

Abstract
Recent genome-wide association studies have reported a FAM13A variant on chromosome 4q22.1 is associated with lung function and COPD. We examined this variant in a case-control study of current or former smokers with chronic obstructive pulmonary disease (COPD, n = 458), lung cancer (n = 454), or normal lung function (n = 488). Sex, age, and smoking history were comparable between groups. We confirmed the FAM13A variant (rs7671167) confers a protective effect on smoking-related COPD alone (C allele odds ratio [OR] = 0.79, P = 0.013, and CC genotype OR = 0.71, P = 0.024) and those with COPD, both with and without lung cancer (C allele OR = 0.80, P = 0.008, and CC genotype OR = 0.70, P = 0.007). The FAM13A variant also confers a protective effect on lung cancer overall (C allele OR = 0.75, P = 0.002, and CC genotype OR = 0.64, P = 0.003) even after excluding those with co-existing COPD (C allele OR = 0.67, P = 0.0007, and CC genotype OR = 0.58, P = 0.006). This was independent of age, sex, height, lung function, and smoking history. This protective effect was confined to those with nonsmall cell lung cancer (C allele OR = 0.72, P = 0.0009, and CC genotype OR = 0.61, P = 0.003). This study suggests that genetic predisposition to COPD is shared with lung cancer through shared pathogenetic factors such as the 4q22.1 locus implicating the Rho-kinase pathway.
Keywords: lung cancer, chronic obstructive pulmonary disease, FAM13A, association study, polymorphism, GTPase
Introduction
Cigarette smoking is the major risk factor for chronic obstructive pulmonary disease (COPD) and lung cancer, and accounts for 85% to 90% of cases. Genetic predisposition may explain why only ≈10% to 20% of smokers are diagnosed with these conditions.1,2 Recent studies have shown that COPD is present in 50% to 70% of lung cancer cases when spirometry is performed,3–5 compared with a COPD prevalence of 15% to 20% among smokers randomly recruited from the community.3,6,7 The presence of COPD is associated with a 4- to 6-fold increased risk of lung cancer compared with smoking controls with normal lung function6 or community-recruited smokers.3 The heritability of COPD and lung cancer is estimated to be 40% to 77% and 15% to 25%, respectively.8,9 These observations suggest that not only is COPD an important phenotype which affects many people with lung cancer (Figure 1a), but that genes conferring a propensity to COPD may also be significant for lung cancer susceptibility.10 Further supporting this claim are the results of recently published genome-wide association (GWA) studies of COPD, lung function (Forced Expiratory Volume in one second, FEV1), and lung cancer.11–16 Despite these studies investigating different disease phenotypes, they have reported associations at several overlapping loci (Table 1). This suggests that, among smokers, some loci that determine susceptibility to COPD may also be important in the susceptibility to lung cancer.
Figure 1.

Overlapping relationship between COPD and lung cancer in current or former smokers. A) Lung function to define COPD subphenotype and healthy smoking (“resistant”) controls,3,17,18 current study. B) No lung function to define COPD subphenotype – lung cancer genome-wide association studies to date.11–13
Table 1.
Chromosomal loci and candidate genes associated with COPD and lung cancer from genome-wide association studies to date11–16
| Disease | Candidate genes | Chromosomal loci | Referencea |
|---|---|---|---|
| COPD (FEV1) | IL6R | 1q21 | 14 |
| FAM13A | 4q22 | 15,23 | |
| GSTCD | 4q24 | 15, 16 | |
| HHIP/GYPA | 4q31 | 13–16 | |
| HTR4/ADAM19 | 5q33 | 15, 16 | |
| BAT3/AGER | 6p21 | 15, 16 | |
| GPR126 | 6q24 | 15, 16 | |
| CHRNA 3/5 | 15q25 | 13, 14 | |
| Lung cancer | CRP | 1q21–23 | 11 |
| GYPA | 4q31 | 11 | |
| CRR9 (TERT) | 5p15 | 2, 11, 12 | |
| BAT3 | 6p21 | 2, 11, 12 | |
| CHRNA 3/5 | 15q25 | 2, 11–13 |
Note:
Available at www.genome.gov/gwastudies. Accessed 25/03/2010
Abbreviations: IL6R, interleukin-6 receptor; FAM13A, family with sequence familiarity 13, member A; GSTCD, glutathione S-transferase; C-terminal domain containing; HHIP, hedgehog interacting protein; GYPA, glycophorin A; HTR4, 5-hydroxytryptamine receptor 4; ADAM19, a disintegrin and metalloproteinase domain 19; BAT3, HLA-B associated transcript 3; AGER, advanced glycosylation end product-specific receptor; GPR126, G protein-coupled receptor 126; CHRNA315, locus containing both cholinergic receptor, nicotinic, alpha 3 and 5 genes; CRP, C-reactive protein; CRR9, cisplatin resistance related protein; TERT, telomerase reverse transcriptase.
