

Abstract
The importance of ubiquitin E3 ligases in neurodegeneration is being increasingly recognized. The crucial role of NEDD4-1 in neural development is well appreciated; however, its role in neurodegeneration remains unexplored. Herein, we report increased NEDD4-1 expression in the degenerated tissues of several major neurodegenerative diseases. Moreover, its expression is upregulated in cultured neurons in response to various neurotoxins, including zinc and hydrogen superoxide, via transcriptional activation likely mediated by the reactive oxygen species (ROS)-responsive FOXM1B. Reduced protein levels of the insulin-like growth factor receptor (IGF-1Rβ) were observed as a consequence of upregulated NEDD4-1 via the ubiquitin-proteasome system. Overexpression of a familial mutant form of superoxide dismutase 1 (SOD1) (G93A) in neuroblastoma cells resulted in a similar reduction of IGF-1Rβ protein. This inverse correlation between NEDD4-1 and IGF-1Rβ was also observed in the cortex and spinal cords of mutant (G93A) SOD1 transgenic mice at a presymptomatic age, which was similarly induced by in vivo-administered zinc in wild-type C57BL/6 mice. Furthermore, histochemistry reveals markedly increased NEDD4-1 immunoreactivity in the degenerating/degenerated motor neurons in the lumbar anterior horn of the spinal cord, suggesting a direct causative role for NEDD4-1 in neurodegeneration. Indeed, downregulation of NEDD4-1 by shRNA or overexpression of a catalytically inactive form rescued neurons from zinc-induced cell death. Similarly, neurons with a NEDD4-1 haplotype are more resistant to apoptosis, largely due to expression of higher levels of IGF-1Rβ.Together, our work identifies a novel molecular mechanism for ROS-upregulated NEDD4-1 and the subsequently reduced IGF-1Rβ signaling in neurodegeneration.
Introduction
Neurodegenerative disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) represent the most devastating diseases in the developing world. Despite a diverse array of causative factors for these diseases, several common pathogenic mechanisms have been identified, including aberrant protein aggregation/deposition, elevated oxidative stress (ROS), mitochondrial dysfunction, and neuronal death.
Impaired insulin/insulin growth factor (IGF)-1 signaling has been associated with all of these major diseases (Gasparini and Xu, 2003; Trejo et al., 2004; de la Monte and Wands, 2005; Freude et al., 2009). The biological effects of IGF-1 are mediated by binding to specific IGF receptors present in target tissues. The IGF-1 receptor (IGF-1R) is a ligand-dependent tyrosine kinase activated by phosphorylation of the tyrosine residues in the tyrosine kinase domain of the β-subunit, thereby stimulating several intracellular signal transduction cascades (Butler et al., 1998). Fine-tuning of IGF-1Rβ regulation is crucial for brain functions:IGF-1Rβ levels on the cell surface are mainly controlled by internalization and degradation via the ubiquitin-proteasome system (UPS). In many cancer cell types, IGF-1Rβ ubiquitination is mediated by the HECT family of E3 ligases such as NEDD4-1 (LeRoith et al., 1995). NEDD4-1 has been found to play key roles in various aspects of neuronal development in vertebrates and invertebrates (Kumar et al., 1992; Yang and Kumar, 2010). For example, NEDD4-1−/− embryos are characterized by the small size of skeletal muscle fibers and the reduced number of motor neurons (MNs; Liu et al., 2009). In addition, NEDD4-1−/− mice die during or shortly after birth and display severe intrauterine growth retardation (Cao et al., 2008; Liu et al., 2009). This same phenotype is also exhibited by mice carrying an Igf1r-null mutation (Baker et al., 1993; Liu et al., 1993). More recently, there are reports that NEDD4-1 is also required for axonal branching and that its overexpression promotes neuritic outgrowth (Drinjakovic et al., 2010; Kawabe et al., 2010). Although widely expressed in the vertebrate CNS (Kumar et al., 1992), the role of NEDD4-1 in adult brains has remained largely unexplored.
Zinc has been increasingly recognized as an important molecule that plays both physiological and pathological roles in a variety of biological processes as well as in chronic neurodegenerative diseases such as AD and ALS via elevated oxidative stress (Cuajungco and Lees, 1997; Koh, 2001; Lee, 2002; Friedlich, 2004; Frederickson et al., 2005; Smith and Lee, 2007); a high concentration of zinc is known to cause acute neuronal death through a mechanism similar to excitotoxicity. We recently demonstrated that zinc induces UPS activation by upregulating NEDD4-1 expression in neurons (Kwak et al., 2010a). Herein, we report that NEDD4-1 is upregulated by a variety of neurotoxins that elicit oxidative stress in neurons, leading to IGF-1R protein degradation via UPS. Elevated NEDD4-1 expression is also found in the brain tissues of AD, PD, HD, and the spinal cord tissues of ALS and mutant SOD1 mice. Moreover, downregulation/inactivation of NEDD4-1 expression/activity completely rescued neurons from zinc-induced cell death. Together, our work discovers a novel molecular mechanism underlying neurodegeneration.
Materials and Methods
Cell culture, treatment of Zn2+, and transfection.
Primary cultured cortical neurons were prepared as described previously (Kwak et al., 2010a) using E17 pregnant rats. For the majority of experiments, zinc was added at 300 μm to 2-week-old cultured neurons for 4 h before cell lysates were prepared. In the present study, we used ZnSO4 instead of ZnCl2 because it has less cellular toxicity. Neuroblastoma N2a cell cultures and transient transfections were performed as described previously (Kwak et al., 2010a). Transient transfections with vector plasmids for pRc-CMV-T7-NEDD4-1-WT, pRc-CMV-T7-NEDD4-1-CS, pSUPER-shNEDD4-1, pCDNA3-EGFP-SOD1-WT, pCDNA3-EGFP-SOD1-G93A, and Stealth-negative siRNA (Invitrogen) were performed with Lipofectamine 2000 (Invitrogen) on subconfluent N2a cells in 6-well culture plates according to the manufacturer's protocol.
