

Abstract
Epidemiological studies indicate that tobacco smoke exposure accounts for nearly 90% of cases of chronic obstructive pulmonary disease (COPD) and lung cancer. However, genetic factors may explain why 10%–30% of smokers develop these complications. This perspective reviews the evidence suggesting that COPD is closely linked to susceptibility to lung cancer and outlines the potential relevance of this observation. Epidemiological studies show that COPD is the single most important risk factor for lung cancer among smokers and predates lung cancer in up to 80% of cases. Genome-wide association studies of lung cancer, lung function, and COPD have identified a number of overlapping “susceptibility” loci. With stringent phenotyping, it has recently been shown that several of these overlapping loci are independently associated with both COPD and lung cancer. These loci implicate genes underlying pulmonary inflammation and apoptotic processes mediated by the bronchial epithelium, and link COPD with lung cancer at a molecular genetic level. It is currently possible to derive risk models for lung cancer that incorporate lung cancer-specific genetic variants, recently identified “COPD-related” genetic variants, and clinical variables. Early studies suggest that single nucleotide polymorphism-based risk stratification of smokers might help better target novel prevention and early diagnostic strategies in lung cancer.
Keywords: lung cancer, chronic obstructive pulmonary disease, association study, single nucleotide polymorphism, risk model
Introduction
Lung cancer and chronic obstructive pulmonary disease (COPD) are pulmonary diseases that result from the dual effects of smoking exposure and genetic susceptibility.1,2 Lung cancer was rare before 1900,3 but by 2008 accounted for nearly 30% of all cancer-related deaths in the US, ie, more than the combined deaths from cancers of the breast, colon, prostate, and ovary.4 Despite decades of various chemotherapy regimens, survival for lung cancer remains poor, with over 160,000 deaths per annum in the US alone.4 COPD is believed to affect more than 15 million people in the US, and is predicted to become the third leading cause of death by 2020. Citing past and present scientific literature, this perspective paper challenges current dogma and proposes that COPD and lung cancer might be related through overlapping pathogenetic pathways activated by smoking. This has relevance in the development and clinical utility of gene-based risk assessment for smoking-related lung disease.
Link between COPD and lung cancer
Epidemiological studies suggest that although tobacco smoke exposure accounts for nearly 80%–90% of COPD and lung cancer cases, only 10%–15% of smokers develop lung cancer while 20%–30% develop clinically significant COPD.5–7 Genetic factors may underlie these observations because the heritability (genetic contribution) of lung cancer and reduced forced expiratory volume in one second (FEV1) that defines COPD is estimated to be 15%–25% and 40%–77%, respectively.8,9 It has been shown that smokers with COPD (irreversible airflow limitation based on reduced FEV1) have a 4–6 fold-increased risk of lung cancer when compared with smokers with normal lung function10 or smokers randomly recruited from the community.11 Studies dating back 30 years show that the distribution of FEV1 is bimodal or trimodal in heavy smokers and unimodal in light smokers.12–14 This observation suggests genetic susceptibility, rather than smoking exposure alone, underlies the development of COPD,15 and the maladaptive lung remodeling (FEV1) response to chronic smoking exposure (Figure 1).12–14 Importantly, 50%–80% of those diagnosed with lung cancer have pre-existing COPD compared with a COPD prevalence of 15%–20% in randomly selected community-based smokers.10,16,17 This means lung cancer is not just a “complex genetic disease” resulting from smoking exposure in genetically susceptible people, but that it is disease of mixed phenotype that includes spirometrically defined COPD as a subphenotype.10 These observations show that most smokers with lung cancer have underlying COPD and suggest that smokers who are susceptible to getting COPD are also susceptible to getting lung cancer (Figure 1). Conversely, smokers who maintain normal lung function (resistant smokers) are unlikely to get lung cancer. Studies of molecular pathways suggest that the inflammatory, repair, and remodeling processes underlying COPD might be linked to lung cancer through excessive release of matrix metalloproteinases and growth factors that lead to epithelial–mesenchymal transition. Epithelial–mesenchymal transition, which has recently been shown to be present in smokers with COPD,18 is a known precursor to lung cancer.19–21 The COPD-lung cancer association may stem from either the matrix remodeling of emphysema, airway inflammation associated with small airways disease, or both. It is very possible that genetic epidemiological studies will help answer this question. Given the observations described above, one asks “Do genetic effects underlying spirometry defined COPD also confer susceptibility to lung cancer?”
Figure 1.

Distribution of FEV1 among light smokers and heavy smokers, COPD, and lung cancer.10,12–14 (A) Light or never smokers (0–20 pack-years). (B) Heavy smokers (60+ pack-years) – COPD versus “resistant smokers”. (C) Heavy smokers (60+ pack-years) – Lung cancer.
Abbreviations: COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in one second.
Genetics of COPD and lung cancer
Recently published genome-wide association studies of lung cancer, lung function, and COPD have identified several chromosomal regions and candidate genes, including 1q21–23 (CRP, IL-6R),22,23 4q22 (FAM13A),24 4q24 (GSTCD),24,25 4q31 (HHIP, GYPA23–27), 5p15 (CRR9),1,23,28 5q32–33 (HTR4, ADAM1924,25, 6p21 (BAT3, AGER1,23–25), 6q24 (GPR126),24 and 15q25 (CHRNA3/5,1,23,25,28,29Table 1). Interestingly, several of the COPD and lung cancer disease loci in this table overlap. In a case-control study, where lung cancer cases and controls were further phenotyped for COPD according to spirometry, the CHRNA3/5 (Chr 15q25) locus was found to be associated with susceptibility to both COPD and lung cancer.30 Using the same approach, variants in the HHIP (Hedgehog interacting protein, Chr 4q31) and FAM13A gene (Family with sequence similarity 13, member A, Chr 4q22) loci, previously associated with lung function,24,27 have been linked to a reduced risk of COPD and lung cancer.31,32 These findings suggest the possibility that shared pathogenetic pathways may underlie “susceptibility” to these two smoking-related diseases (Figure 2). Based on the candidate genes implicated to date, it appears that epithelial-based receptors may be orchestrating some of the downstream effects on inflammation33 and apoptosis32,34 that underlie both COPD and lung cancer. Given the increased risk of other inflammatory extrapulmonary diseases in smokers with COPD,15 these genetic variants may have effects beyond the lungs. The FAM13A (Chr 4q22) link is particularly interesting because it suggests that activity of the intracellular signaling molecule, Rho A, implicated in both COPD35 and lung cancer,36 may represent a possible chemopreventive target.32 In this regard, it is very interesting that statins (inhibitors of Rho A) have been shown in observational studies to improve outcomes for smokers with respect to both COPD and lung cancer.37
Table 1.