Lung cancer GWA studies have successfully identified several novel susceptibility loci,11–13 but these studies may have severely underestimated the contribution of COPD and its underlying genetic determinants of lung cancer (“COPD-related” genes, Figure 1).3 Due to the high prevalence of COPD in lung cancer, as well as its greater heritability, genetic studies in lung cancer might improve their power to identify these COPD-related genes by using a control group of smokers who have a comparable level of smoking exposure but normal lung function (“resistant” smoker, see Figure 1a).17,18 This type of control group would represent the majority of smokers19,20 and those least likely to develop either COPD or lung cancer.3–7 It has recently been demonstrated that genetic variants that confer a protective or “resistant” phenotype might be better identified using healthy smokers as controls.18 Significant differences in COPD prevalence may exist in lung cancer GWA studies between lung cancer cases and controls,11–13 (Figure 1b) which could go unrecognized as it appears spirometry was not used to examine this possibility.3,17,18 Furthermore, spirometry can be utilized to “subphenotype” smokers (with comparable smoking histories) into smokers with normal lung function, those with COPD, and those who have lung cancer with or without co-existing COPD.3
Using the approach described above, re-examination of the associations between genetic variants at the 15q25 and 4q31 loci have shown overlapping effects with COPD and lung cancer not previously reported.17,18 Specifically the chromosome 15q25 locus, which was initially associated with lung cancer in GWA studies,11,12 was also shown to be independently associated with susceptibility to COPD by Young et al.17 Subsequent GWA and candidate gene studies of COPD have confirmed this finding.13,21 In addition, Young et al have shown that the chromosome 4q31 locus, strongly associated with a reduced risk of COPD in GWA studies,14,15 also confers a reduced risk to lung cancer independent of COPD.18
Recently, two GWA studies reported variants at the 4q22 locus, within the FAM13A (family with sequence similarity 13, member A) gene, are associated with normal lung function (FEV1/forced vital capacity, FVC)15 and reduced risk of COPD.22 To date this gene has been poorly characterized (http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=613299, last accessed 8th October, 2010), although sequence analysis has indicated the presence of a Rho GTPase-activating protein (Rho-GAP) domain on exons 2–5. Based on the presence of this domain,23 it is thought that FAM13A may have tumor suppressor activity through Rho-GAP mediated inhibition of the intracellular signal transduction molecule Rho A.24 The rs7671167 SNP, first described by Cho et al,22 is situated on intron 4 of the FAM13A gene and in linkage disequilibrium with SNPs in the Rho-GAP domain region. The current study examines the association of the FAM13A rs7671167 SNP in similarly exposed smokers (current or former) with normal lung function, COPD, and lung cancer (where lung cancer subjects are subphenotyped for COPD) to consider the presence of shared genetic effects, such as those reported for chromosome 15q25 and 4q31.17,18
Materials and methods
Study subjects
All subjects recruited were of Caucasian ancestry based on their grandparents’ descent (all 4 grandparents of Caucasian descent). Subjects recruited into the study were aged 40 to 80 years, with a minimum smoking history of 15 pack-years and COPD confirmed by a respiratory specialist based on prebronchodilator spirometric criteria. All subjects were recruited between 2001 and 2007. Control subjects were recruited based on the following criteria: aged 45 to 80 years and with a minimum smoking history of 15 pack-years. Control subjects were volunteers who were identified through either a community postal advertisement or while attending community-based retired military/servicemen’s clubs located in the same patient catchment as those serving the lung cancer and COPD hospital clinics. Lung cancer cases were aged .40 years and in 95% of cases, their diagnosis was confirmed through histological or cytological specimens. Nonsmokers with lung cancer were excluded from the study and only primary lung cancer cases with the following pathological diagnosis were included: adenocarcinoma, squamous cell cancer, small cell cancer, and nonsmall cell cancer (generally large cell or bronchoalveolar subtypes). Lung function measurement (prebronchodilator) was performed within 3 months of lung cancer diagnosis, prior to surgery, and in the absence of pleural effusions or lung collapse on plain chest radiographs.
Spirometry was done in 100% of those with COPD and the smoking controls, while 93% of those with lung cancer underwent spirometry. Lung function conformed to American Thoracic Society standards for acceptability and reproducibility, with the highest value of the best three blows used for classification of COPD status. Spirometry was performed using a portable spirometer (Easy-One™; ndd Medizintechnik AG, Zurich, Switzerland). COPD was defined according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) spirometric criteria of II or more (FEV1/FVC < 70% and FEV1% predicted ≤ 80%) although using prebronchodilator measurements (www.goldcopd.com). For lung cancer cases that had already undergone surgery, preoperative lung function performed by the hospital lung function laboratory was sourced from medical records. Controls with COPD based on spirometry (GOLD stage I or more) constituted 50% of the community volunteers and were excluded from further analysis. At the time of recruitment all participants gave written informed consent, and underwent blood sampling for DNA extraction, spirometry, and an investigator-administered questionnaire. The study was approved by the Multi Centre Ethics Committee (New Zealand).
Study design
The present retrospective case-control study compared smokers of the same ethnicity and comparable demographic variables (specifically age, sex, and smoking history). The healthy (resistant) controls in the current study were carefully chosen to be representative of the majority of smokers (60%–80%) who have maintained normal or near-normal lung function despite decades of smoking.19,20 Accordingly, such a group allows the association with COPD to be independently assessed and COPD-related genetic associations to be identified as they (“resistant” smokers) best reflect those smokers least likely to develop lung cancer or COPD. This approach resembles a pharmacogenetic approach and minimizes phenotype misclassification of controls, thereby improving the power to detect differences between affected and unaffected smokers.25
Genotyping
Genomic DNA was extracted from whole blood samples using standard salt-based methods and purified genomic DNA was aliquoted (10 ng μL−1 concentration) into 96-well plates. Samples were genotyped using Taqman® SNP genotyping assays (Applied Biosystems, Foster City, CA, USA) utilizing minor groove-binder probes as previously described.17,18 The present study investigated the genotype frequencies of the FAM13A SNP in the 4q22 region; rs7671167 (assay ID: C_1143656_10, Applied Biosystems) which was identified by the Cho et al GWA study.22 The rs7671167 SNP lies in intron 4 of the FAM13A gene (Figure 2a) and exhibits linkage disequilibrium (LD) with the other top SNPs identified in this region (rs1903003: D′ = 1.0 r2 = 0.852; rs2869967: D′ = 1.0 r2 = 0.64; rs6830970: D′ = 0.926 r2 = 0.365 – Figure 2b). Call rates of ≥99% for this SNP were achieved in each group and genotyping replicated in a random sample of 150 subjects (11%).