Quantitative reverse transcriptase PCRs.
Quantitative reverse transcriptase PCR (qRT-PCR) on NEDD4-1 and a glyceraldehyde-3-phosphat dehydrogenase (GAPDH) control were performed as described previously (Kwak et al., 2010a). Primers used for NEDD4-1 and GAPDH amplification were NEDD4-1(+): 5′-CCAGGTTGGGAAGAAAAACA-3′ and NEDD4-1 (−): 5′-ATTTCAGATGGCTGGGTCAC-3′ and GAPDH (+): 5′-CCCTTCATTGACCTCAACTA-3′ and GAPDH (−): 5′-CCTTCTCCATGGTGGTGAA-3′, respectively.
Chromatin immunoprecipitation assay.
HEK293 cells (2 × 106) were treated with 50 or 100 μm H2O2 for 6 h and then were prepared for the assay according to the manufacturer's instructions from the ChIP assay kit (Millipore). The resulting DNA complexes were first immunoprecipitated using the indicated anti-FOXM1B antibodies (Santa Cruz Biotechnology) and then DNA was purified from the complexes and subjected to PCR to amplify the putative FOXM1B binding region 1 (−745 to −560 bp) of the NEDD4-1 promoter using the primers 5′-TCTTCGTCACTGCCTTCTG-3′ and 5′-TTGGACTCGGGATCTGAAA-3′.
NEDD4-1 heterozygous and SOD1-Tg mice.
NEDD4-1+/− mice were generously provided by Mutant Mouse Regional Resource Centers (B6;129S5-Nedd4Gt(IRESBetageo)249Lex/Mmucd; stock #011742-UCD) and genotyped following the instruction provided by NIH-0931 Genotyping Strategies. Cortical neurons were isolated and prepared from P0 pups following quick genotyping to separate heterozygous from wild-type (WT) pups. The G93A SOD1-Tg mice (stock #004435) were obtained from the The Jackson Laboratory, maintained by crossing with C57BL/6 mice, and genotyped by genomic PCR following the procedure provided by the vendor.
Immunoprecipitation and Western blot analysis.
IP-Western experiments were performed as described previously (Kwak et al., 2010a). The following antibodies (Abs) were used: mouse anti-monopoly-Ub Ab (Enzo), rabbit anti-NEDD4-1 Ab (Millipore), rabbit anti-NEDD4–2 (Abcam), mouse anti-enhanced green fluorescent protein (EGFP) (Cell Signaling Technology), rabbit anti-IGF-1Rβ (Cell Signaling Technology), mouse α-tubulin (Sigma), mouse anti-β-actin Ab (Sigma), and anti-mouse IgG and anti-rabbit IgG horseradish peroxidase-conjugated Abs (Millipore Bioscience Research Reagents). For immunoprecipitation, cell lysates (300 μg) were immunoprecipitated with specific antibody against IGF-1Rβ using protein A-Sepharose (GE Healthcare Bioscience), followed by standard procedures described in our previous work (Kwak et al., 2010a).
Immunocytochemistry and immunohistochemistry.
For neuronal morphology (MAP2) and propidium iodide staining, experiments were performed as described previously (Kwak et al., 2010a), e.g., SOD1-Tg mouse spinal cord histology. Briefly, paraffin sections (5 μm) were deparaffinized by passing through 100% xylene and rehydrated through serial dilutions of ethanol (100, 95, and 75%). Antigen retrieval was accomplished by incubating the sections for 4–5 min in 10 mm citrate buffer at 95°C. Endogenous peroxidase activity was suppressed with 3% H2O2 (in dH2O) for 30 min. Following washing with PBS and blocking with 5% goat serum [1 h; room temperature (RT)], sections were incubated with IGF-1Rβ (1:100; Cell Signaling Technology, #3027) or NEDD4-1 (1:400 to 1:2000; Millipore, #07-049) Ab in PBST at 4°C overnight. After incubation with the first antibody and proper washing, sections were incubated with biotinylated secondary antibody (1:200 in PBST; Vector Laboratories, Vectastain Elite ABC Kit; #PK-6200) for 2 h at RT. After washing, the sections were incubated with ABC reagent (1:100 in PBST; Vector Laboratories, Vectastain Elite ABC Kit) for 1 h at RT. The colored reaction product was developed with a diaminobenzidine peroxidase substrate kit (Vector Laboratories, #SK-4100). Sections are finally dehydrated and mounted. Images are obtained under a bright-field light microscope (Olympus).
The TUNEL assay was performed by a kit (Promega, category #G3250). Cells were cultured in poly-l-lysine-coated coverslip in a 24-well plate. To control for the effects of cell density, equal numbers of cells were plated per well. TUNEL+ cells were counted from digital images using PicCnt software. To select TUNEL nuclei, Cell-counting software was applied that accounted for clustering, but excluded very small nuclear fragments. Positive controls are performed by incubating with DNase 1.
Human tissue samples.
Human brain samples were provided by University of California San Diego and were analyzed with institutional permission under California and National Institutes of Health (NIH) guidelines. Informed consent was obtained following the procedures of the Institutional Review Boards of the Sanford-Burnham Institute for Medical Research. Human lumbar spinal cord segments from five patients that died of respiratory failure caused by ALS were provided by the Human Brain and Spinal Fluid Resource Center, West Los Angeles Healthcare Center, Los Angeles, CA, sponsored by National Institute of Neurological Disorders and Stroke–National Institute of Mental Health, National Multiple Sclerosis Society, and the Department of Veterans Affairs. Control samples were obtained from six individuals who died from AD and lung cancer. The age, gender, and postmortem interval of the subjects are described in Table 1.