Chromosomal loci associated with COPD, reduced lung function, and lung cancer identified by GWA studies and overlap suggested by case-control study, modified with permission from PLoS One. Young RP, Hopkins RJ, Whittington CF, et al. Individual and cumulative effects of GWAS loci in lung cancer: associations after sub-phenotyping for COPD. PLoS One. 2011;6:e16476.44
| Disease | GWA study* | Chromosomal region | Candidate genes |
|---|---|---|---|
| COPD (FEV1) | Wilk et al27 | 1q23 | IL6R |
| Hancock et al24 | 4q22 | FAM13A | |
| Cho et al52 | |||
| Repapi et al25 | 4q24 | GSTCD | |
| Hancock et al24 | |||
| Wilk et al27 | 4q31 | HHIP/GYPA | |
| Pillai et al26 | |||
| Repapi et al25 | |||
| Hancock et al24 | |||
| Repapi et al25 | 5q33 | HTR4/ADAM19 | |
| Hancock et al24 | |||
| Repapi et al25 | 6p21 | BAT3/AGER | |
| Hancock et al24 | |||
| Hancock et al24 | 6q24 | GPR126 | |
| Pillai et al26 | 15q25 | CHRNA 3/5 | |
| Lung cancer | Amos et al23 | 1q21 | CRP |
| Amos et al23 | 4q31 | GYPA | |
| Amos et al23 | 5p15 | CRR9 (TERT) | |
| Hung et al28 | |||
| Amos et al23 | 6p21 | BAT3 | |
| Hung et al28 | |||
| Young et al88 | 6q24 | RGS17§ | |
| Amos et al23 | 15q25 | CHRNA 3/5 | |
| Hung et al28 | |||
| Thorgeirsson et al29 | |||
| COPD and lung cancer overlap | Case-control studies | ||
| Young et al30 | 15q25 | CHRNA 3/5 | |
| Young et al31 | 4q31 | HHIP/GYPA | |
| Young et al32 | 4q22 | FAM13A | |
Notes:
Available at www.genome.gov/gwastudies. Accessed 16/03/2011;
associated with familial lung cancer.88
Abbreviations: COPD, chronic obstructive pulmonary disease; GWAS, genome-wide association study; IL6R, interleukin-6 receptor; FAM13A, family with sequence similarity 13, member A; GSTCD, glutathione s-transferase, C-terminal domain containing; HHIP, Hedgehog interacting protein; GYPA, glycophorin A; HTR4, 5-hydroxytryptamine receptor 4; ADAM19, a disintegrin and metalloproteinase domain 19; BAT3, HLA-B associated transcript 3; AGER, advanced glycosylation end product-specific receptor; GPR126, G protein-coupled receptor 126; CHRNA3/5, locus containing both cholinergic receptor, nicotinic, alpha 3 and 5 genes; CRP, C-reactive protein; CRR9, cisplatin resistance-related protein; TERT, telomerase reverse transcriptase; RGS17, regulator of G-protein signaling 17.
Figure 2.
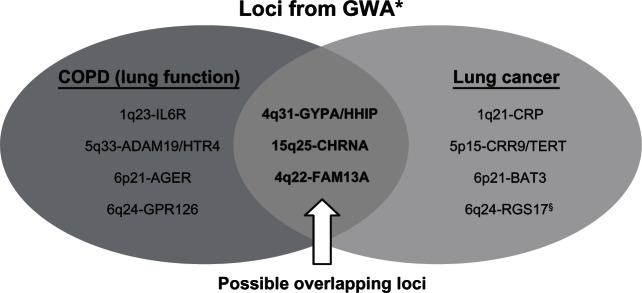
Chromosomal loci associated with COPD (reduced lung function) and lung cancer from GWA studies22–29 with possible overlapping loci.30–32
Notes: *Available at www.genome.gov/gwastudies. Accessed October 23, 2010; §associated with familial lung cancer.88
Abbreviations: COPD, chronic obstructive pulmonary disease; GWA, genome-wide association; IL6R, interleukin-6 receptor; FAM13A, family with sequence familiarity 13, member A; GSTCD, glutathione S-transferase, C-terminal domain containing; HHIP, Hedgehog interacting protein; GYPA, glycophorin A; HTR4, 5-hydroxytryptamine receptor 4; ADAM19, a disintegrin and metalloproteinase domain 19; BAT3, HLA-B associated transcript 3; AGER, advanced glycosylation end product-specific receptor; GPR126, G protein-coupled receptor 126; CHRNA3/5, locus containing both cholinergic receptor, nicotinic, alpha 3 and 5 genes; CRP, C-reactive protein; CRR9, cisplatin resistance-related protein; TERT, telomerase reverse transcriptase; RGS17, regulator of G-protein signaling 17.
The aforementioned epidemiological, molecular, and genetic findings suggest that lung cancer and COPD may not be discrete diseases related only through smoking exposure, but that many of the smokers who are susceptible to COPD are also susceptible to lung cancer through overlapping molecular pathways.10–14,19–21,30–39 Such a hypothesis was proposed by Petty five years ago38 and recently reviewed by Punturieri et al.41 Assuming susceptibility or protective loci have overlapping effects, it is possible that some of the genetic factors implicated in COPD might also be relevant in lung cancer.38,41 This may be analogous to the overlapping pathogenetic pathways underlying obesity and type 2 diabetes, where the fat mass and obesity-associated gene has been implicated in the propensity to developing both obesity and diabetes.42 In this context, body mass index is the physiological biomarker used to define the subphenotype of obesity (heritability 50%–70%) in those with type 2 diabetes (heritability 20%–30%), just as the spirometry measure of FEV1 defines COPD in those with a propensity to develop lung cancer. In other words, because body mass index reflects a propensity (or susceptibility) to obesity following a high calorie diet, FEV1 reflects an inherent genetic susceptibility to COPD (or exaggerated pulmonary inflammation) and lung cancer after chronic smoke exposure.43 If there is an overlap in genetic susceptibility between COPD and lung cancer,44 “Should an alternative approach to current case-control study designs be considered to help better identify COPD-related genes in lung cancer?”
Genetic epidemiology of COPD and lung cancer: choice of controls
The above observations suggest that an alternate genetic approach to current case-control studies could be used for genetic association studies in lung cancer.40,44 This approach would use “healthy” smoker controls (Figure 3A)23,28,29 in preference to the unselected smokers used in the recent genome-wide association case-control studies, where genetic effects are explored in lung cancer cases and smoking controls with unknown but likely different lung function (Figure 3B). As stated earlier, studies to date suggest the prevalence of COPD in randomly selected smokers is 15%–20% and in lung cancer cases 50%–80%. In studies aiming to understand better the complex relationship between COPD and lung cancer, all subjects (smokers in both cases and controls) should ideally be matched for smoking exposure and have COPD status confirmed by spirometry testing (Figure 3A). This is because the prevalence of COPD in smokers used as controls is highly dependent on the recruitment method and population sampled (range 10%–40%).10,31,45–48
Figure 3.
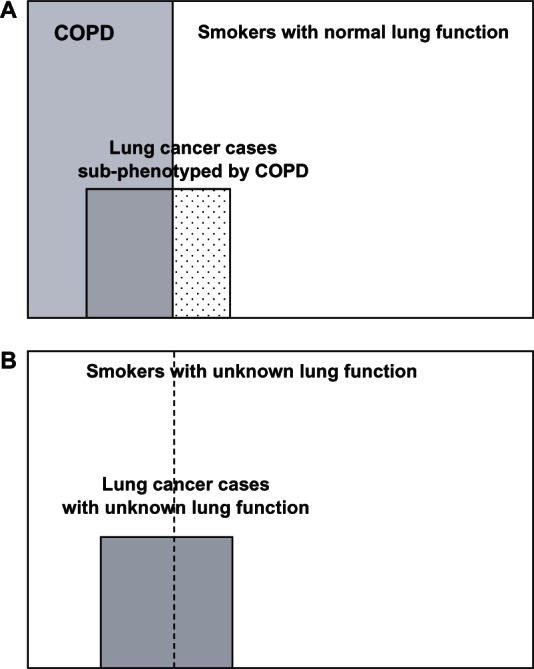
Case-control study design for lung cancer genetic association studies. (A) subphenotyping for COPD in lung cancer study.30–32 Current and former smokers with >15 pack-year smoking history and spirometry performed to allow subphenotyping in order to establish associations better. (B) COPD status of cases and controls unknown – GWAS approach.23,28,29 Current and former smokers with variable smoking exposure and unknown COPD status could result in spurious associations (confounding/modifying effect) or no association (dilution effect).
Abbreviations: COPD, chronic obstructive pulmonary disease; GWAS, genome-wide association study.