Figure 2.
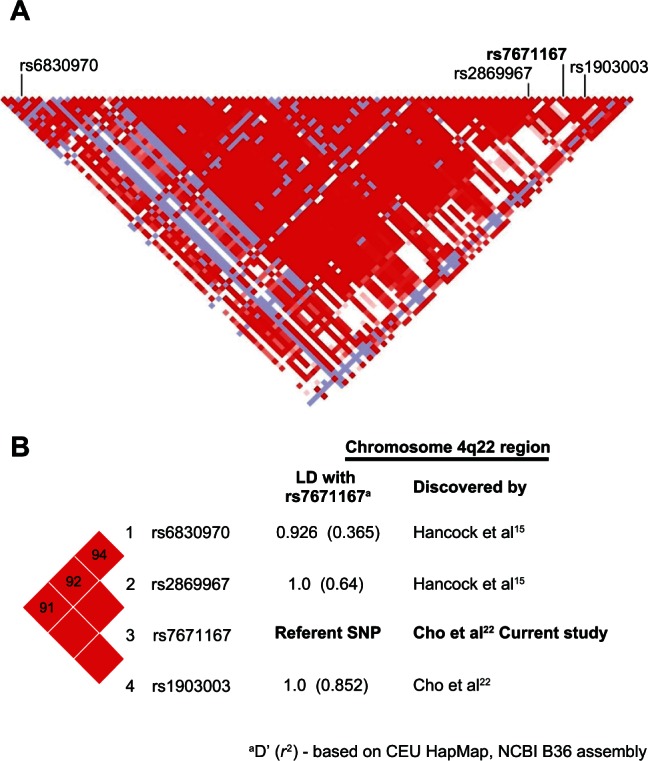
Linkage disequilibrium map of the chromosome 4q22 region including the FAM13A SNPs reported to date.
Analysis
Patient characteristics in the cases and controls were compared by ANOVA for continuous variables and χ2 test for discrete variables (Mantel-Haenszel, odds ratio [OR]). Genotype and allele frequencies for the cases and controls were examined separately for each SNP by Hardy-Weinberg Equilibrium). Population admixture across groups was performed using structure analysis on genotyping data from 40 unrelated SNPs.26 Distortions in the genotype and allele frequencies were identified between cases and controls using two-by-two contingency tables. Both the additive- (allelic) and genotype-based genetic models were tested although the latter is preferred.27 Bonferroni correction was used for the genetic models tested above but not for the single FAM13A SNP analysis, as this study was testing for replication only.
Results
Demographic variables
Characteristics of the lung cancer cases, COPD cases and healthy control smokers are summarized in Table 2 and described in detail elsewhere.3,17,18 The demographic variables and histological subtypes of the lung cancer cases (identified in 94% of cases) are comparable with previously published data.28 The COPD cases have higher pack-year exposure than the lung cancer cases and healthy control smokers (P < 0.05). This reflects outliers with high smoking histories in the COPD group (skewed distribution) and no difference exists after log transformation of pack-years (data not shown). All groups are comparable for age started smoking, years smoked, years since quitting, and cigarettes/day (Table 2). Overall, we believe the 3 groups have comparable smoking exposure. The lower frequency of current smokers in the lung cancer and COPD cases, compared with the healthy smoker group (35% vs 40% vs 48%, respectively), likely reflects the presence of symptoms from pulmonary complications of smoking (primarily breathlessness from COPD) stimulating higher quit rates.
Table 2.
Summary of the characteristics for the lung cancer, COPD, and control smokers
| Parameter mean (1 SD) | Lung cancer N = 454 | COPD N = 458 | Control smokers N = 488 |
|---|---|---|---|
| % male | 53% | 59% | 60% |
| Age (years) | 69 (10) | 66 (9) | 65 (10) |
| Height (m)* | 1.67 (0.08) | 1.68 (0.09) | 1.69 (0.09) |
| Smoking history | |||
| Current smoking (%) | 35% | 40% | 48% |
| Age started (year) | 18 (4) | 17 (3) | 17 (3) |
| Years smoked | 41 (12) | 42 (11) | 35 (11) |
| Pack-years* | 41 (25) | 47(20)a | 40 (19) |
| Cigarettes/day | 20 (10) | 23 (9) | 24 (11) |
| Years since quitting | 11.4 (6.7) | 9.8 (7.4) | 13.9 (8.1) |
| History of other exposures | |||
| Work dust exposure* | 63% | 59% | 47% |
| Work fume exposure | 41% | 40% | 38% |
| Asbestos exposure* | 23% | 22% | 16% |
| Family history (FHx) | |||
| FHx of COPD | 33% | 37% | 28% |
| FHx of lung cancer* | 19% | 11% | 9% |
| Lung function | |||
| FEV1 (L)* | 1.86 (0.48) | 1.25 (0.48) | 2.86 (0.68) |
| FEV1 % predicted* | 73% | 46% | 99% |
| FEV1/FVC* | 64% (13) | 46% (8) | 78% (7) |
| Spirometric COPDb,* | 51% | 100% | 0% |
Notes:
P < 0.05.