Table 1.
Patient sample information
| Age | Sex | Braak_Combo | Diagnosis | |
|---|---|---|---|---|
| 1 | 69 | Male | 1.2 | Normal |
| 2 | 102 | Female | 1.2 | Normal |
| 3 | 97 | Female | 1.2 | Normal, lacunar infarct, chronic (old), ischemia |
| 4 | 74 | Female | 1.2 | Normal |
| 5 | 84 | Male | 0.2 | Alzheimer's changes, infarct, chronic (old), other (unclassified) |
| 6 | 84 | Male | 1.2 | Alzheimer's changes, mild |
| 7 | 91 | Female | 1.2 | Alzheimer's changes, moderate |
| 8 | 83 | Male | 5.2 | Alzheimer's changes, progressive multifocal leukoencephalopathy |
| 9 | Male | 1 | Parkinson's disease, idiopathic | |
| 10 | 74 | Male | 0.2 | Parkinson's disease, idiopathic |
| 11 | 89 | Female | 4.2 | Parkinson's disease, idiopathic/Alzheimer's disease |
| 12 | 74 | Male | Parkinson's disease, idiopathic/Alzheimer's change, moderate/Amyloid angiopathy | |
| 13 | 73 | Male | 0 | Parkinson's disease, idiopathic/neocortical Lewy body/amyloid angiopathy |
| 14 | 47 | Male | 0 | Huntington's disease |
| 15 | Female | 0 | Huntington's disease | |
| 16 | Male | 6.2 | Huntington's disease | |
| 17 | 35 | Female | 0 | Huntington's disease |
| 18 | 75 | Female | 0 | Huntington's disease |
Administration of ZnSO4 to C57BL/6 mice.
Mouse handling was performed according to the standard procedures approved by the Animal Research Committee at the Sanford-Burnham Medical Research Institute and NIH Guidelines. A small volume (200 μl, 600 μm) of ZnSO4 or saline was administered to 12-month-old female C57BL/6 mice by intraperitoneal injection at different time points (0, 6, 12, 18, and 24 h). To examine protein expression of NEDD4-1/IGF-1Rβ, the expression levels of these proteins were analyzed in the brain and spinal cord samples from these mice.
Statistics.
All quantitative data were presented as means ± SDV. Comparisons between groups were analyzed with unpaired ANOVA using GraphPad PRIZM software and values of p < 0.05 were considered to be significant.
Results
Increased expression of NEDD4-1 protein in degenerated human specimens
To investigate the potential involvement of the E3 ligase NEDD4-1 in the process of neurodegeneration, we examined protein expression levels using Western blot analysis on a panel of specimens taken from patients diagnosed with end-stage AD, PD, HD, and ALS and compared them to age-matched control specimens (NC: patients died from disorders not related to CNS). The patient cohort and information are summarized in Table 1. From semiquantitative profiling of NEDD4-1 in 33 human specimens, we found that NEDD4-1 protein levels were elevated in all of the degenerated specimens we examined, with the highest levels seen in the forebrains of HD patients (Fig. 1A) and four of the five spinal cord ALS samples (Fig. 1B), while nearly undetectable in the spinal cords of four controls (AD). We also noticed that the same polyclonal anti-NEDD4-1 antibody we used in these Western blot assays revealed doublets in cortical samples and single species of the NEDD4-1 protein in the spinal cord samples. Interestingly, the NEDD4-1 protein expression level was also found to be increased in the normal aged brains of C57BL/6 mice, though levels increased slower (data not shown).
Figure 1.

Quantitative analysis of NEDD4-1 protein levels in human specimens. A, NEDD4-1 was detected by immunoblot analysis using 15 μg protein lysates prepared from frontal regions of clinical patients. NC, normal age-matched control; AD, Alzheimer's disease; PD, Parkinson's disease; HD, Huntington's disease. Densitometry analysis reveals a statistically significant elevation of total NEDD4-1 levels in AD, PD, and HD brains compared with NC brains. ***p < 0.001; *p < 0.05. B, Increased NEDD4-1 in the lumbar spinal cord specimens from ALS patients. Age-matched AD cases and two NC (patients died from lung cancer) were used as control for ALS.
NEDD4-1 expression is upregulated by various neurotoxins via elevated oxidative stress
Since ROS is the common denominator in neurodegenerative diseases and in normal aging processes, we suspected that ROS induces the upregulation of NEDD4-1. Indeed, using cultured primary rat cortical neurons, we found that NEDD4-1 is rapidly induced in neurons after treatment with a variety of neurotoxic agents (Fig. 2A); all these agents are known to induce ROS and are thus relevant to the aforementioned neurodegenerative diseases. These agents include the soluble form of β-amyloid peptides secreted from a natural source (7PA2/CHO cells stably transfected with two familial AD mutants of amyloid precursor protein (APP; Walsh et al., 2002), the classic apoptotic stimulus (camptothecin), H2O2, and zinc. Furthermore, pretreatment of neurons with several antioxidants for 1 h completely prevented neurons from zinc-induced NEDD4-1 upregulation; these included diphenylene iodonium at 10 μm, N-acetyl cysteine at 0.5 or 1 mm, and catalase at 1000 U (data not shown).
Figure 2.