In lung cancer association studies, subphenotyping for spirometry-defined COPD44 is relevant for two reasons. First, significant differences in the prevalence of COPD among comparator populations might result in a modifier/confounder effect from COPD and explain why several associations were not replicated both within and across cohorts in the genome-wide association studies of lung cancer.23,28,29 The possibility that coexisting COPD in lung cancer cases might introduce a modifier or confounder effect in lung cancer association studies has been previously discussed.30,49 Spirometry, which is inexpensive and safe to perform, is necessary to define the presence of COPD because airflow limitation characterizing COPD is insidious in onset and is underdiagnosed in 50%–80% of cases10,45,46 due to widespread underutilization of spirometry. By subphenotyping for reduced FEV1, three smoking cohorts would be defined among the smokers, ie, those with normal or near-normal FEV1 (“resistant” controls), those with reduced FEV1 (COPD), and those with lung cancer subphenotyped for coexisting COPD (Figure 3A). Using this approach, studies have shown that the chromosome 15q25 locus, originally associated with susceptibility to lung cancer in genome-wide association studies,23,28,29 is also associated with susceptibility to COPD.30,44 This finding has recently been replicated in both genome-wide association25 and candidate gene studies.50 It has also been verified in a post hoc analysis of one of the original lung cancer genome-wide association studies.51
The second reason for using healthy smoking controls in the lung cancer studies, and examining lung cancer cases with COPD separately, is that it may be a better approach to identifying genes conferring a “protective” effect. Using this approach, it has recently been reported that the chromosome 4q31 locus and 4q22 locus, associated with a reduced risk of COPD (protective effect),24–27,52 are also independently associated with a reduced risk of lung cancer (Figure 4).31,32,44 This apparent protective effect might be overlooked in studies where lung cancer cases are compared with controls that include a high proportion of subjects with COPD. This possibility is best illustrated by comparing the allele and genotype frequencies for the protective variants of HHIP and FAM13A between COPD and lung cancer cases where they appear very similar (Table 2), and might explain why lung cancer genome-wide association studies to date have not identified these protective loci.
Figure 4.
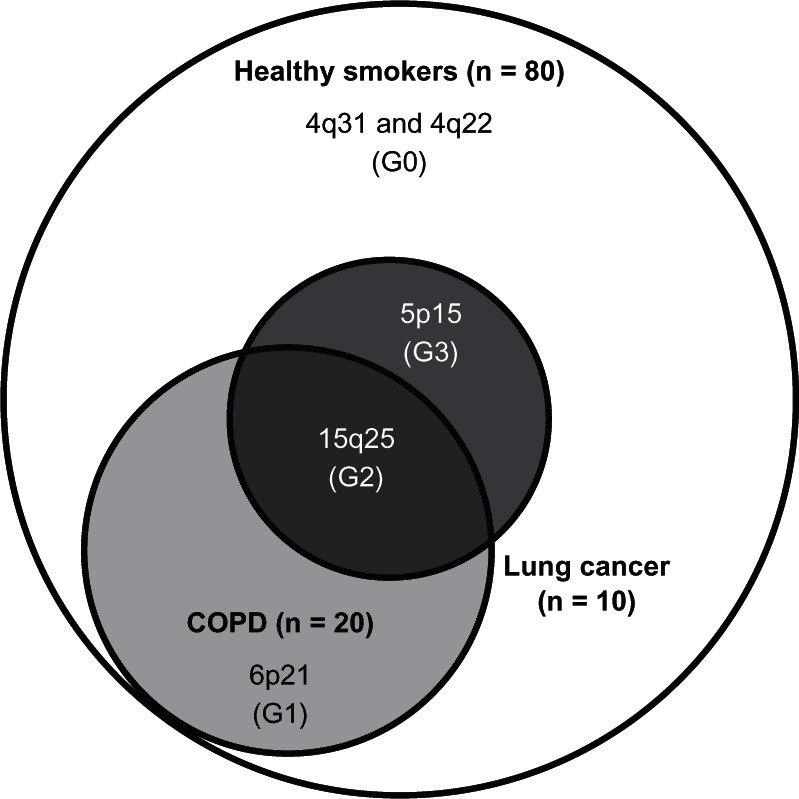
Loci conferring a nonresponder phenotype (resistant smoker, G0) and responder phenotype (susceptibility to COPD [G1], lung cancer [G3] or both [G2]): a pharmacogenetic approach to chronic smoke exposure,* modified with permission from PLoS One. Young RP, Hopkins RJ, Whittington CF, et al. Individual and cumulative effects of GWAS loci in lung cancer: associations after sub-phenotyping for COPD. PLoS One. 2011;6:e16476.44
Note: *Assumes 20% of smokers get COPD and 10% get lung cancer where 70% of the latter have pre-existing COPD in 100 chronic smokers. G0 = genes primarily conferring protection (or resistance) to both COPD and lung cancer (eg, 4q31-HHIP, 4q22 – FAM13A),31,32,44 G1 = genes primarily conferring protection or susceptibility to COPD only (eg, 6p21-AGER),24,25,44 G2 = genes primarily conferring susceptibility to COPD and lung cancer (eg, 15q25-CHRNA),1,23,25,28–30,44 G3 = genes primarily conferring protection or susceptibility to lung cancer only (eg, 5p15-CRR9).1,23,28,44
Abbreviation: COPD, chronic obstructive pulmonary disease.
Table 2.
Comparison of allele and genotype frequencies of candidate SNPs30–32 reported by GWA studies for COPD (lung function) and lung cancer,23–29 and examples of overlapping effects44
| Cohorts | Alleles | Genotypes | |||||
|---|---|---|---|---|---|---|---|
| FAM13A32 (rs7671167) | C | T | ORPvalue | CC | CT | TT | ORPvalue |
| Resistant | 530 | 440 | – | 145 | 240 | 100 | – |
| (55%) | (45%) | – | (30%) | (49%) | (21%) | – | |
| COPD | 448 | 468 | 0.79 | 107 | 234 | 117 | 0.71 |
| (49%) | (51%) | 0.01 | (23%) | (51%) | (26%) | 0.02 | |
| Lung cancer | 427 | 471 | 0.75 | 96 | 235 | 118 | 0.64 |
| (48%) | (52%) | 0.002 | (21%) | (52%) | (26%) | 0.003 | |
| Lung cancer with COPD# | 212 | 218 | 0.81 | 47 | 118 | 50 | 0.66 |
| (49%) | (51%) | 0.07 | (22%) | (55%) | (23%) | 0.03 | |
| Lung cancer only# | 185 | 229 | 0.67 | 41 | 103 | 63 | 0.58 |
| (47%) | (53%) | 0.0007 | (20%) | (50%) | (30%) | 0.006 | |
| HHIP31 (rs1489759) | G | A | ORPvalue | GG | AG | AA | ORPvalue |
| Resistant | 389 | 579 | – | 83 | 223 | 178 | – |
| (40%) | (60%) | – | (17%) | (46%) | (37%) | – | |
| COPD | 320 | 594 | 0.80 | 50 | 220 | 187 | 0.59 |
| (35%) | (65%) | 0.02 | (11%) | (48%) | (41%) | 0.006 | |
| Lung cancer | 327 | 563 | 0.86 | 56 | 215 | 174 | 0.70 |
| (37%) | (63%) | 0.13 | (13%) | (48%) | (39%) | 0.05 | |
| Lung cancer with COPD# | 134 | 292 | 0.68 | 24 | 86 | 103 | 0.61 |
| (31%) | (69%) | 0.002 | (11%) | (40%) | (48%) | 0.05 | |
| Lung cancer only# | 136 | 276 | 0.73 | 27 | 82 | 97 | 0.73 |
| (33%) | (67%) | 0.01 | (13%) | (40%) | (47%) | 0.18 | |
| CHRNA3/5α30 (rs16969968) | A | G | ORPvalue | AA | AG | GG | ORPvalue |
| Resistant | 295 | 655 | – | 45 | 205 | 225 | – |
| (31%) | (69%) | – | (9%) | (43%) | (47%) | – | |
| COPD | 339 | 551 | 1.37 | 60 | 219 | 166 | 1.47 |
| (38%) | (62%) | 0.002 | (14%) | (49%) | (37%) | 0.06 | |
| Lung cancer | 335 | 539 | 1.38 | 68 | 199 | 170 | 1.76 |
| (38%) | (62%) | 0.001 | (16%) | (46%) | (39%) | 0.005 | |
| Lung cancer with COPD# | 225 | 297 | 1.68 | 50 | 125 | 86 | 2.26 |
| (43%) | (57%) | 0.000004 | (19%) | (48%) | (33%) | 0.002 | |
| Lung cancer only# | 105 | 231 | 1.01 | 18 | 69 | 81 | 1.15 |
| (31%) | (69%) | 0.95 | (11%) | (41%) | (48%) | 0.64 | |
Notes:
COPD defined according to prebronchodilator GOLD 2+ spirometry criteria, OR comparing referent allele and genotypes (underlined) of resistant smokers to other groups.
