. Of the 580 multiplex genotypings, 576 agreed with the monoplex results. However, duplicate sites of one sample were not in agreement with each other, requiring repetition, upon which, discrepancies were resolved. One problem with this technique was not obtaining an allele designation due to suboptimal PCR amplification, improper desalting, and increased background. This multiplex array can be extended to accommodate additional mutations. The cost of the system is primarily related to the cartridge. The authors reported that their multiplex protocol costs €1.55/genotype (without including reagent cost); if additional mutations in the same amplicon (four mutations have been observed in Greeks) can be detected, the cost per genotype could be reduced to €0.66/genotype.64
Another option to provide a rapid means of mutation screening and reduce costs is described by Taylor et al.65 The authors developed an amplification refractory mutation system (ARMS) specifically designed to detect 11 mutations in the LDLR gene, one common mutation in the APOB gene (p.Arg3527Gln, formally R3500Q) and one mutation in the PCSK9 gene (p.Asp374Tyr, D374Y). These mutations were selected on the basis of their frequency in a cohort of 400 UK patients with FH. The authors compared the utility of this assay in a cohort of patients with definite or possible diagnosis of FH. The ARMS method is based on having a set of primer pairs for each mutation to be detected, one of which has, at its 3′ end, the mutation-specific base; under appropriate conditions, amplification can occur if the DNA sample carries at least one copy of the mutation. This method detected mutations in 54 of 400 suspected FH patients. Each detected mutation had to be further tested by either sequence or restriction digestion to confirm the result and determine the zygosity status. The ARMS was complemented by extended mutation screening of the 18 exons and promoter region of the LDLR gene by SSCP or DHPLC analysis. No false-positive or false-negative results were detected from the kit. Design and optimization of the primers require a substantial amount of time, and a well-designed ARMS assay needs balanced primer sequences that are both sensitive and specific on DNA of different qualities and extraction methods. ARMS primers matches/mismatches must be chosen to ensure good discrimination of target DNA sequences, avoiding ambiguity of detection. In addition, melting temperature of different amplifying primers must be similar, and the final concentration of primers in the mix must be optimized for best results. The ARMS test is efficient and seems costeffective, although no formal cost comparisons have been carried out. The test is simple, reliable, does not require specialized equipment, and results can be obtained in 2 days. Large multiplex ARMS containing more primer mixes are feasible, increase the number of mutations detected, but agarose gel detection of PCR limits the useful size of the multiplex. The method is suitable for first approach and can be used for a wider patient group.
Among the techniques that aim to evaluate the entire LDLR gene, a rapid and accurate method for the detection of all the mutations is sequencing the whole cDNA for the LDLR gene. This method employs a combination of DNA sequence analysis programs to allow a quantitative estimation of sequence quality, visual display of integrated sequence information, detection of the zygosity status, and permits identification of both new and already described mutations in patients with FH, as reported by Liguori et al.57 The RNA extracted is used as a template for RT-PCR, generating a set of overlapping fragments that cover the whole coding region. All fragments are sequenced on both strands, and an automated, computer-assisted analysis follows, in which the complete cDNA sequence is reassembled and compared with the wild-type sequence to identify known and new mutations. This method is integrated by a standard validation procedure, based on direct DNA sequencing the corresponding genomic DNA exon to identify or confirm the nature and location of the mutations identified during the primary scan. Deletions or insertions can be detected with accuracy. An advantage of this method is the relatively low cost and the reduction in time for results compared with other methods, allowing an overnight diagnosis (2 days including validation by independent exon sequencing). Another advantage is the possibility of high automation in all the steps, eliminating the need of human intervention. This simplifies diagnostic testing, is suitable for large-scale population screenings, and allows molecular diagnosis of genetic disorder at a presymptomatic early age.
The development of novel molecular methods, such as DHPLC and its combination with direct sequencing, has substantially aided in the diagnosis of many genetic disorders involving large genes. Bodamer et al55 have evaluated patients with a definite diagnosis of FH to assess the analytical conditions of DHPLC followed by direct sequencing. PCR was carried out with already described primers sequences and respective annealing temperatures. The PCR products were denatured at 95°C, and the temperature at which heteroduplex detection occurred was deducted from a software, which analyzes the melting profile of the specific DNA fragment. Subsequently, PCR fragments from individuals with FH presenting abnormal DHPLC heteroduplex formation compared with controls were sequenced. The sensitivity and specificity of DHPLC appear to be >96% irrespective of sequence variations with the exception of high-melting domains surrounded by lower-melting sequences. The optimum amplicon size varies between 150 and 700 bp, depending on GC content. Therefore, large deletions beyond the amplicon or rearrangements in the LDLR gene cannot be detected by this method, and require other methods, such as Southern blotting. DHPLC may be cost-effective in the diagnosis of FH compared with other analytical methods, such as sequencing all exons without prior screening.