Oxidative stress transcriptionally upregulates NEDD4-1 protein expression levels in primary cultured cortical neurons. A, Increase in NEDD4-1 protein levels induced by various neurotoxins. Two-week-old cultured mature neurons were exposed to various concentrations of agents for 4 h followed by Western blot analysis. 7PA2 represents a source of soluble A from cultured conditioned media collected from mutant APP-transfected CHO cells (Walsh et al., 2002): “1/10” indicates 10-fold dilution, which contains ∼200 pm Aβ. Quantification bar graph is based on densitometry analysis of three independent experiments; data presented as means ± SD. **p < 0.005; *p < 0.05. B, H2O2 upregulates NEDD4-1 transcription in primary neurons dose dependently as determined by qRT-PCR 6 h after treatment. Data presented as means ± SD from three independent experiments. ***p < 0.001. C, The schematic structure of NEDD4-1 promoter region. “+1” indicates the transcription initiation site. The putative binding sites for several transcription factors were predicted by the online program Genomatix Matinspector using Matix Family Library Version 8.3 (October 2010) with parameters of core/matrix sim, 0.75/Optimized. The DNA sequence of the promoter region (−753 to −553 bp) containing the putative FOXM1B binding site (underlined) is presented. Two primer sequences used in a ChIP assay are highlighted in green. D, H2O2-induced binding of FoxM1B to the NEDD4-1 promoter as detected by a ChIP assay. HEK293 cells were treated with 50 μm or 100 μm H2O2 for 6 h followed by the ChIP procedure. The micrograph shows the PCR results from the DNA purified after immunoprecipitation with anti-FOXM1B antibody; 200 bp bands represent the amplified DNA fragment containing the FOXMB1 binding site. Input and IgG (used in immunoprecipitation) serve as positive and negative controls for the samples treated with H2O2, which were immunoprecipitated by specific FOXMB1 antibody.
Oxidative stress (H2O2) induces transcriptional activation of NEDD4-1 through a factor FOXM1B
Using qRT-PCR, we then demonstrated that H2O2 induced NEDD4-1 transcription in a dose-dependent manner, with the maximum induction of ∼2.5-fold increase in NEDD4-1 mRNA achieved at 100 μm (Fig. 2B). To identify the key transcription factor(s) mediating this ROS response, the NEDD4-1 promoter region was analyzed using MatInspector software (Genomatix), which predicted a ∼2 kb 5′-flanking NEDD4-1 promoter region containing several putative regulatory elements, such as heat shock transcription factor (HSF), activating protein (AP1), specificity protein 1 (SP1), and a member of FOXM1B (Fig. 2C). Using a chromatin immunoprecipitation assay, we further demonstrated that H2O2 induced FOXM1B binding to the human NEDD4-1 promoter (Fig. 2D; at 6 h after H2O2 treatment). It should be pointed out that H2O2-induced NEDD4-1 transcription reached a plateau after 1 h treatment and lasted for 24 h (data not shown). As controls, no enhanced binding of the other putative transcription factors (HSF-1, AP1, or SP1) to the NEDD4-1 promoter region was detected upon H2O2 treatment (data not shown), suggesting FOXM1B as the major factor mediating the ROS response in upregulating NEDD4-1 transcription; whether the timing of induced FOXM1B binding correlates with NEDD4-1 transcriptional activation warrants further study.
H2O2/zinc induces NEDD4-1, which downregulates the IGF-1Rβ protein in cultured neurons
We next examined the effect of oxidative stress caused by H2O2 or zinc on the expression levels of NEDD4-1 and one of its major substrates from the tyrosine receptor family, IGF-1Rβ. Using primary cultured neurons, we found that H2O2 treatment (10, 50, and 100 μm) resulted in reduced IGF-1Rβ levels, which correlated dose dependently with the increased NEDD4-1 levels (Fig. 3A). In contrast to the elevated NEDD4-1 transcription induced by H2O2, no increase in IGF-1Rβ messenger RNA was found (data not shown). Similarly, cytotoxic zinc increased NEDD4-1 protein levels, but not NEDD4–2, while decreasing IGF-1Rβ levels in both a dose-dependent (Fig. 3B) and time-dependent manner (data not shown). We found that this effect was only achieved by zinc and not by other heavy metals such as magnesium and copper. Moreover, zinc also slightly reduced levels of insulin receptor (IR), another substrate of NEDD4-1, but exerted no effect on the other two E3 ligases (e.g., Mdm2 and c-Cbl) that are known to mediate IR/IGF-1Rβ degradation in several non-neuronal cell models (Girnita et al., 2003; Sehat et al., 2008) (data not shown). We therefore speculated that NEDD4-1 is the major E3 ligase that mediates the IGF-1Rβ protein degradation induced by zinc in neurons. In support of this, we found that overexpression of NEDD4-1 in N2a cells gradually reduced IGF-1Rβ with complete ablation of the receptor protein achieved by 0.5 μg/ml of plasmid (Fig. 3C).
Figure 3.

Neurotoxic zinc promotes ubiquitination of IGF-1Rβ/protein degradation via upregulating NEDD4-1. A, B, H2O2 and zinc induce NEDD4-1 expression and reduce IGF-1Rβ levels in a dose-dependent manner in primary cultured neurons. Protein levels were determined by Western blot analysis after a 4 h treatment. Representative data are shown from four sets of independent experiments. C, Overexpression of NEDD4-1 in N2a mouse neuroblastoma cells results in reduction (by 0.1 μg/ml plasmid) and elimination (by 0.5 μg/ml plasmid) of IGF-1Rβ protein as detected 48 h after transfection. D, Zinc-induced IGF-1Rβ reduction is due to UPS-mediated protein degradation. Neurons were pretreated with MG132 at 25 μm for 1 h before zinc treatment (300 μm) and the protein level of IGF-1Rβ was determined 4 h later by Western blot analysis. E, NEDD4-1 mediates zinc-induced IGF-1Rβ ubiquitination. Treatment with zinc specifically promoted ubiquitination of IGF-1Rβ as detected by a polyclonal antibody against ubiquitin after IGF-1Rβ immunoprecipitation. NEDD4-1-specific shRNA or a scramble control was transfected into N2a cells to test whether NEDD4-1 is the E3 ligase responsible for the zinc-induced reduction of IGF-1Rβ. F, Neurons were pretreated with TPEN at various concentrations for 1 h, followed by zinc treatment for an additional 4 h before IGF1Rβ protein was determined by Western blot analysis.