Abbreviations: COPD, chronic obstructive pulmonary disease; GOLD, Global initiative for Chronic Obstructive Lung Disease; OR, odds ratio; SNPs, single nucleotide polymorphisms; GWA, genome-wide association.
In designing the most appropriate study to identify genetic susceptibility genes, it is noteworthy that COPD and lung cancer are distinctive from other complex genetic diseases, especially other cancers. The gene–environment interaction in COPD and lung cancer is considerably strengthened by the fact that smoking exposure, almost a prerequisite to developing these pulmonary diseases, is measurable, albeit retrospectively. Quantifying this exposure is very important because gene penetrance may be significantly affected by the degree of exposure, and it enables stratification, matching, or adjusting for tobacco exposure in the study design or analysis.53,54 A good example of the gene–environment (penetrance) effect is seen in α1-antitrypsin deficiency where disease status (COPD phenotype) is highly dependent on the degree of smoking exposure.55 In the majority of cases, the genotype conferring α1-antitrypsin deficiency is not expressed (low penetrance) in lifelong nonsmokers.
Furthermore, the potential to misclassify “randomly recruited” controls is problematic when comparing unselected (or unscreened) control smokers with cases because the prevalence (and lifetime risk) of COPD among these chronic smokers is estimated to be 20%–30% and that 20%–30% of smokers with COPD will get lung cancer.6,7,56 It has been estimated that the prevalence of COPD is in the order of 30%–40% in community-based smoking volunteers or random hospital/ clinic based controls.10,31,46 The true prevalence of COPD has been shown to be about 10%–20% when spirometry is used in truly randomly selected populations.48 On this basis, it could be argued that smokers matched for ethnicity, gender, and pack-years, with normal or near-normal lung function would minimize any potential to misclassify controls up to 30% of the time. Although the healthy smoker control group might be described as an “extreme” phenotype, and their use as controls contravenes traditional epidemiological views of controls that requires a “representative” sample, they are actually representative of the majority of chronic smokers (60%–70%)6,7 who are “resistant” or “low responders”. Importantly, they can be unambiguously phenotyped as normal by spirometry (see discussion earlier on underdiagnosis of COPD).45,46 Moreover, it is this group of chronic smokers who can be defined as “normal” (resistant) in the bimodal/trimodal distribution of FEV1 (Figure 1) with a low risk of lung cancer.14 It should not be overlooked that genetic factors might actually define this “low responder” but well defined group (“resistant smokers”,15Figures 1 and 3) rather than those susceptible to disease. For example, genetic effects selected through evolutionary pressures57 conferring a “protective (hyporesponsive) phenotype” might emerge as an alternative to a “susceptibility (hyper-responsive) phenotype”. Unlike the susceptibility phenotype, which confers an excessive pulmonary inflammatory response (eg, pneumonia complicated by acute respiratory distress syndrome), the “protective (hyporesponsive) phenotype” minimizes the inflammatory response.15,55,58 In circumstances of chronic inflammation from an exposure like smoking, these genetically determined “phenotypes” determine which smokers have aberrant lung remodeling (“hyper-responsive”) and develop COPD and which smokers maintain near-normal lung function (“hyporesponsive”). This is somewhat analogous to the sickle cell trait which was initially “selected” for its protective effect on survival from malaria but can cause mild forms of hemolytic anemia at times of acute stress or drug exposure.
Lastly, if smoking was considered a drug and FEV1 the physiological marker of smoking susceptibility (or exaggerated inflammatory response),14,43 then it would make good methodological sense in a pharmacogenetic experiment to distinguish hyporesponders (those with normal lung function) from hyper-responders (those with COPD, see Figure 4). In this instance, it would be preferable to exclude people who have not taken the drug (nonsmokers) or those with little drug exposure (light smokers). Nonsmokers and light smokers have been used as controls in the lung cancer case-control genome-wide association studies,23,28,29 but this may obscure or attenuate any genetic effects due to insufficient exposure (reducing penetrance, odds ratios, and statistical significance). Importantly, there are preliminary findings to suggest different genes may be relevant in nonsmokers with lung cancer59 or COPD.60 A further reason for using “resistant” (or hyporesponsive) controls is that minimizing misclassification of controls increases the power of the study to detect significant effects or associations,61 particularly as the contribution of “protective” (hyporesponsive) genes may be as relevant as the susceptibility genes31,32,44,58 (Figure 4). In the COPD gene study (www.copdgene.org) smokers with normal lung function and no radiological evidence of emphysema (“super controls”) are being studied separately from those with normal lung function but varying degrees of emphysema on computerized tomography (CT). However, the heritability of this “emphysema only” phenotype is poorly understood, and the epidemiological, pathological, and clinical relevance of this phenotype remains somewhat unclear.62 Most studies suggest that the presence of CT-based emphysema is related to a risk of lung cancer independent of airflow limitation.16,62 However, recent prospective CT-based studies suggest mild emphysema, in the absence of airflow limitation, may lead to overinflation (from air trapping) that heralds subsequent accelerated FEV1 decline.62 Regardless, the vast majority of those with emphysema on CT have underlying airflow limitation characterized by a reduced FEV1.16 We conclude that subphenotyping of lung cancer cases and controls using spirometry, and possibly CT, will help minimize misclassification of controls in the COPD and lung cancer studies, and further strengthen the likelihood that robust associations will be identified. However, it is accepted that effect sizes conferring risk determined by comparing cases with “resistant” or super controls may be reduced in population studies where controls are unselected. Although this is relevant in the clinical utility of risk prediction according to the population under study, it is outweighed by the potential to identify important pathogenetic pathways that might be used to target preventive therapies. We conclude that COPD and lung cancer represent unique disease models for study from a complex genetics perspective. This is because the susceptibility phenotype, defined by reduced FEV1. with high heritability and a strong association with lung cancer susceptibility, can be compared with a nonresponsive or resistant phenotype confirmed by spirometry and well matched for smoking exposure.10–17,63
Genetic association and risk modeling
Currently, COPD and lung cancer account for approximately 50% of annual deaths from smoking in the US, nearly twofold that of deaths from cardiovascular disease (26% for heart attack and stroke).4 Diagnosis is generally late in the natural history of both respiratory complications, where survival five years after diagnosis is 50% in severe COPD and only 15% in lung cancer. Both are potentially preventable diseases, particularly if smokers quit before 40 years of age.64,65 Although the clinical utility of identifying smokers at greatest risk of lung cancer and COPD remains to be confirmed, possible benefits may include targeted prevention-based approaches or more cost-effective screening approaches. There is preliminary data to suggest genetic testing might help some smokers quit.66,67 Using a risk model (or test) to stratify smokers may make an important contribution to tobacco control that currently relies solely on nonpersonalized public health messaging or disincentives. Over the last three decades, there has been a considerable reduction in coronary artery disease mortality through risk assessment and lifestyle modification. In contrast, there is no personalized or targeted risk assessment in the prevention of smoking-related lung disease. This is important because smoking, through the smoking-related disorders of COPD and lung cancer, directly accounts for the 2nd and 4th leading causes of death in the US.4 The clear potential for prevention of COPD and lung cancer, through lifestyle modification, is another unique feature of these complex diseases where environment (smoking exposure) plays such a significant role. This contrasts strongly with the other common cancers where the preventive effect of lifestyle choice is in most cases unknown. We suggest that identification of single nucleotide polymorphism associations in COPD and lung cancer to develop risk models may offer very important clinical utilities in risk prediction and risk stratification to enhance early detection or prevention strategies.