HRM analysis is the next generation of mutation scanning technology and can be cost-effective, requiring less time for results when compared to other scanning techniques. Post-PCR amplicons in the presence of a saturating double-stranded DNA-binding dye are slowly heated with concomitant fluorescence measurement. In samples heterozygous for a mismatch mutation, the presence of heteroduplexes will result in a lower melting temperature of the amplicon, yielding an altered melting curve compared with normal samples which have only homoduplexes. Therefore, HRM analysis seems to be an attractive method of screening for LDLR mutations. Laurie and George66 evaluated samples of patients with a definite diagnosis of heterozygous FH due to mutations in LDLR gene. All samples were amplified, and PCR products visualized on agarose gel to confirm a single amplicon for each primer were obtained. Following amplification, PCR products were analyzed in a melting step (75°C–95°C) with 20 data acquisitions/°C. Melting data were analyzed using specific software, incorporating normalization of pre- and post-melt regions, temperature shift adjustment, and calculation of difference curves. Primers were selected to have 200–300 bp size, with only one melting domain, using the primers and conditions that had been previously optimized for DHPLC. With the exception of exon 4b, which was problematic to analyze due to the high GC content, 96% (52/54) of the sequence variants were detected by HRM. Another problem is the presence of benign polymorphisms in several exons or in flanking intronic regions that can be detected by melting analyses, when compared to normal samples, but the difference curve cannot be used to discriminate samples from heterozygous individuals with such mutations or those carrying pathogenic mutations in the same exon. This technique is not suitable for scanning amplicons with a high frequency of polymorphisms, for HRM is not 100% sensitive. However, mutation scanning has the advantage of being rapid. A plate of 96 samples can be melted in 10 min, which is 100 times faster than DHPLC; mutation scanning by HRM avoids the use of solvents, maintenance of pumps and injectors, and the challenges with DNASep column.
Finally, the method which could be considered the gold-standard approach for detecting mutations in LDLR gene is nucleotide sequencing of all exons and the flanking splicing regions, combined with multiplex ligation probe amplification to detect large insertions or deletions. Sequencing, although expensive, presents almost 100% sensitivity in the regions examined. As capillary sequencing platforms become available in many laboratories, the balance often favors sequencing.
Analysis of cost-effectiveness of screening close relatives followed by treatment
Fifty percent of men and 20% of women with heterozygous FH who do not receive appropriate treatment will present an acute coronary episode in the fifth decade. There is an increased risk of fatal coronary events also in people with FH below 40 years of age. After the era of statins, mortality rates in FH have been markedly decreased. Early detection and initiation of therapy are essential and responsible for these changes. Many studies have demonstrated that screening close relatives of affected members diagnosed with FH is the most cost-effective preventive strategy. After diagnosis, the majority of patients initiate therapy and therefore will be less susceptible to cardiovascular events. A study evaluated the cost-effectiveness of genetic screening for first-degree relatives of patients with FH, followed by treatment when necessary, and compared with the alternative of no screening. Genetic screening for first-degree relatives of people diagnosed with FH, and subsequent treatment, was found to be an efficient alternative when compared with the alternative of no screening. The strategy of genetic screening plus treatment in first-degree relatives of people diagnosed with FH would have a 95% probability of being efficient. Genetic screening identifies patients at a younger age and improves treatment and treatment compliance. The results obtained provide support for the implementation of a plan for the detection of FH.62
Ethical implications of genetic screening and consequences for life and disability insurance
Patients with FH have age- and sex-standardized cardiovascular mortality rates four to five times higher than the general population. The active identification of FH by DNA testing started an intense debate focused on themes such as cost-effectiveness, right to privacy, and ethical dilemmas like access to insurance.