No significant difference after log transformation of pack-years due to skewed distribution;
According to GOLD II or more criteria.
The lung cancer cases, COPD cases, and smoking controls were also comparable for other aero-pollutant exposures. The lung cancer cases reported higher rates of a family history of lung cancer compared to the COPD cases and healthy smokers (19% vs 11% vs 9%). Mean height was slightly lower in those with lung cancer compared to those in the control group (P < 0.05) but height was not different when sex adjustment was made. As expected, lung function was worse in the lung cancer and COPD cases compared to the healthy smoker controls. Testing of lung function (as described above) was achieved in 93% of lung cancer cases and allows stratification of results to test for an interactive or confounding effect of COPD.
Genotype and allele frequencies
Genotype and allele frequencies for the FAM13A rs7671167 SNP are shown in Tables 3 and 4. The genotype frequencies were consistent with those reported in the literature22 (personal communication, EK Silverman) and from the International Hapmap Project (www.hapmap.org). As SNP genotypes were confirmed with careful examination of amplification plots, replicated in 150 samples with 100% concordance and in Hardy–Weinberg equilibrium for each phenotypic group, we believe significant genotyping error can be excluded. We found no evidence for population stratification between the case and control groups using 40 unlinked SNPs from unrelated genes (mean χ2 = 3.3, P = 0.58).26
Table 3.
Genotype and allele frequencies for the rs7671167 FAM13A SNP in COPD and lung cancer (total and subgrouped by COPD) cohorts compared with smoking controls
| Primary cohorts (call rate %) | C | T | Odds ratioa (95% CI)a P valuea | CC | CT | TT | Odds ratiob (95% CI)b P valueb |
|---|---|---|---|---|---|---|---|
| Controls N = 485 | 530 | 440 | – | 145 | 240 | 100 | – |
| (99%) | (55%) | (45%) | (30%) | (49%) | (21%) | ||
| COPD* N = 458 | 448 | 468 | 0.79 | 107 | 234 | 117 | 0.71 |
| (100%) | (49%) | (51%) | (0.66–0.96) 0.013 |
(23%) | (51%) | (26%) | (0.53–0.97) 0.024 |
| Lung cancer (LC) N = 449 | 427 | 471 | 0.75 | 96 | 235 | 118 | 0.64 |
| (99%) | (48%) | (52%) | (0.62–0.91) 0.002 |
(21%) | (52%) | (26%) | (0.47–0.87) 0.003 |
| Lung cancer subgroup analysesc | |||||||
| LC with COPD* N = 215 | 212 | 218 | 0.81 | 47 | 118 | 50 | 0.66 |
| (49%) | (51%) | (0.64–1.02) 0.065 |
(22%) | (55%) | (23%) | (0.44–0.97) 0.028 |
|
| LC only N = 207 | 185 | 229 | 0.67 | 41 | 103 | 63 | 0.58 |
| (45%) | (55%) | (0.53–0.85) 0.0007 |
(20%) | (50%) | (30%) | (0.38–0.87) 0.006 |
|
| COPD subgroup analysis | |||||||
| COPD* and LC with | 660 | 686 | 0.80 | 154 | 352 | 167 | 0.70 |
| COPD* N = 673 | (49%) | (51%) | (0.67–0.95) 0.008 |
(23%) | (52%) | (25%) | (0.53–0.91) 0.007 |
Notes:
C vs T compared with matched smoking controls (Mantel–Haenszel);
CC vs CT/TT compared to matched smoking controls (Mantel–Haenszel);
Spirometry available in 422/454 (93%);
COPD* = GOLD II or more criteria.
Abbreviation: 95% CI, 95% confidence interval.
Table 4.
Genotype and allele frequencies for the rs7671167 FAM13A SNP in the lung cancer cases (subgrouped by histology) compared with smoking controls
| Primary cohorts | C | T | Odds ratioa (95% CI)a P valuea | CC | CT | TT | Odds ratiob (95% CI)b P valueb |
|---|---|---|---|---|---|---|---|
| Controls N = 485 | 530 | 440 | – | 145 | 240 | 100 | – |
| (55%) | (45%) | (30%) | (49%) | (21%) | |||
| Lung cancer (LC) N = 449 | 427 | 471 | 0.75 | 96 | 235 | 118 | 0.64 |
| (48%) | (52%) | (0.62–0.91) 0.002 |
(21%) | (52%) | (26%) | (0.47–0.87) 0.003 |
|
| Lung cancer histology subanalysisc | |||||||
| Small cell | 86 | 70 | 1.02 | 23 | 40 | 15 | 0.98 |
| N = 78 (18% of total LC) | (55%) | (45%) | (0.72–1.45) 0.91 |
(29%) | (51%) | (19%) | (0.56–1.70) 0.94 |
| Non small cell unspecifiedc | 43 | 47 | 0.76 | 9 | 25 | 11 | 0.59 |
| N = 45 (11%) | (48%) | (52%) | (0.48–1.20) 0.21 |
(20%) | (56%) | (24%) | (0.26–1.31) 0.16 |
| Adenocarcinomac | 175 | 207 | 0.70 | 36 | 103 | 52 | 0.54 |
| N = 191 (45%) | (46%) | (54%) | (0.55–0.90) 0.003 |
(19%) | (54%) | (27%) | (0.35–0.84) 0.004 |
| Squamous cellc | 101 | 115 | 0.73 | 26 | 49 | 33 | 0.74 |
| N = 108 (26%) | (47%) | (53%) | (0.54–0.99) 0.036 |
(24%) | (45%) | (31%) | (0.45–1.23) 0.23 |
| Nonsmall cell lung cancerc combination analysis | |||||||
| All nonsmall cell N = 344 | 319 | 369 | 0.72 | 71 | 177 | 96 | 0.61 |
| (82% of total LC, N = 422) | (46%) | (54%) | (0.59–0.88) 0.0009 |
(23%) | (52%) | (25%) | (0.43–0.86) 0.003 |
Notes:
C vs T compared with matched smoking controls (Mantel–Haenszel);
CC vs CT/TT compared with matched smoking controls (Mantel–Haenszel);
Histology available in 422/454 (93%) of lung cancer cases.