There are several existing risk models for lung cancer that include clinical variables such as age, smoking history, family history, and past history of pneumonia.40,44,68–70 Only three of these models incorporate COPD as a risk variable40,44,70 despite the finding that reduced FEV1 confers a greater risk than age and smoking exposure.63 As more single nucleotide polymorphism associations are being identified for complex diseases, attention has turned to the utility of using single nucleotide polymorphism markers in combination to risk stratify people according to genotype.71 The offering of tests using single nucleotide polymorphism markers has been appropriately criticized due to their potential to provide misleading information when used in isolation and in the absence of important environmental factors or other clinical risk factors.72 In contrast, adding single nucleotide polymorphism markers to established risk models which are based on clinical variables has, in some cases, been shown to add, albeit modestly, to the clinical performance of these models.71,72–74 With this in mind, two research groups have developed risk models for lung cancer that combine clinical variables (age, family history, and prior diagnosis of COPD) with a panel of 20 single nucleotide polymorphisms,40 three single nucleotide polymorphisms,75 and nine single nucleotide polymorphisms.44 The 20 single nucleotide polymorphism panel of Young et al included candidate single nucleotide polymorphisms previously associated with COPD and incorporates both susceptibility and protective single nucleotide polymorphisms identified using healthy smoker controls (area under the curve [AUC] 0.79).40 This model, which assigns smokers to either moderate, high, or very high risk of lung cancer, has been validated in a small prospective study76 but requires further validation in other populations. The three single nucleotide polymorphism model of Spitz et al includes three susceptibility genome-wide association-related single nucleotide polymorphisms and was based on current or former smokers who volunteered as controls (AUC 0.67).75 This panel shares the CHRNA3/5 (15q25) loci with the 20 single nucleotide polymorphism panel (the single nucleotide polymorphisms are in tight linkage disequilibrium with the same allele frequency). A recently reported nine single nucleotide polymorphism panel includes genome-wide association-related single nucleotide polymorphisms only and used the same model (clinical variables and algorithm) as the 20 single nucleotide polymorphism model (AUC 0.69). This model shares two of the three loci (CHRNA3/5 [15q25] and TERT [5p15] single nucleotide polymorphisms) with the three single nucleotide polymorphism model.44 In contrast with the three single nucleotide polymorphism model of Spitz et al, the 20 and nine single nucleotide polymorphism models proposed by Young et al use genotypes rather than alleles, and in order to maximize the discriminatory utility, combine susceptible and protective single nucleotide polymorphisms in a simple additive model to distinguish best lung cancer cases from healthy (“resistant”) smoking controls.40,44 Further studies in diverse populations will be required to validate these single nucleotide polymorphism-based approaches.
Although “risk alleles” may be used in risk models to define high-risk subgroups,73,74 there are several reasons why “risk genotypes” might be a useful alternative. Although an additive allelic model might explain variation across populations for physiological phenotypes like lung function (“passive” phenotype),24,25 the important effect that environmental exposures like smoking has on penetrance, might be of over-riding importance. The disease α1-antitrypsin deficiency is very illustrative of this point because the serum level of α1-antitrypsin is allele-dependent but the disease phenotype of COPD, expressed after smoking exposure (“dynamic” phenotype), is genotype-dependent (autosomal recessive inheritance).55 It has been argued that a per- genotype analysis of association makes fewer assumptions about interallelic effects and presence of Hardy–Weinberg equilibrium.77 Indeed, most single gene diseases are autosomal recessive rather than dominant, just as most pharmacogenetic effects (also “dynamic” phenotype) result from the effects of genotypes rather than alleles on drug responsiveness.78 Interestingly, all of the single nucleotide polymorphism-based lung cancer risk models described above include single nucleotide polymorphisms underlying susceptibility to COPD,40,44,70 just as the panels for type 2 diabetes include obesity-related single nucleotide polymorphisms.73,74 It is likely that future single nucleotide polymorphism-based models will incorporate either alleles or genotypes depending on how each contributes to optimizing the discriminatory utility of each model.
In contrast with single gene diseases, where the genotype result is dichotomized to the presence or absence of a disease-related mutation (eg, BRCA1 for risk of future breast cancer), multi/single nucleotide polymorphism tests allow a score to be derived over a continuous linear scale.40,44,71 This offers several advantages which include the following: it allows many disease-related variants to contribute to risk assessment (increasing total attributable risk), it allows for genetic heterogeneity where any one person’s lung cancer may be due to a variable combination of single nucleotide polymorphisms; risk is not limited to yes or no outcomes (or high and low risk) based on genetic profile; clinical variables can be added to genotype data; and, most importantly, it allows optimization of the test through receiver-operator curve (ROC) analyses (sensitivity and specificity) according to the inclusion of different single nucleotide polymorphisms or clinical variables.40,44,70 Family history is one such clinical variable that has been incorporated in the risk tests for prostate cancer and diabetes71,73,74 as it has in the lung cancer models.40,44,70 A similar approach is taken by the Framingham risk score for cardiovascular disease or Gail score for breast cancer risk, each incorporating a number of independent risk factors to derive a global risk score. What is consistently found in these models is that clinical variables contribute considerably to the risk and that the single nucleotide polymorphisms provide additional discriminatory utility, particularly if they measure effects not already accounted for by the clinical risk variables. Importantly, the risk associated with specific single nucleotide polymorphisms is very important in younger subjects where, due to the natural history of the disease, the clinical variables (eg, family history or diagnosis of COPD) have not yet become manifest.40,44 Other genetic variants associated with risk, including structural variation (copy number variants, deletions, duplications, and microsatellites) or epigenetic variation, could also contribute to risk models in the future.