The goal to identify all FH carriers through a large-scale molecular screening program does not seem to be limited by a restriction in terms of access to insurance due to special guidelines for insurance companies on how to deal with risk assessment and genetic screening for FH.67 A large-scale screening program for FH offers the opportunity to serve as a model for screening for other hereditary diseases. Excess mortality rates were not detected in other homozygous conditions, such as haemochromatosis.
In summary, FH is a serious and common disorder that results in premature atherosclerosis. Diagnosis of FH is based on clinical criteria, but confirmation requires genetic testing. With the advent of statins, early diagnosis and initiation of treatment have modified disease progression and outcomes. Therefore, identification of affected individuals and screening of their relatives is an important issue. Molecular and metabolic differences in disorders producing the FH phenotype can have different impact on disease, and the knowledge of such differences can enable health professionals in the clinical management of affected patients. Genetic screening of the LDLR gene is challenging to achieve at a feasible cost, especially in people who do not have a founder effect. Nucleotide sequencing of all exons and flanking splicing regions in combination with multiplex ligation probe amplification to detect large insertions or deletions is considered the gold-standard approach to screen for LDLR mutations. Multiplex analysis can be appropriate for populations with founder effects or a low number of different mutations. There are many techniques for a mutation scanning approach, which have some benefits over sequencing, and also with the potential for detecting known and novel mutations. Cost-effectiveness of genetic screening, ethical aspects, and insurance policies still need to be discussed and demand implementation worldwide.
Footnotes
Disclosure
The authors declared no conflict of interest.
References
- 1.Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med. 1964;37:402–407. doi: 10.1016/0002-9343(64)90196-2. [DOI] [PubMed] [Google Scholar]
- 2.Ose L. Müller-Harbitz disease – familial hypercholesterolemia. Tidsskr Nor Laegeforen. 2002;122(9):924–925. [PubMed] [Google Scholar]
- 3.Müller C. Xanthomata, hypercholesterolemia and angina pectoris. Acta Med Scand. 1938;89:75–84. [Google Scholar]
- 4.Brown MS, Goldstein JL. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor bound low density lipoprotein. Cell. 1976;9(4 Pt 2):663–674. doi: 10.1016/0092-8674(76)90130-6. [DOI] [PubMed] [Google Scholar]
- 5.Brown MS, Goldstein JL. Receptor-mediated control of cholesterol metabolism. Science. 1976;191(4223):150–154. doi: 10.1126/science.174194. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 7.Russell DW, Yamamoto T, Schneider WJ, Slauther CJ, Brown MS, Goldstein JL. c-DNA cloning of the bovine low density lipoprotein receptor: feedback regulation of a receptor mRNA. Proc Natl Acad Sci U S A. 1983;80(24):7501–7505. doi: 10.1073/pnas.80.24.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Motulski AG. Genetic aspects of familial hypercholesterolemia and its diagnosis. Arteriosclerosis. 1989;9( Suppl I):I3–I7. [PubMed] [Google Scholar]
- 9.Garcia CK, Wilund K, Arca M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292(5520):1394–1398. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- 10.Morganroth J, Levy RI, McMahon AE, Gotto AM., Jr Pseudohomozygous type II hyperlipoproteinemia. J Pediatr. 1974;85(5):639–643. doi: 10.1016/s0022-3476(74)80506-8. [DOI] [PubMed] [Google Scholar]
- 11.Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, Mc Carthy BJ. Association between a specific apoprotein B mutation and familial defective apo B 100. Proc Natl Acad Sci U S A. 1989;86(2):587–591. doi: 10.1073/pnas.86.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharyya AK, Connor WE. Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest. 1974;53(4):1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel SB, Salen G, Hidaka H, et al. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest. 1998;102(5):1041–1044. doi: 10.1172/JCI3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH. Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations. Arch Neurol. 2002;59(4):527–529. doi: 10.1001/archneur.59.4.527. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein JL, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Bases of Inherited Diseases. New York: McGraw-Hill; 1989. pp. 1215–1250. [Google Scholar]
- 17.Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. pp. 2863–2913. [Google Scholar]
- 18.Rauh G, Keller C, Schuster H, Wolfgram G, Zöllner N. Familial defective apolipoprotein B-100: a common cause of primary hyperlipemia. Clin Investig. 1992;70(1):77–84. doi: 10.1007/BF00422946. [DOI] [PubMed] [Google Scholar]
- 19.Miserez AR, Laager R, Chiodetti N, Keller U. High prevalence of familial defective apolipoprotein B-100 in Switzerland. J Lipid Res. 1994;35(4):574–583. [PubMed] [Google Scholar]
- 20.Civeira F. International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173(1):55–68. doi: 10.1016/j.atherosclerosis.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 21.NICE. Clinical Guideline on Familial Hypercholesterolaemia. [Accessed 2010 Jun 8.]. Available from: http://guidance.nice.org.uk/CG71/NICEGuidance/doc/English.