We found the FAM13A variant (rs7671167) confers a protective effect on the development of COPD (N = 458; C allele OR = 0.79, P = 0.013, and CC genotype OR = 0.71, P = 0.024) (Table 3), consistent with previously reported studies.15,22 We also show for the first time this FAM13A variant has a protective effect on lung cancer (N = 449; C allele OR = 0.75, P = 0.002, and CC genotype OR = 0.64, P = 0.003), which remained significant even after excluding those with co-existing COPD (GOLD II or more) (N = 207; C allele OR = 0.67, P = 0.0007, and CC genotype OR = 0.58, P = 0.006) (Table 3). The association with FAM13A was also found in lung cancer where the diagnosis of COPD predated lung cancer diagnosis (N = 215; C allele OR = 0.81, P = 0.065, and CC genotype OR = 0.66, P = 0.028). When these cases (lung cancer with COPD) were combined with COPD cases a significant association with FAM13A was also found (N = 673; C allele OR = 0.80, P = 0.008, and CC genotype OR = 0.70, P = 0.007). We found the associations between COPD and lung cancer with FAM13A best fitted an autosomal recessive model (COPD: OR = 0.72 [95% CI 0.54–0.97], P = 0.037 and Lung Cancer: OR = 0.65 [95%CI 0.48–0.88], P = 0.0058) although this was marginal when compared with an additive model (COPD: OR = 0.77 [95% CI 0.57–1.04], P = 0.088 and lung cancer: OR = 0.73 [95% CI 0.54–0.99], P = 0.049). Only the recessive model for lung cancer was significant after Bonferroni correction (P = 0.01). Across all 3 FAM13A genotypes (Table 3) the 2×3 χ2 comparison for COPD and lung cancer, compared with controls, was significantly different (P = 0.041 and P = 0.006, respectively). The FAM13A association was still statistically significant regardless of smoking status (ie, stratification by current and former smoker status). There was no association with the FAM13A variant and sex, lung function, age, or height.
When the FAM13A association was examined in the lung cancer cases subdivided according to histological subtype (Table 4), we found the protective effect conferred by the FAM13A SNP was confined to those with nonsmall cell lung cancer (defined as adenocarcinoma, nonsmall cell carcinoma – unspecified, and squamous cell carcinoma) (C allele OR = 0.72, P = 0.0009, and CC genotype OR = 0.61, P = 0.003) and lost in those with small cell carcinoma (C allele OR = 1.02, P = 0.91, and CC genotype OR = 0.98, P = 0.94). We also found no association between this variant and lung function or smoking exposure in the lung cancer cases, the only group recruited independent of lung function criteria (Table 5). As expected, no evidence of concordance was found when genotypes from all 3 groups for the FAM13A rs7671167 SNP (Chr 4q22) were compared with those from the HHIP rs1489759 SNP (Chr 4q31),18 which have respective positions 55 megabases apart on the long arm of chromosome 4.
Table 5.
Relationship between FAM13A genotype, smoking exposure, and lung function in the lung cancer cases
| Gene | Genotype | Lung function (1 SD)
|
Pack-years (1 SD) | ||
|---|---|---|---|---|---|
| FEV1 (L) | FEV1 % predicted | FEV1/FVC | |||
| FAM13A1 | CC | 1.85 (0.67) | 72.2% (22.2) | 65.7% (12.2) | 39.1 (18.1) |
| (rs7671167) | CT | 1.84 (0.71) | 72.8% (23.6) | 63.4% (13.3) | 42.6 (16.2) |
| TT | 1.90 (0.68) | 73.1% (22.0) | 64.3% (12.7) | 40.1(13.6) | |
| P (ANOVA) | 0.71 | 0.97 | 0.35 | 0.65 | |
Discussion
This study replicates the findings of Cho et al,22 showing the C allele of the FAM13A variant rs7671167 is associated with a reduced risk of COPD (C allele OR = 0.79, P = 0.013, and CC genotype OR = 0.71, P = 0.024). In addition, the study shows that this protective effect exists in those with COPD combined with those with lung cancer who also had pre-existing COPD (C allele OR = 0.80, P = 0.008, and CC genotype OR = 0.70, P = 0.007). Of greater significance, this study shows for the first time that this variant is associated with a reduced risk of lung cancer (C allele OR = 0.75, P = 0.002, and CC genotype OR = 0.64, P = 0.003), even after subphenotyping to exclude those with co-existing COPD (C allele OR = 0.67, P = 0.0007, and CC genotype OR = 0.58, P = 0.006) (Table 3). The latter, together with the observation that FAM13A genotype was not related to lung function nor pack-year exposure (Table 5), thereby excludes a confounding effect by COPD, low lung function, or smoking exposure. These findings suggest that this FAM13A variant on 4q22 is independently associated with a reduced risk of both COPD and lung cancer. This observation adds the 4q22 locus to those loci which also appear to be shared (or overlapping) between COPD and lung cancer on chromosome 15q25 (CHRNA 3/5 susceptible) and 4q31 (HHIP protective) (Table 111–16). Shared genetic susceptibility may explain why there appears to be such a strong relationship between COPD and lung cancer among smokers even after correcting for smoking exposure.3–7