Most importantly, a linear composite score allows all smokers (and exsmokers) to be assigned some level of risk of lung cancer whether it is moderate, high, or very high.76 Given the multitude of other lethal complications of smoking, there is no reason to suggest a moderate risk level might somehow condone continued smoking or falsely reassure smokers,82 just as a normal fasting cholesterol in an obese person would not condone a diet high in unsaturated fats. A risk stratification approach to tobacco control might strongly augment the waning effect of the current public health approach, typified by a recent study that reported over 50% of smokers with lung cancer had considered themselves at no risk.79 An additional clinical utility of a gene-based risk assessment, assigned according to a linear score, is the ability to define cutoff criteria according to the clinical use, specifically to optimize sensitivity (low cutoff) for smoking cessation and closer surveillance (eg, spirometry) for smokers given the broad benefits and optimize specificity (high cutoff) for early detection or chemoprevention given the potential harms. It is accepted that where risk tests (or models) can achieve a reasonable level of discrimination (sensitivity and specificity with area under a ROC approximating 0.70) then the test has utility for screening purposes.80
Utility of a single nucleotide polymorphism-based risk model for lung cancer
A gene-based risk model (or test) for stratifying lung cancer susceptibility has several possible clinical utilities, including targeted prevention and targeted screening for early diagnosis. Such a test would also have several potential clinical advantages over other tests of common complex diseases (summarized in Table 3). The group requiring risk stratification is easily defined by their smoking history. Given that nearly 50% of those diagnosed with lung cancer are exsmokers, this would include current or former smokers with chronic (over 20 pack-years) smoking exposure. In lung cancer there is an irrefutable intervention that will mitigate the risk, namely smoking cessation. Indeed, the very basis of smoking today, despite public health initiatives, is the high degree of optimistic bias (denial of risk) and low motivational tension to quit smoking.79,81 A lung cancer risk test, educating smokers about their personalized risk, aims to undermine this barrier to quitting (see above) and in preliminary studies shows promise in improving smoking cessation.66,67,81,82 Preliminary results from a feasibility study found that 32% of randomly recruited smokers who took a single nucleotide polymorphism-based risk test for lung cancer had quit smoking six months after testing.82 That gene-based risk prediction might assist smokers to quit was first suggested by Dr Francis Collins in 1999,83 who described a 26-year-old smoker called John attending his primary care physician who, based on John’s genetic profile indicating susceptibility to smoking, strongly recommended John quit smoking. Other clinical applications where identifying smokers at greatest risk of lung cancer is beneficial include CT screening84 or chemoprevention,85 where the costs, risks, and benefits require careful appraisal. Results from the National Lung Cancer Screening Trial were reported in November 2010, showing a 20% reduction in lung cancer mortality in current and former smokers undergoing yearly low-dose CT screening compared with yearly chest x-rays. Preliminary data using a single nucleotide polymorphism-based risk model for lung cancer estimates that over 50% of all lung cancer could be identified by CT screening only in those smokers at greatest risk (20% of eligible smokers).86 The cost implications of such an approach are enormous. Recently, a study from the Centers for Disease Control and Prevention found that genetic testing prompted changes in management by doctors in 75% of cases.87 Current or former smokers in the higher-risk categories for lung cancer might benefit from closer observation by their physician, with more use of routine spirometry and earlier referral for CT imaging when symptoms persist despite aggressive treatment for COPD.79 The factors suggesting that a lung cancer risk stratification test may have clinical utility in the general screening of smokers are summarized in Table 3.
Table 3.
Lung cancer as a complex genetic disease, and utility of screening
| Feature | Relevance to lung cancer |
|---|---|
| Polygenic | Segregation and pedigree studies suggest lung cancer results from smoking and the effects of many genes. Twin studies suggest concordance of only 15%–25%. |
| Environment | Approximately 90% of lung cancer can be attributed to smoking exposure although other environmental factors are relevant in a minority of cases. |
| Gene-environment | Genetic associations can be examined in people with smoking exposures which can be quantified to allow for matching, adjustment or stratification. |
| Subphenotyping | Lung cancer can be subphenotyped into subgroups according to age of onset, histology, or coexisting COPD (spirometry or CT emphysema). COPD, characterized by a low FEV1, is highly heritable and can be quantified by spirometry, identifying smokers at greatest risk of COPD. The distribution of FEV1 in heavy smokers is bimodal or trimodal, suggesting factors other than smoking exposure alone determines susceptibility to COPD. |
| Family history | Among smokers, the presence of a first-degree relative with lung cancer increases the risk by approximately two-fold. This increases to three-fold if there are more than two affected first-degree relatives. Family history alone has low sensitivity. |
| Identifiable group | Current or former smokers at greatest risk of lung cancer include those over 40 years old with >20 pack years of smoking exposure. |
| Risk modeling | In addition to chronic smoke exposure, studies show that age, FEV1, family history, asbestos exposure, and lung disease are associated with an increased risk of lung cancer. |
| Genetic tests and patient responses | Preliminary studies suggest that genetic testing improves smoking cessation. Testing personalizes risk and undermines optimistic bias (the tendency to underestimate personal risk). |
| Risk mitigation and early diagnosis | Quitting smoking has been shown in prospective studies to reduce the risk of lung cancer substantially. The National Lung Cancer screening Trial has shown a 20% reduction in lung cancer deaths in those undergoing annual low-dose CT screening compared with chest x-ray. |
Abbreviations: CT, computed tomography; COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in one second.
Summary
Here we have reviewed the scientific literature suggesting that COPD and lung cancer may share overlapping molecular genetic pathways and genetic “susceptibility” genes. We have also reviewed recent literature suggesting that single nucleotide polymorphism-based risk stratification of smokers is possible, and may have clinical utility as suggested a decade ago by Dr Francis Collins, director of the National Institutes of Health. We conclude that smoking causes the vast majority of COPD and lung cancer in most developed countries, and remains a public health problem despite decades of public health initiatives. Recently, the link between COPD and lung cancer has been identified at a molecular18–21 and genetic level,30–32 identifying novel pathways with potential for chemopreventive strategies. Smoking cessation remains the single most important intervention at a personal and population level, and can significantly reduce the burden of disease from smoking. While a better understanding of the molecular genetic pathways underlying COPD and lung cancer is emerging, it is clear that the selection of carefully phenotyped controls is critical to this understanding and the ability to identify relevant genetic associations.30–32,44 With the identification of novel single nucleotide polymorphism associations, along with the development of clinically useful risk prediction tools for risk stratification,40,44,70 a new era in preventive medicine may unfold where some smokers may benefit from novel preventive and/or diagnostic approaches.66,67,81–83 Preliminary data suggest that gene-based risk tools have utility in this setting.81,82,84,87 We suggest that single nucleotide polymorphism-based risk models, derived from well designed epidemiological studies, may allow the translation of recent advances in genomic medicine to personalized medicine to address what remains the single biggest and most preventable health issue of the 21st century.