- 22.Damgaard D, Larsen ML, Nissen PH, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis. 2005;180(1):155–160. doi: 10.1016/j.atherosclerosis.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Winder AF, Jolleys JC, Day LB, Butowski PF. Corneal arcus, case finding and definition of individual clinical risk in heterozygous familial hypercholesterolaemia. Clin Genet. 1998;54(6):497–502. doi: 10.1111/j.1399-0004.1998.tb03770.x. [DOI] [PubMed] [Google Scholar]
- 24.Koivunen-Niemela T, Alanen A, Viikari J. Sonography of the Achilles tendon in hypercholesterolaemia. J Intern Med. 1993;234(4):401–405. doi: 10.1111/j.1365-2796.1993.tb00762.x. [DOI] [PubMed] [Google Scholar]
- 25.Junyent M, Gilabert R, Zambon D, et al. The use of Achilles tendon sonography to distinguish familial hypercholesterolemia from other genetic dyslipidemias. Arterioscler Thromb Vasc Biol. 2005;25(10):2203–2208. doi: 10.1161/01.ATV.0000183888.48105.d1. [DOI] [PubMed] [Google Scholar]
- 26.Kotze MJ, Peeters AV, Loubser O, et al. Familial hypercholesterolemia: potential diagnostic value of mutation screening in a pediatric population of South Africa. Clin Genet. 1998;54(1):74–78. doi: 10.1111/j.1399-0004.1998.tb03698.x. [DOI] [PubMed] [Google Scholar]
- 27.Koivunen-Niemela T, Viikari J, Niinikoski H, Simell O, Alanen A. Sonography in the detection of Achilles tendon xanthomata in children with familial hypercholesterolaemia. Acta Paediatr. 1994;83(11):1178–1181. doi: 10.1111/j.1651-2227.1994.tb18277.x. [DOI] [PubMed] [Google Scholar]
- 28.Burnett JR, Ravine D, van Bockxmeer FM, Watts GF. Familial hypercholesterolaemia: a look back, a look ahead. Med J Aust. 2005;182(11):552–553. [PubMed] [Google Scholar]
- 29.Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA. Screening for hypercholesterolaemia versus case finding for familial hypercholesterolaemia: a systematic review and cost effectiveness analysis. Health Technol Assess. 2000;4(29):1–123. [PubMed] [Google Scholar]
- 30.Hill JS, Hayden MR, Frohlich J, Pritchard PH. Genetic and enviromental factors affecting the incidence of coronary artery disease in heterozygous familial hypercholesterolemia. Arterioscler Thromb. 1991;11(2):290–297. doi: 10.1161/01.atv.11.2.290. [DOI] [PubMed] [Google Scholar]
- 31.Ferrieres J, Lambert J, Lussier-Cacan S, Davignon J. Coronary artery disease in heterozygous familial hypercholesterolemia patients with the same LDL receptor gene mutation. Circulation. 1995;92(3):290–295. doi: 10.1161/01.cir.92.3.290. [DOI] [PubMed] [Google Scholar]
- 32.Scientific Steering Committee (SSC) on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolemia. BMJ. 1991;303(6807):893–896. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldman L, Goldman PA, Williams LW, Weinstein MC. Cost effectiveness considerations in the treatment of heterozygous familial hypercholesterolemia with medications. Am J Cardiol. 1993;72(10):75D–79D. doi: 10.1016/0002-9149(93)90015-5. [DOI] [PubMed] [Google Scholar]
- 34.Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. BMJ. 2007;335(7620):599. doi: 10.1136/bmj.39300.616076.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodenburg J, Vissers MN, Wiegman A, et al. Statin treatment in children with familial hypercholesterolemia the younger, the better. Circulation. 2007;116(6):664–668. doi: 10.1161/CIRCULATIONAHA.106.671016. [DOI] [PubMed] [Google Scholar]
- 36.Chmara M, Wasag B, Zuk M, et al. Molecular characterization of Polish patients with familial hypercholesterolemia: novel and recurrent LDLR mutations. J Appl Genet. 2010;51(1):95–106. doi: 10.1007/BF03195716. [DOI] [PubMed] [Google Scholar]
- 37.Berge KE, Ose L, Leren TP. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol. 2006;26(5):1094–1100. doi: 10.1161/01.ATV.0000204337.81286.1c. [DOI] [PubMed] [Google Scholar]