To further investigate whether the zinc-induced reduction in IGF-1Rβ is mediated by the UPS, neurons were cotreated with zinc with or without a proteasomal inhibitor (MG132) (Fig. 3D). We found that zinc-induced degradation of IGF-1Rβ was completely prevented by blocking the UPS. In line with this finding, IGF-1Rβ was found to be abundantly ubiquitinated upon zinc treatment (Fig. 3E); downregulation of NEDD4-1 gene expression by a specific shRNA resulted in ∼50% reduction of NEDD4-1 expression as previously reported (Kwak et al., 2010a) and inhibition of receptor ubiquitination to the basal level, even in the presence of zinc. A cell-permeant metal ion chelator (TPEN) that is commonly used in zinc treatment studies to deplete intracellular and extracellular zinc consistently increased the steady-state level of the endogenous IGF-1Rβ protein dose dependently and partially preserved IGF-1Rβ after treatment with neurotoxic zinc (Fig. 3F). Collectively, these findings indicate that NEDD4-1 is the major E3 ligase for the degradation of IGF-1Rβ in response to zinc in cultured neurons.
SOD1 plays an important role in counteracting zinc-induced IGF-1Rβ downregulation
SOD1s are a class of enzymes that catalyze the dismutation of superoxide into oxygen and hydrogen peroxide and thus represent the most important enzymes that protect cells from oxidative stresses. SOD1 mutations are frequently found in familiar ALS cases and certain SOD1 mutants render neurons more vulnerable to zinc-mediated toxicity due to an altered binding affinity to copper and zinc ions (Zimmerman et al., 2007; Ilieva et al., 2009). Moreover, an elevated intracellular zinc level is found in primary patient tissues and in the spinal cord MNs of SOD1-G93A-Tg mice (Kim et al., 2009). To elucidate the pathological relevance of a zinc-induced reduction of IGF-1Rβ in our cultured neurons, we examined whether SOD1 can rescue the reduction in IGF-1Rβ. We found that overexpression of WT SOD1 by transient transfection prevented the zinc-induced reduction of IGF-1Rβ in mouse neuroblastoma N2a cells while the mutant SOD1-G93A further downregulated IGF-1Rβ (Fig. 4A,B). The effect of SOD1 on the IGF-1Rβ level was also tested by measuring the level of ubiquitinated IGF-1Rβ. In the absence of zinc, the basal receptor ubiquitination levels were further reduced by WT SOD1 transfection but significantly enhanced by the mutant SOD1 (Fig. 4C,D). As predicted, treatment with zinc increased ubiquitination of IGF-1Rβ (mostly polyubiquitination), while overexpression of SOD1-WT dramatically blocked this zinc-induced polyubiquitination. Interestingly, overexpression of SOD1-G93A enhanced IGF-1Rβ ubiquitination but at a lower range of molecular weight species, which may represent some degradative protein products.
Figure 4.

Differential effects of WT and mutant SOD1 gene transfection on IGF-1Rβ levels upon zinc treatment. A, N2a neuroblastoma cells overexpressing WT and mutant (G93A) SOD1 were treated with or without zinc for 48 h after transient transfection with 0.4 μg/ml plasmid. The IGF-1Rβ protein level was determined by Western blot analysis 4 h after zinc treatment. Anti-EGFP antibody was used to determine the transfection efficiency. B, Quantification was based on three sets of experiments. ***p < 0.001; **p < 0.005. C, Mutant SOD1 increases basal IGF-1Rβ ubiquitination levels in the absence of zinc. Experiments were conducted in a similar manner to those described in Figure 3E. D, Quantification of ubiquitination levels of IGF-1Rβ based on three experiments. ***p < 0.001.
Encouraged by the findings from the cell model, we further investigated the relationship between NEDD4-1 and IGF-1Rβ in cortical and spinal cord specimens taken from G93A SOD1 transgenic and control mice. At a presymptomatic age (30-d-old mice), we found that the NEDD4-1 expression level was much higher in the samples from mutant mice than from nontransgenic (non-Tg) littermate control mice in all the tissues examined, including cortex, cervical, and lumbar cords (Fig. 5A). Spinal cord samples express higher levels of NEDD4-1 protein than cortex. However, a significant reduction of IGF-1Rβ levels was only observed in the cortical samples (containing layer V). On the contrary, IGF-1Rβ protein levels appeared to be unchanged in the cervical cord samples (C5) and even slightly increased in the lumbar cord samples (L5) of the SOD1-Tg mice. At a late symptomatic age (125 d), NEDD4-1 protein levels were increased by 3- to 4-fold in the lumbar cord samples (p < 0.001); however, there was no significant reduction of IGF-1Rβ (p > 0.05) (Fig. 5B). Histological analysis consistently showed that a basal level of NEDD4-1 immunoreactivity was usually associated with neuronal cells in the spinal cords of non-Tg control mice. A higher immunoreactivity to NEDD4-1 was associated with degenerating or degenerated MNs in the lumber anterior horn regions taken from end-stage G93A SOD1-Tg mice (Fig. 5C,D). These results strongly indicate the involvement of upregulated NEDD4-1 in neurodegeneration.