Identification of the FAM13A association with lung cancer was made possible by using smokers with normal lung function, well matched for ethnicity, age, sex, and most importantly, smoking exposure. This control group is representative of the majority of “resistant” smokers (60%–80%) who maintain normal or near normal lung function and therefore best represents a “low responder” phenotype.19,20 Had the smoking controls for this study been recruited from hospital or community based volunteers, in the absence of spirometric screening (ie, unscreened controls), the prevalence of COPD may have been as high as 30% or more.29,30 Such an excess of COPD subjects in the unscreened controls would be expected to dilute the effect of protective variants (such as the FAM13A gene) given comparable allele and genotype frequencies in COPD and lung cancer cases compared with unscreened controls (see Figure 1 and Table 3). If this hypothesis were true, it might explain why none of the 3 lung cancer GWA studies11–13 reported an association with the 4q22 locus. In a similar vein, only 1 of the 3 lung cancer GWA studies identified that the 4q31 locus was implicated in lung cancer.11 This further supports the view that unscreened controls in these studies were heterogeneous and may have included many smokers with COPD (Figure 1b), thereby diluting the “protective” effect of the 4q31 locus. This would be analogous to including a high proportion of obese subjects in the controls of a genetic epidemiology study of type 2 diabetes, thereby reducing the study’s power to detect relevant obesity-related genes.31
The rs7671167 SNP has no known biological function, indeed the FAM13A gene itself is also poorly understood. FAM13A is an interesting candidate gene because of the Rho-GAP domain it encodes23 and its associated tumor suppressor activity through inhibition of the intracellular signal transduction molecule Rho A.24 This Rho-GAP activity implicates FAM13A in carcinogenesis.32 Furthermore, other proteins with Rho-GAP catalytic activity (eg, Deleted in Liver Cancer-1) function as tumor suppressors in many cancers,33,34 including nonsmall cell lung cancer.35–37 The importance of Rho-GAPs in the development of nonsmall cell lung cancer is of interest as this study found the FAM13A association was limited to this histological subtype. The relevance of this putative Rho-GAP function (inhibition of Rho A) of FAM13A extends to COPD, where increased Rho A activity has been implicated in oxidative stress and impaired apoptosis underlying COPD.38 For both COPD and lung cancer, differential expression of FAM13A has been observed during pulmonary type II cell differentiation39 and in the epithelium of cystic fibrosis patients, another inflammatory based lung disease.40 Whether the rs7671167 variant is itself functional is not known although given its location (intron 4 of the Rho-GAP domain), effects on splice variants are possible. The possibility that variable expression (or altered function) of FAM13A plays a role in COPD and lung cancer, mediated through its putative role as an inhibitor of Rho A activity (Rho-GAP), opens the door to several possible preventive or chemotherapeutic therapies.32,41–43 Specifically, the authors suggest that targeting FAM13A gene expression, or simulating its inhibitory function, might also have therapeutic benefit or preventive potential in COPD and lung cancer. Inhibition of Rho A and simulation of Rho-GAP function has been shown to inhibit carcinogenesis32–34 and has been suggested to have therapeutic utility in the treatment of many cancers including lung cancer.35–37 In this regard, it is intriguing that Rho-GAP activity appears to inhibit the HMGCoA reductase enzyme, thereby simulating the effects of HMGCoA reductase inhibitors (statins),32,41–43 and provides a plausible explanation as to why statins may reduce the tendency to COPD and lung cancer.44 An exaggerated inflammatory/remodeling response to smoking mediated through GTPases, and the development of epithelial mesenchymal transition, are thought to link COPD and lung cancer.37,38,44–46 Based on the results of this study, it is proposed that augmented Rho-GAP activity from variable expression (or function), conferred by FAM13A variants (or SNPs in linkage disequilibrium), may be relevant in reducing the risk of both COPD and lung cancer. Further studies will be needed to replicate these findings and ascertain the specific genetic variant/s (and functional effects) underlying the FAM13A association reported here.