- 38.Fokkema IFAC, Den Dunnen JT, Taschner PEM. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-Box” approach. Hum Mutat. 2005;26(2):63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 39.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1(6):445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 40.Hobbs HH, Brown MS, Russell DW, Davignon J, Goldstein JL. Deletion in the gene for the low-density-lipoprotein receptor in a majority of French Canadians with familial hypercholesterolemia. N Engl J Med. 1987;317(12):734–737. doi: 10.1056/NEJM198709173171204. [DOI] [PubMed] [Google Scholar]
- 41.Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common low-density lipoprotein receptor mutations in the French Canadian population. J Clin Invest. 1990;85(4):1014–1023. doi: 10.1172/JCI114531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leitersdorf E, van der Westhuyzen DR, Coetzee GA, Hobbs HH. Two common low density lipoprotein receptor gene mutations cause familial hypercholesterolemia in Afrikaners. J Clin Invest. 1989;84(3):954–961. doi: 10.1172/JCI114258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotze MJ, Warnich L, Langenhoven E, du Plessis L, Retief AE. An exon 4 mutation identified in the majority of South African familial hypercholesterolaemics. J Med Genet. 1990;27(5):298–302. doi: 10.1136/jmg.27.5.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lehrman MA, Schneider WJ, Brown MS, et al. The Lebanese allele at the low density lipoprotein receptor locus. Nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J Biol Chem. 1987;262(1):401–410. [PubMed] [Google Scholar]
- 45.Aalto-Setälä K, Koivisto UM, Miettinen TA, et al. Prevalence and geographical distribution of major LDL receptor gene rearrangements in Finland. J Intern Med. 1992;231(3):227–234. doi: 10.1111/j.1365-2796.1992.tb00528.x. [DOI] [PubMed] [Google Scholar]
- 46.Seftel HC, Baker SG, Jenkins T, Mendelsohn D. Prevalence of familial hypercholesterolemia in Johannesburg Jews. Am J Med Genet. 1989;34(4):545–547. doi: 10.1002/ajmg.1320340418. [DOI] [PubMed] [Google Scholar]
- 47.Oppenheim A, Friedlander Y, Dann EJ, Berkman N, Schwartz SP, Leitersdorf E. Hypercholesterolemia in five Israeli Christian-Arab kindreds is caused by the “Lebanese” allele at the low density lipoprotein receptor gene locus and by an additional independent major factor. Hum Genet. 1991;88(1):75–84. doi: 10.1007/BF00204933. [DOI] [PubMed] [Google Scholar]
- 48.Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228(4701):815–822. doi: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphries SE, Gudnason V, Whittall R, Day IN. Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection in familial hypercholesterolemia. International Federation of Clinical Chemistry Scientific Division: Committee on Molecular Biology Techniques. Clin Chem. 1997;43(3):427–435. [PubMed] [Google Scholar]
- 50.Whittall R, Gudnason V, Weavind GP, Day LB, Humphries SE, Day IN. Utilities for high throughput use of the single strand conformational polymorphism method: screening of 791 patients with familial hypercholesterolaemia for mutations in exon 3 of the low density lipoprotein receptor gene. J Med Genet. 1995;32(7):509–515. doi: 10.1136/jmg.32.7.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reshef A, Nissen H, Triger L, et al. Molecular genetics of familial hypercholesterolemia in Israel. Hum Genet. 1996;98(5):581–586. doi: 10.1007/s004390050263. [DOI] [PubMed] [Google Scholar]
- 52.Day IN, Whittall RA, O’Dell SD, et al. Spectrum of LDL receptor gene mutations in heterozygous familial hypercholesterolemia. Hum Mutat. 1997;10(2):116–127. doi: 10.1002/(SICI)1098-1004(1997)10:2<116::AID-HUMU4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 53.Nissen H, Guldberg P, Hansen AB, Petersen NE, Horder M. Clinically applicable mutation screening in familial hypercholesterolemia. Hum Mutat. 1996;8(2):168–177. doi: 10.1002/(SICI)1098-1004(1996)8:2<168::AID-HUMU9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 54.Ekstrom U, Abrahamson M, Sveger T, Lombardi P, Nilsson-Ehle P. An efficient screening procedure detecting six novel mutations in the LDL receptor gene in Swedish children with hypercholesterolemia. Hum Genet. 1995;96(2):147–150. [Google Scholar]