Figure 5.
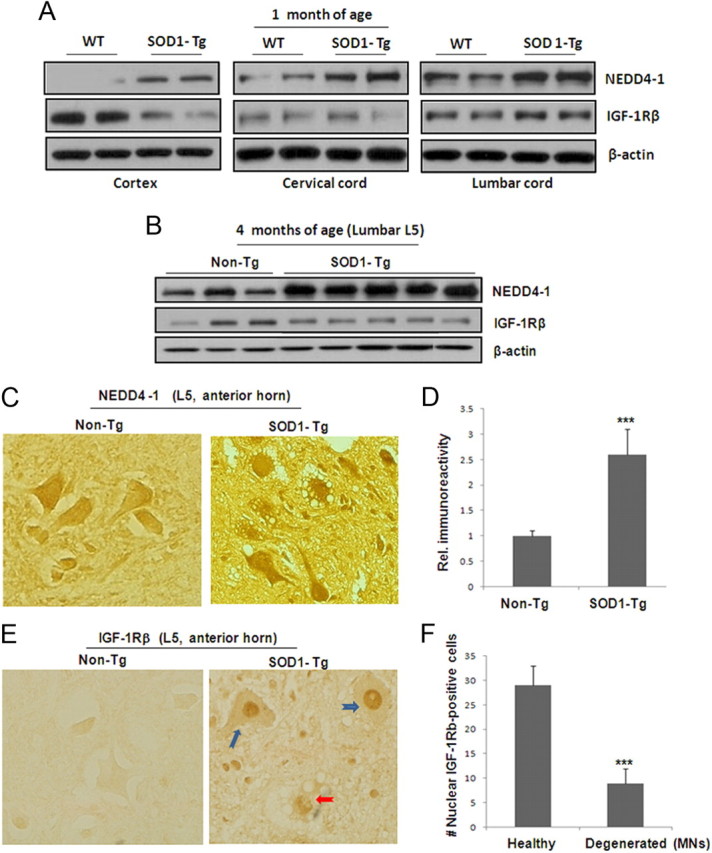
Altered protein expression levels in the cortex and spinal cords of the mutant G93A SOD1 mice. A, Protein levels of NEDD4-1 and IGF-1Rβ in presymptomatic G93 SOD1-Tg and littermate control mice. Two pairs of mice at 30 d old were used for determination of protein expression using Western blot analysis on whole tissue lysates (15 μg proteins) prepared from the frontal cortex and spinal cord (cervical C5 and lumbar L5 sections). All panels shown are from the same Western blot analysis, which makes it feasible for comparison of protein expression levels. B, Protein levels from L5 lumbar section of SOD1-Tg mice at the symptomatic stage (125 d) and control mice. C, Increased immunoreactivity of NEDD4-1 in SOD1-Tg spinal cord. Immunohistochemical analysis was conducted on paraffin sections of spinal cords (L5 section) of 5 μm thickness. Representative images of large MNs identified by morphology are shown from the anterior horn regions of the late stage SOD1-Tg and control mice (2 pairs of mice). D, Quantification of NEDD4-1 immunoreactivity from 50 large (25–50 μm) MNs from each mouse from two pairs of spinal cord specimens. Data presented as mean ± SD; ***p < 0.001. E, Nuclear IGF-1Rβ immunoreactivity in SOD1-Tg spinal cord samples. Immunohistochemical analysis of the spinal cord L5 section from the same two pairs of mice revealed massive IGF-1Rβ-reactive signals in the nuclei of the SOD1-Tg samples. The normal littermate control samples show evenly distributed receptor on cells. F, Quantification of nuclear-IGF-1Rβ-positive large MNs from one SOG1-Tg mouse indicates that a majority of them are healthy looking MNs (E, blue arrows) while a portion (∼25%) of MNs showing signs of degeneration (E, red arrow).
Interestingly, IGF-1Rβ expression levels are much lower in the spinal cords than in cortical neurons at early stages (Fig. 5A), consistent with their messenger RNA levels (data not shown). Although the receptor protein levels did not show a significant difference between SOD1-Tg and control mice at late stages based on Western blot analysis using whole tissue lysates (Fig. 5B), we found striking nuclear localization of the receptor with higher densities found in the majority of the large lumbar anterior horn MNs, as compared with the homogeneous membranous staining of the MNs in non-Tg tissue (Fig. 5E). Moreover, the morphology of most of these MNs with nuclear receptors appeared normal (∼75%) with only a small percentage of neurons (∼25%) showing signs of degeneration (Fig. 5F). Recently, IGF-1R is shown to be present in the cell nucleus of several human cancer lines and primary tumors (Larsson et al., 2007; Sehat et al., 2010; Sarfstein et al., 2012). However, its nuclear localization in neuronal lineage has not been reported. It is therefore not clear whether this nuclear IGF-1Rβ immunoreactivity represents functional or dysfunctional receptors.
Increased NEDD4-1 expression and decreased IGF-1Rβ in vivo
To answer the question of whether the NEDD4-mediated protein degradation of IGF-1Rβ we observed in cultured cortical neurons, in response to zinc, is valid in various brain regions in vivo, we systemically administered zinc (200 μl, 600 μm) to young adult C57BL/6 mice and analyzed the IGF-1Rβ and NEDD4-1 levels in the brain and spinal cord lysates prepared at several time points (0, 6, 12, and 24 h). As in cultured neurons, we observed an inverse correlation between NEDD4-1 and IGF-1Rβ levels in the frontal brain region in response to zinc in a time-dependent manner (Fig. 6A), suggesting that zinc can induce IGF-1Rβ degradation in vivo via upregulation of NEDD4-1 levels. Surprisingly, we did not see significantly inversed correlation between NEDD4-1 and IGF-1Rβ protein levels in the lumbar spinal cord specimen after systemic zinc administration of up to 24 h, despite a slight increase in NEDD4-1 levels (Fig. 6B). It appears that the IGF-1Rβ receptors from the cortex are more sensitive to protein degradation by the zinc-induced NEDD4-1 than those in the spinal cord.