Acknowledgment
This study was supported by funding grants from the University of Auckland, Auckland Medical Research Foundation, Health Research Council of New Zealand, and by Synergenz BioScience New Zealand Ltd. This work was also supported in part by a grant from the National Heart, Lung and Blood Institute.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Broderick P, Wang Y, Vijayakarishnan V, et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69:6633–6641. doi: 10.1158/0008-5472.CAN-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molfino NA. Genetics of COPD. Chest. 2004;125:1929–1940. doi: 10.1378/chest.125.5.1929. [DOI] [PubMed] [Google Scholar]
- 3.Witschi H. A short history of lung cancer. Toxicol Sci. 2001;64:4–6. doi: 10.1093/toxsci/64.1.4. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society Available from: http://www.cancer.org/Accessed July 26, 2010
- 5.Mattson ME, Pollack ES, Cullen JW. What are the odds that smoking will kill you? Am J Pub Health. 1987;77:425–431. doi: 10.2105/ajph.77.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohansal R, Martinez-Camblor P, Agusti A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham Offspring Cohort. Am J Respir Crit Care Med. 2009;180:3–10. doi: 10.1164/rccm.200901-0047OC. [DOI] [PubMed] [Google Scholar]
- 7.Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61:935–939. doi: 10.1136/thx.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer: analyses of cohorts of twins from Sweden, Denmark and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 9.Hubert HB, Fabsitz RR, Feinleib M, Gwinn C. Genetic and environmental influences on pulmonary function in adult twins. Am Rev Respir Dis. 1982;125:409–415. doi: 10.1164/arrd.1982.125.4.409. [DOI] [PubMed] [Google Scholar]
- 10.Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. COPD prevalence is increased in lung cancer independence of age, gender and smoking history. Eur Respir J. 2009;34:380–386. doi: 10.1183/09031936.00144208. [DOI] [PubMed] [Google Scholar]
- 11.Mannino DM, Aguayo SM, Petty TL, Redd SC. Low lung function and incident lung cancer in the United States: data from the first NHANES follow-up. Arch Intern Med. 2003;163:1475–1480. doi: 10.1001/archinte.163.12.1475. [DOI] [PubMed] [Google Scholar]
- 12.Burrows B, Knudson RJ, Cline MG, Lebowitz MD. Qualitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis. 1977;115:195–205. doi: 10.1164/arrd.1977.115.2.195. [DOI] [PubMed] [Google Scholar]
- 13.Dockery DW, Speizer FE, Ferris BG, Jr, Ware JH, Louis TA, Spiro A. Cumulative and reversible effects of lifetime smoking on simple tests of lung function in adults. Am Rev Respir Dis. 1988;137:286–292. doi: 10.1164/ajrccm/137.2.286. [DOI] [PubMed] [Google Scholar]
- 14.Givelber RJ, Couropmitree NN, Gottlieb DJ, et al. Segregation analysis of pulmonary function among families in the Framingham study. Am J Respir Crit Care Med. 1998;157:1445–1451. doi: 10.1164/ajrccm.157.5.9704021. [DOI] [PubMed] [Google Scholar]
- 15.Young RP, Hopkins RJ, Eaton TE. Forced expiratory volume in one second: not just a lung function test but a marker of premature death from all causes. Eur Respir J. 2007;30:616–622. doi: 10.1183/09031936.00021707. [DOI] [PubMed] [Google Scholar]
- 16.Wilson DO, Weissfeld JL, Balkan A, et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am J Respir Crit Care Med. 2008;178:738–744. doi: 10.1164/rccm.200803-435OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young R, Hopkins R. Lung function predicts lung cancer. Eur Respir J. 2010;35:1421–1422. doi: 10.1183/09031936.00009410. [DOI] [PubMed] [Google Scholar]
- 18.Sohal S, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15:930–938. doi: 10.1111/j.1440-1843.2010.01808.x. [DOI] [PubMed] [Google Scholar]
- 19.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dasari V, Gallup M, Lemjabbar H, Maltseva I, MacNarmara N. Epithelial-mesenchymal transition in lung cancer: is tobacco the “ smoking gun”? Am J Respir Cell Mol Biol. 2006;35:3–9. doi: 10.1165/rcmb.2006-0051SF. [DOI] [PubMed] [Google Scholar]
- 21.Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ. Collagen 1 promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-β signalling. Am J Respir Cell Mol Biol. 2008;38:95–104. doi: 10.1165/rcmb.2007-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilk J, Walter R, Laramie J, Gottlieb DJ, O’Connor GT. Framingham Heart Study genome-wide association: results for pulmonary function measures. BMC Med Genet. 2007;8(Suppl 1):S8. doi: 10.1186/1471-2350-8-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2009;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2009;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilk JB, Chen T, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 29.Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young RP, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Lung cancer gene associated with COPD: triple whammy or possible confounding effect? Eur Respir J. 2008;32:1–7. doi: 10.1183/09031936.00093908. [DOI] [PubMed] [Google Scholar]
- 31.Young RP, Whittington CF, Hopkins RJ, et al. Chromosome 4q31 locus in COPD also associated with lung cancer. Eur Respir J. 2010;36:1375–1382. doi: 10.1183/09031936.00033310. [DOI] [PubMed] [Google Scholar]
- 32.Young RP, Hopkins RJ, Hay BA, et al. FAM13A locus in COPD independently associated with lung cancer – evidence of a molecular genetic link between COPD and lung cancer. Appl Clin Genet. 2011;4:1–10. doi: 10.2147/TACG.S15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gwilt CR, Donnelly LE, Rogers DF. The non-neuronal cholinergic system in the airways: an unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther. 2007;115:208–222. doi: 10.1016/j.pharmthera.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 35.Richens TR, Linderman DJ, Horstmann SA, et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of Rho A. Am J Respir Crit Care Med. 2009;179:1011–1021. doi: 10.1164/rccm.200807-1148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asnaghi L, Vass WC, Quadri R, et al. E-Cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of GTPases. Oncogene. 2010;29:2760–2771. doi: 10.1038/onc.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: potential utility in COPD. Eur Respir Rev. 2009;18:222–232. doi: 10.1183/09059180.00005309. [DOI] [PubMed] [Google Scholar]
- 38.Petty TL. Are COPD and lung cancer two manifestations of the same disease? Chest. 2005;128:1895–1897. doi: 10.1378/chest.128.4.1895. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz AG, Ruckdeschel JC. Familial lung cancer: genetic susceptibility and relationship to chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:16–22. doi: 10.1164/rccm.200502-235PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young RP, Hopkins RJ, Hay BA, et al. A gene based risk score for lung cancer susceptibility in smokers and ex-smokers. Postgrad Med J. 2009;85:515–524. doi: 10.1136/pgmj.2008.077107. [DOI] [PubMed] [Google Scholar]
- 41.Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM. Lung cancer and chronic obstructive pulmonary disease: needs and opportunities for integrated research. J Natl Cancer Inst. 2009;101:554–559. doi: 10.1093/jnci/djp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–662. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 43.Young RP, Hopkins RJ. Link between COPD and lung cancer. Respir Med. 2010;104:758–759. doi: 10.1016/j.rmed.2009.11.025. [DOI] [PubMed] [Google Scholar]
- 44.Young RP, Hopkins RJ, Whittington CF, et al. Individual and cumulative effects of GWAS loci in lung cancer: associations after sub-phenotyping for COPD. PLoS One. 2011;6:e16476. doi: 10.1371/journal.pone.0016476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stav D, Raz M. Prevalence of chronic obstructive pulmonary disease among smokers aged 45 and up in Israel. Isr Med Assoc J. 2007;9:800–802. [PubMed] [Google Scholar]
- 46.Zaas D, Wise R, Wiener C, et al. Airways obstruction is common but unsuspected in patients admitted to a general medicine service. Chest. 2004;125:106–111. doi: 10.1378/chest.125.1.106. [DOI] [PubMed] [Google Scholar]