- 55.Bodamer OA, Bercovich D, Schlabach M, Ballantyne C, Zoch D, Beaudet AL. Use of denaturing HPLC to provide efficient detection of mutations causing familial hypercholesterolemia. Clin Chem. 2002;48(11):1913–1918. [PubMed] [Google Scholar]
- 56.Naoumova RP, Neuwirth C, Pottinger B, Whittal R, Humphries SE, Soutar AK. Genetic diagnosis of familial hypercholesterolaemia: a mutation and a rare non-pathogenic amino acid variant in the same family. Atherosclerosis. 2004;174(1):67–71. doi: 10.1016/j.atherosclerosis.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Liguori R, Argiriou A, Simone VD. A rapid method for detecting mutations of the human LDL receptor gene by complete cDNA sequencing. Mol Cell Probes. 2003;17(1):15–20. doi: 10.1016/s0890-8508(02)00113-5. [DOI] [PubMed] [Google Scholar]
- 58.Sozen M, Whittall R, Humphries SE. Mutation detection in patients with familial hypercholesterolaemia using heteroduplex and single strand conformation polymorphism analysis by capillary electrophoresis. Atheroscler Suppl. 2004;5(5):7–11. doi: 10.1016/j.atherosclerosissup.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Alharbi KK, Aldahmesh MA, Spanakis E, et al. Mutation scanning by meltMADGE: validations using BRCA1 and LDLR, and demonstration of the potential to identify severe, moderate, silent, rare, and paucimorphic mutations in the general population. Genome Res. 2005;15(7):967–977. doi: 10.1101/gr.3313405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tejedor D, Castillo S, Mozas P, et al. Spanish FH Group. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem. 2005;51(7):1137–1144. doi: 10.1373/clinchem.2004.045203. [DOI] [PubMed] [Google Scholar]
- 61.Tejedor D, Castillo S, Mozas P, et al. Comparison of DNA array platform vs DNA sequencing as genetic diagnosis tools for familial hypercholesterolemia. Clin Chem. 2006;52(10):1971–1972. doi: 10.1373/clinchem.2006.073957. [DOI] [PubMed] [Google Scholar]
- 62.Oliva J, López-Bastida J, Moreno SG, Mata P, Alonso R. Costeffectiveness analysis of a genetic screening program in the close relatives of Spanish patients with familial hypercholesterolemia. Rev Esp Cardiol. 2009;62(1):57–65. [PubMed] [Google Scholar]
- 63.Blesa S, Garcia-Garcia AB, Martinez-Hervas S, et al. Analysis of sequence variations in the LDL receptor gene in Spain: general gene screening or search for specific alterations? Clin Chem. 2006;52(6):1021–1025. doi: 10.1373/clinchem.2006.067645. [DOI] [PubMed] [Google Scholar]
- 64.Laios E, Drogari E. Analysis of LDLR mutations in familial hypercholesterolemia patients in Greece by use of the NanoChip microelectronic array technology. Clin Chim Acta. 2006;374(1–2):93–99. doi: 10.1016/j.cca.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 65.Taylor A, Tabrah S, Wang D, et al. Multiplex ARMS analysis to detect 13 common mutations in familial hypercholesterolaemia. Clin Genet. 2007;71(6):561–568. doi: 10.1111/j.1399-0004.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- 66.Laurie AD, George PM. Evaluation of high-resolution melting analysis for screening the LDL receptor gene. Clin Biochem. 2009;42(6):528–535. doi: 10.1016/j.clinbiochem.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 67.Homsma SJ, Huijgen R, Middeldorp S, Sijbrands EJ, Kastelein JJ. Molecular screening for familial hypercholesterolaemia: consequences for life and disability insurance. Eur J Hum Genet. 2008;16(1):14–17. doi: 10.1038/sj.ejhg.5201940. [DOI] [PubMed] [Google Scholar]