In contrast to the GWA studies,15,22 this study found no correlation between the FAM13A variant and lung function in the lung cancer cases. This may be due to several factors specific to this study including the modest size of the groups used for this analysis, the subject disease status (lung cancer cases of which 51% had co-existing COPD), the heavy smoking exposure (mean 41 pack-years) or modifying effects of other smoking-responsive genes on lung function decline, eg, Chr 15q25 and Chr 4q31 loci.17,18 Based on our analysis and the distortions in genotype and allele frequencies, we suggest the FAM13A effect in COPD and lung cancer is most consistent with a recessive model, the former in agreement with Cho et al.22 The modest cohort size together with the case-control cross-sectional design may be considered as limitations of this study. However all subjects were well matched for all facets of smoking exposure (see Table 2) and the potential for confounding by COPD was carefully excluded. There was no interactive effect between the FAM13A variant with height, sex, and pack-years which is also consistent with Cho et al.22
In summary, there is growing evidence that lung cancer results from exposure to smoking and the net effect of genetic factors conferring beneficial or harmful pathogenic effects on the predisposition to developing lung cancer.47 Moreover, some of these genetic effects have relevance to COPD susceptibility10,47 where loci on chromosomes 15q25, 4q31, and 4q22 have been implicated in this overlap.17,18 The authors suggest that these 3 genetic variants represent the first examples of genetic susceptibility loci shared between COPD and lung cancer,17,18 and demonstrate the importance of using healthy smoking (“resistant”) controls to identify both susceptibility (15q25) and protective (4q31,4q22) loci for lung cancer. The association with the 4q22 locus connecting COPD and lung cancer is of particular interest as it implicates the Rho-kinase pathway at a pathogenetic level for the first time and further supports the chemopreventive potential of existing drugs, such as statins, which inhibit Rho kinase activity.38,44 Lung cancer, rare before the 20th century, remains the single greatest cause of cancer death in the United States with approximately 440 deaths per day (28% of all cancer deaths, 2009 figures) and in Europe with 910 deaths per day (2006 figures). Over the past 5 years, GWA studies and candidate gene studies have begun to elucidate the pathogenetic basis linking COPD and lung cancer as proposed by Dr Tom Petty in 2005.10 Although reduction in smoking rates must remain a public health priority, a better understanding of the genetic factors underlying susceptibility might offer new opportunities to develop novel targeted strategies for prevention or early diagnosis of lung cancer in the future.44,47
Acknowledgments
This paper is dedicated to the memory of Dr Tom Petty and to the smokers with lung cancer who agreed to participate in this study. We also acknowledge the support of Dr Peter Black who died suddenly in January 2010. This study was supported by grants from the University of Auckland, Auckland Medical Research Foundation, and Health Research Council of New Zealand.
Glossary
Abbreviations
- COPD
chronic obstructive pulmonary disease
- FAM13A
family with sequence familiarity 13
- FEV1
forced expiratory volume in 1 second
- FVC
forced vital capacity
- GOLD
Global Initiative for Chronic Obstructive Lung Disease
- GTPase
guanosine triphosphatase
- GWA
genome-wide association
- HMGCoA reductase
3-hydroxy-3-methylglutaryl-CoA reductase
- OR
odds ratio
- Rho-GAP
Rho GTPase-activating protein
- SNP
single nucleotide polymorphism.
Footnotes
Disclosure
The authors declare no conflicts of interest.
References
- 1.Molfino NA. Genetics of COPD. Chest. 2004;125:1929–1940. doi: 10.1378/chest.125.5.1929. [DOI] [PubMed] [Google Scholar]
- 2.Broderick P, Wang Y, Vijayakarishnan V, et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69:6633–6641. doi: 10.1158/0008-5472.CAN-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. COPD prevalence is increased in lung cancer independent of age, sex and smoking history. Eur Respir J. 2009;34:380–386. doi: 10.1183/09031936.00144208. [DOI] [PubMed] [Google Scholar]
- 4.De Torres J, Bastarrika G, Wisnivesky JP, et al. Assessing the relationship between lung cancer risk and emphysema detected on low dose CT of the chest. Chest. 2007;132:1932–1938. doi: 10.1378/chest.07-1490. [DOI] [PubMed] [Google Scholar]
- 5.Wilson DO, Weissfeld JL, Balkan A, et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am J Respir Crit Care Med. 2008;178:738–744. doi: 10.1164/rccm.200803-435OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mannino DM, Aguayo SM, Petty TL, et al. Low lung function and incident lung cancer in the United States: data from the first NHANES follow-up. Arch Int Med. 2003;163:1475–1480. doi: 10.1001/archinte.163.12.1475. [DOI] [PubMed] [Google Scholar]
- 7.Tockman MS, Anthonisen NR, Wright EC, et al. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–518. doi: 10.7326/0003-4819-106-4-512. [DOI] [PubMed] [Google Scholar]
- 8.Hubert H, Fabsitz R, Feinleib M, et al. Genetic and environmental influences on pulmonary function in adult twins. Am Rev Respir Dis. 1982;125:409–415. doi: 10.1164/arrd.1982.125.4.409. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer: analyses of cohorts of twins from Sweden, Denmark and Finland. N Eng J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 10.Petty TL. Are COPD and lung cancer two manifestations of the same disease? Chest. 2005;128:1895–1897. doi: 10.1378/chest.128.4.1895. [DOI] [PubMed] [Google Scholar]
- 11.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 13.Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilk JB, Chen T, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2009;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young RP, Hopkins RJ, Hay BA, et al. Lung cancer gene associated with COPD: triple whammy or possible confounding effect? Eur Respir J. 2008;32:1158–1164. doi: 10.1183/09031936.00093908. [DOI] [PubMed] [Google Scholar]