Figure 6.
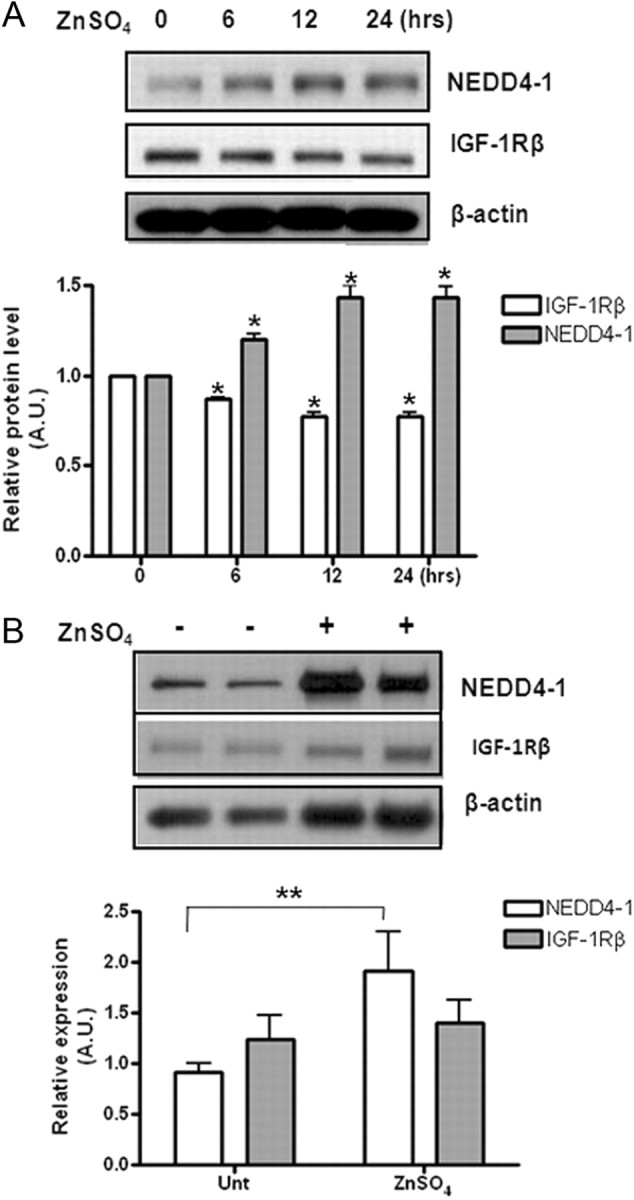
Zinc-induced NEDD4-1 upregulation in vivo correlated with reduced IGF-1Rβ levels. A, Zinc induces inversely correlated upregulation of NEDD4-1 and IGF-1Rβ protein expression in mice. Protein expression levels were determined at various time points as indicated after one single intraperitoneal injection of zinc (200 μl, 600 μm) into young adult C57BL/6 mice. Quantitative data are presented as means ± SD from n = 3 mice/time point. *p < 0.05. B, Effects of intraspinal cord-injected zinc (5 μl 1 mm) on the protein expression levels of NEDD4-1 and IGF-1Rβ of WT C57BL/6 mice. **p < 0.005.
ROS-induced NEDD4-1 upregulation contributes directly to neuronal death in N2a cultured model
We then sought to investigate whether upregulated NEDD4-1 contributes to neurodegeneration or is a consequence of neurodegeneration. Since many previous reports indicated that elevated zinc induces cell death through oxidative stress, we used zinc treatment to induce apoptosis in two culture models. In mouse neuroblastoma N2a, 100 μm zinc caused neurotoxicity over the course of 24 h, with 300 μm resulting in massive neuronal death/apoptosis after 4–8 h (Fig. 7A). As shown in Figure 3B with primary neurons, zinc induced NEDD4-1 upregulation and IGF-1Rβ reduction in both cell lines (data not shown); overexpression of IGF-1Rβ greatly prevented cells from zinc toxicity. Notably, downregulation of NEDD4-1 by specific shRNA greatly rescued neurons from zinc-induced apoptosis while inactivation of the E3 ligase of NEDD4-1 by a dominant-negative form (CS) expressing catalytically inactive enzyme resulted in a similar effect in both cellular models (Fig. 7B). Although overexpression of NEDD4-1 by transient transfection was not sufficient to cause significant neuronal death without zinc, it appeared to exert a similar effect as 100 μm zinc treatment in reducing cytosolic α-tubulin (Fig. 7C), resulting in slightly swollen cell morphology as compared with the shrinkage effect by zinc. Furthermore, primary cortical neurons with NEDD4-1 haplotype were consistently found to be more resistant to zinc-induced cell death (Fig. 7D). Moreover the neurons prepared from NEDD4 haplotype P0 pups (NEDD4-1+/−) displayed increased IGF-1Rβ levels (Fig. 7E). Together, these results indicate that zinc or ROS-induced NEDD4-1 upregulation directly contribute to neurodegeneration.
Figure 7.