- 47.Hill K, Goldstein RS, Guyatt GH, et al. Prevalence and under-diagnosis of chronic obstructive pulmonary disease among patients at risk in primary care. CMAJ. 2010;182:673–678. doi: 10.1503/cmaj.091784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koshiol J, Rotunno M, Consonni D, et al. Chronic obstructive pulmonary disease and altered risk of lung cancer in a population-based case-control study. Plos One. 2009;4:e7380. doi: 10.1371/journal.pone.0007380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang P, Yafei L, Jiang R, Cunningham JM, Zhang F, de Andrade M. A rigorous and comprehensive validation: common genetic variations and lung cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:240–244. doi: 10.1158/1055-9965.EPI-09-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambrechts D, Buysschaert I, Zanen P, et al. The 15q24/25 susceptibility variant for lung cancer and chronic obstructive pulmonary disease is associated with emphysema. Am J Respir Crit Care Med. 2010;181:486–493. doi: 10.1164/rccm.200909-1364OC. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Spitz MR, Amos CI, et al. Mediating effects of smoking and chronic obstructive pulmonary disease on the relation between the CHRNA5-A3 genetic locus and lung cancer. Cancer. 2010;116:3458–3462. doi: 10.1002/cncr.25085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janssens ACJW, van Duijn CM. Genome-based prediction of common disease: methodological considerations for future research. Genome Med. 2010;1:20. doi: 10.1186/gm20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA. 2008;299:1335–1344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 55.Piitulainen E, Eriksson S. Decline in FEV1 related to smoking status in individuals with severe alpha1-antitrypsin deficiency (PiZZ) Eur Respir J. 1999;13:247–251. doi: 10.1183/09031936.99.13224799. [DOI] [PubMed] [Google Scholar]
- 56.Vilkman S, Keistinen T, Tuuponen T, Kivela SL. Survival and cause of death among elderly chronic obstructive pulmonary disease patients after first admission to hospital. Respiration. 1997;64:281–284. doi: 10.1159/000196687. [DOI] [PubMed] [Google Scholar]
- 57.Marciniak SJ, Lomas DA. What can naturally occurring mutations tell us about the pathogenesis of COPD. Thorax. 2009;64:359–364. doi: 10.1136/thx.2008.099408. [DOI] [PubMed] [Google Scholar]
- 58.Young RP, Hopkins R, Black PN, et al. Functional variants of antioxidant genes in smokers with COPD and in those with normal lung function. Thorax. 2006;30:394–399. doi: 10.1136/thx.2005.048512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Sheu CC, Ye YQ, et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol. 2010;11:321–330. doi: 10.1016/S1470-2045(10)70042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sampsonas F, Kaparianos A, Lykouras D, Karkoulias K, Spiropoulos K. DNA sequence variations of metalloproteinases: their role in asthma and COPD. Postgrad Med J. 2007;83:244–250. doi: 10.1136/pgmj.2006.052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moskvina V, Holmans P, Schmidt KM, Craddock N. Design of case-controls studies with unscreened controls. Ann Hum Genet. 2005;68:566–576. doi: 10.1111/j.1529-8817.2005.00175.x. [DOI] [PubMed] [Google Scholar]
- 62.Sin DD, Leipsic J, Man SFP. CT in COPD: just a pretty picture or really worth a thousand words (or dollars)? Thorax. 2011 May 19; doi: 10.1136/thx.2011.161430. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 63.Tockman MS, Anthonisen N, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–518. doi: 10.7326/0003-4819-106-4-512. [DOI] [PubMed] [Google Scholar]
- 64.Anthonisen NR, Skeans MA, Wise RA, Manfreda J, Kanner RE, Connett JE. The effects of smoking cessation intervention on 14.5 year mortality: a randomized clinical trial. Ann Intern Med. 2005;142:233–239. doi: 10.7326/0003-4819-142-4-200502150-00005. [DOI] [PubMed] [Google Scholar]
- 65.Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321:323–329. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McBride CM, Bepler G, Lipkus IM, et al. Incorporating genetic susceptibility feedback into a smoking cessation program for African-American smokers with low income. Cancer Epidemiol Biomarkers Prev. 2002;11:521–528. [PubMed] [Google Scholar]
- 67.Carpenter MJ, Strange C, Jones Y, et al. Does genetic testing result in behavioral health change? Changes in smoking behavior following testing for alpha-1 antitrypsin deficiency. Ann Behav Med. 2007;33:22–28. doi: 10.1207/s15324796abm3301_3. [DOI] [PubMed] [Google Scholar]
- 68.Bach PB.Lung cancer assessment tool Available from: http://www.mskcc.orgAccessed June 15, 2011
- 69.Cassidy A, Duffy SW, Myles JP, Liloglou T, Field JK. Lung cancer risk prediction: a tool for early detection. Int J Cancer. 2006;120:1–6. doi: 10.1002/ijc.22331. [DOI] [PubMed] [Google Scholar]
- 70.Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Nat Cancer Inst. 2007;99:715–726. doi: 10.1093/jnci/djk153. [DOI] [PubMed] [Google Scholar]
- 71.Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med. 2008;358:910–919. doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
- 72.Wright CF, Kroese M.Evaluation of genetic tests for susceptibility to complex diseases: why, when and how? Hum Genet 2010127125–134. [DOI] [PubMed] [Google Scholar]
- 73.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.He M, Cornelis MC, Franks PW, Zhang C, Hu FB, Qi L. Obesity, genotype score and cardiovascular risk in women with type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2010;30:327–333. doi: 10.1161/ATVBAHA.109.196196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spitz MR, Amos CI, D’Amellio, Dong Q, Etzel C. Re: discriminatory accuracy from single-nucleotide polymorphisms in models. J Natl Cancer Inst. 2009;24:1731–1732. doi: 10.1093/jnci/djp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young RP, Hopkins RJ, Hay BJ, Gamble GD. GWAS and candidate SNPs for COPD and lung cancer combine to identify lung cancer susceptibility: validation in a prospective study; Abstr A3738 presented at the annual meeting of the American Thoracic Society; New Orleans, LA. May-19. 2010. [Google Scholar]
- 77.Thankkinstian A, Thompson JR, Minelli C, Attia J. Choosing between per-genotype, per-allele, and trend approaches for initial detection of gene-disease association. J Appl Stat. 2009;36:633–646. [Google Scholar]
- 78.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348:529–537. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 79.Smith SM, Campbell NC, MacLeod U, et al. Factors contributing to the time taken to consult with symptoms of lung cancer: a cross-sectional study. Thorax. 2009;64:523–531. doi: 10.1136/thx.2008.096560. [DOI] [PubMed] [Google Scholar]
- 80.Wald NJ. When can a risk factor be used as a worthwhile screening test? BMJ. 1999;319:1562–1565. doi: 10.1136/bmj.319.7224.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Young RP, Hopkins RJ, Smith M, Hogarth DK. Smoking cessation: the potential role of risk assessment tools as motivational triggers. Postgrad Med J. 2010;86:26–33. doi: 10.1136/pgmj.2009.084947. [DOI] [PubMed] [Google Scholar]
- 82.Hopkins RJ, Young RP, Hay BA, Gamble GD. Gene-based risk test for lung cancer risk motivates smoking cessation in randomly selected smokers; Abstr P4714 presented at the annual meeting of the European Respiratory Society; Barcelona, Spain. Sep-22. 2010. [Google Scholar]
- 83.Collins FS. Shattuck lecture – Medical and societal consequences of the human genome project. N Engl J Med. 1999;341:28–37. doi: 10.1056/NEJM199907013410106. [DOI] [PubMed] [Google Scholar]
- 84.Bechtel JJ, Kelly WA, Coons TA, et al. Lung cancer detection in patients with airflow obstruction identified in a primary care outpatient practice. Chest. 2005;127:114–115. doi: 10.1378/chest.127.4.1140. [DOI] [PubMed] [Google Scholar]
- 85.Lee G, Walser TC, Dubinett SM. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Curr Opin Pulm Med. 2009;15:303–307. doi: 10.1097/MCP.0b013e32832c975a. [DOI] [PubMed] [Google Scholar]
- 86.Young RP, Hopkins RJ, Field JK, et al. Abstr B31 presented at the annual meeting of the American Association of Cancer Research. Frontiers in the Prevention of Cancer; Philadelphia, PA: Nov-Oct. 2010. Gene-based lung cancer risk test identifies smokers for early detection of lung cancer. [Google Scholar]
- 87.Kolor K, Liu T, St Pierre J, Khoury MJ. Health care provider and consumer awareness, perceptions and use of direct to consumer personal genomic tests, US, 2008. Genet Med. 2009;11:595. doi: 10.1097/GIM.0b013e3181b1cc2c. [DOI] [PubMed] [Google Scholar]
- 88.You M, Wang D, Liu P, et al. Fine mapping of chromosome 6q23–25 region in familial lung cancer families reveals RGS17 as a likely candidate gene. Clin Cancer Res. 2009;15:2666–2674. doi: 10.1158/1078-0432.CCR-08-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]