- 18.Young RP, Whittington CF, Hopkins RJ, et al. Chromosome 4q31 locus in COPD also associated with lung cancer. Eur Respir J. 2010;36:1375–1382. doi: 10.1183/09031936.00033310. [DOI] [PubMed] [Google Scholar]
- 19.Løkke A, Lange P, Scharling H, et al. Developing COPD: A 25 year follow up study of the general population. Thorax. 2006;61:935–939. doi: 10.1136/thx.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohansal R, Martinez-Camblor P, Agusti A, et al. The natural history of chronic airflow obstruction revisited: An analysis of the Framingham Offspring Cohort. Am J Respir Crit Care Med. 2009;180:3–10. doi: 10.1164/rccm.200901-0047OC. [DOI] [PubMed] [Google Scholar]
- 21.Lambrechts D, Buysschaert I, Zanen P, et al. The 15q24/25 susceptibility variant for lung cancer and chronic obstructive pulmonary disease is associated with emphysema. Am J Respir Crit Care Med. 2010;181:486–493. doi: 10.1164/rccm.200909-1364OC. [DOI] [PubMed] [Google Scholar]
- 22.Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen M, Reichenstein M, Everts-van der Wind A, et al. Cloning and characterization of FAM13A1 – a gene near a milk protein QTL on BTA6: evidence for population-wide linkage disequilibrium in Israeli Holsteins. Genomics. 2004;84:374–383. doi: 10.1016/j.ygeno.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 25.Moskvina V, Holmans P, Schmidt KM, et al. Design of case-controls studies with unscreened controls. Ann Hum Genet. 2005;68:566–576. doi: 10.1111/j.1529-8817.2005.00175.x. [DOI] [PubMed] [Google Scholar]
- 26.Pritchard J, Stephens M, Donnelly P. Inference of population structure from multi-locus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thankkinstian A, Thompson JR, Minelli C, et al. Choosing between per-genotype, per-allele, and trend approaches for initial detection of gene-disease association. J App Stat. 2009;36:633–646. [Google Scholar]
- 28.Yang P, Allen MS, Aubry MC, et al. Clinical features of 5,628 primary lung cancer patients; experience at Mayo Clinic from 1997 to 2003. Chest. 2005;128:452–462. doi: 10.1378/chest.128.1.452. [DOI] [PubMed] [Google Scholar]
- 29.Stav D, Raz M. Prevalence of chronic obstructive pulmonary disease among smokers aged 45 and up in Israel. Isr Med Assoc J. 2007;9:800–802. [PubMed] [Google Scholar]
- 30.Zaas D, Wise R, Wiener C, et al. Airways obstruction is common but unsuspected in patients admitted to a general medicine service. Chest. 2004;125:106–111. doi: 10.1378/chest.125.1.106. [DOI] [PubMed] [Google Scholar]
- 31.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–662. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 32.Kandpal RP. Rho GTPase Activating proteins in cancer phenotypes. Curr Protein Pept Sci. 2006;7:355–365. doi: 10.2174/138920306778018025. [DOI] [PubMed] [Google Scholar]
- 33.Kim TY, Vigil D, Der CJ, et al. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 2009;28:77–83. doi: 10.1007/s10555-008-9167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Guan M, Vigil D, et al. p120Ras-GAP binds the DLC-1 Rho-GAP tumor suppressor protein and inhibits its RhoA GTPase and growth-suppressing activities. Oncogene. 2009;28:1401–1409. doi: 10.1038/onc.2008.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan BZ, Jewffersen AM, Baldwin KT, et al. DLC-1 operates as a tumor suppressor gene in non-small cell lung carcinomas. Oncogene. 2004;23:1405–1411. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- 36.Healy KD, Hodgson L, Kim TY, et al. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2007;47:326–337. doi: 10.1002/mc.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asnaghi L, Vass W, Quadri R, et al. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of Rho GTPases. Oncogene. 2010;29:2760–2771. doi: 10.1038/onc.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richens TR, Linderman DJ, Horstmann SA, et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med. 2009;179:1011–1021. doi: 10.1164/rccm.200807-1148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wade KC, Guttentag SH, Gonzales LW, et al. Gene induction during differentiation of human pulmonary type II cells in vitro. Am J Respir Crit Care Med. 2006;34:727–737. doi: 10.1165/rcmb.2004-0389OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright JM, Merlo CA, Reynolds JB, et al. Respiratory epithelial gene expression in patients with mild and severe cystic fibrosis lung disease. Am J Respir Crit Care Med. 2006;35:327–336. doi: 10.1165/rcmb.2005-0359OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riganti C, Aldieri E, Doublier S, et al. Statins-mediated inhibition of Rho GTPases as a potential tool in anti-tumor therapy. Mini Rev Med Chem. 2008;8:609–618. doi: 10.2174/138955708784534436. [DOI] [PubMed] [Google Scholar]
- 42.Walker K, Olson MF. Targeting Ras and Rho GTPases as opportunities for cancer therapeutics. Curr Opin Genet Dev. 2005;15:62–68. doi: 10.1016/j.gde.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Fritz G, Kaina B. Rho GTPases: Promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006;6:1–14. [PubMed] [Google Scholar]
- 44.Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: potential utility in COPD. Eur Respir Rev. 2009;18:222–232. doi: 10.1183/09059180.00005309. [DOI] [PubMed] [Google Scholar]
- 45.Dasari V, Gallup M, Lemjabbar H, et al. Epithelial-mesenchymal transition in lung cancer: Is tobacco the “smoking gun”? Am J Respir Cell Mol Biol. 2006;35:3–9. doi: 10.1165/rcmb.2006-0051SF. [DOI] [PubMed] [Google Scholar]
- 46.Lee G, Walser TC, Dubinett SM. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Curr Opin Pulm Med. 2009;15:303–307. doi: 10.1097/MCP.0b013e32832c975a. [DOI] [PubMed] [Google Scholar]
- 47.Young RP, Hopkins RJ, Hay BA, et al. A gene based risk score for lung cancer susceptibility in smokers and ex-smokers. Postgrad Med J. 2009;85:515–524. doi: 10.1136/pgmj.2008.077107. [DOI] [PubMed] [Google Scholar]