Upregulated NEDD4-1 expression contributes directly to neuronal death in cultured models. A, Zinc induces massive neuronal death (apoptosis) in mouse N2a neuroblastoma cells in a time-dependent manner. At various time points after the zinc treatment of the cells plated in 24-well plates with coverslips, TUNEL assays were performed on fixed cells along with DAPI staining. Apoptosis was assessed and quantified by counting TUNEL-positive cells from three fields of a total of 300 DAPI-positive cells. B, Effects of overexpression, downregulation, and inactivation of NEDD4-1 in zinc-induced cell death. Plasmids encoding rat NEDD4-1, CS-NEDD4-1 (Vecchione et al., 2003), and shRNA (Kwak et al., 2010a) were transiently transfected into N2a cells. Forty-eight hours later, cells were challenged with 100 μm zinc and cell death was determined by TUNEL/DAPI staining. In parallel, two separate sets of cells were transfected with a plasmid containing the cDNA for IGF-1Rβ and challenged with zinc later to see the effect of restored/increased IGF-1Rβ expression on zinc-mediated cell death as a measure for IGF-1 signaling. ***p < 0.005; *p < 0.05. C, Immunocytochemistry on α-tubulin (1:5000 diluted; Sigma) of the N2a cells transfected with NEDD4-1 cDNA or treated with zinc. Representative micrographs shown at 400× magnification. D, Effect of NEDD4-1 haplotype on neuronal survival after zinc challenge. Primary cortical neurons were prepared from P0 pups from the liters of NEDD4-1+/− and WT breeding. Neurons were combined from three to four pups of each liter with genotypes of NEDD4-1+/− or WT littermate control after genotyping. Zinc was added to the 10 days in vitro (DIV) neurons and cell death determined by TUNEL/DAPI staining and quantified. Data presented as means ± SD from three independent experiments. E, Expression levels of NEDD4-1 and IGF-1Rβ from 14 DIV neurons determined by Western blot analysis. N = 4 pups/genotype.
Discussion
Ubiquitination marks and directs a protein for rapid degradation by the proteasome system. Recent findings suggest that the ubiquitin system participates in neuronal cell death and that aberrant UPS functioning contributes to neurodegeneration in various neuronal types. Among the ubiquitin system proteins, E3 ligases have increasingly been recognized as playing particularly important roles in regulating neuronal functions. In this study, we investigated the role of the HECT E3 ligase NEDD4-1 in neurodegeneration. We report for the first time that NEDD4-1 expression is upregulated in postmortem human AD, PD, and HD brains and in the spinal cords of ALS cases; ROS can induce NEDD4-1 transcription, which accounts for the elevated protein expression. In cultured neuronal models, we further demonstrate that downregulation or functional inactivation of NEDD4-1 rescued neurons from zinc-induced apoptosis. Given that ROS is the most common denominator in the major forms of neurodegenerative diseases and that elevated NEDD4-1 occurs early in presymptomatic SOD1-Tg mice, our findings strongly support a direct causative role for NEDD4-1 in neurodegeneration.
In this study, we also validate a regulatory role for NEDD4-1 in the protein stability of IGF-1Rβ in cultured neurons, mouse brains, and spinal cord tissue. We demonstrate that excitotoxic zinc or ROS can induce NEDD4-1 transcription, leading to protein degradation of IGF-1Rβ via the UPS. NEDD4-1 has other CNS substrates such as PTEN; upregulated NEDD4-1 also downregulates PTEN, resulting in activated PI3K/Akt signaling, as shown in our earlier work (Kwak et al., 2010a,b). Thus, the net outcome (e.g., neuroprotective or neurodegenerative) of upregulated NEDD4-1 may be determined by the interplay between the PTEN/Akt and IR/IGF-1R pathways. Our results (Fig. 7) support a direct causative role for NEDD4-1 through downregulation of IGF-1 signaling in the zinc-induced cell death model though overexpression of NEDD4-1 alone appears to be insufficient to cause increased neuronal death. Unlike the Parkin gene in PD, no pathological genetic mutation of NEDD4-1 has been identified, though its binding proteins have been associated with human diseases: Nedd4-WW domain-binding protein 5 is associated with neuronal survival after acute cortical brain injury (Sang et al., 2006) and BEAN is linked to spinocerebellar ataxia (Sato et al., 2009). Recently, NEDD4-1 was reported to be the E3 ligase for α-synuclein aggregates and its upregulation was associated with Lewy bodies, implying its positive role in clearing Parkinson's proteopathy and other α-synucleinopathies (Tofaris et al., 2011).
Our work also demonstrates for the first time that zinc-induced IGF-1Rβ degradation in neurons can be reversed by WT SOD1 and worsened by mutant G93A SOD1 gene overexpression. Our finding adds to the growing body of evidence that alteration of zinc homeostasis impairs the UPS pathway (Chen et al., 2009), which has particularly important implications for ALS. The transition metals zinc and copper regulate protein stability of SOD1and survival MNs (Burnett et al., 2009) via UPS. Imbalance in the homoeostasis of these metals has been implicated in ALS pathogenesis. The mutant G93-SOD1 transgenic mouse is the most widely used ALS model as they develop all the pathological events necessary to fully recapitulate ALS pathology, including a marked elevation of intracellular zinc and oxidative stress (Kruman et al., 1999; Poon et al., 2005; Van Damme et al., 2005; Barber et al., 2006).
Impaired IGF-1 signaling is implicated in the major neurodegenerative diseases such as AD, PD, and ALS (Trejo et al., 2004). Previous positive results from cellular and animal models provided a strong rationale for using IGF-1 in promoting the survival of injured MNs (Kaspar et al., 2003; Ilieva et al., 2009). Moreover, the US Food and Drug Administration approval of the two IGF-1-based drugs, Increlex (Tercica) and IGF-1/IGFBP-3 complex/Iplex (Insmed) in 2005 also provides the proof-of-concept for using an IGF-1-based strategy in treating a primary IGF-1 deficiency. Despite positive results from the first clinical trial, the following two large-scale trials on hrIGF-1 therapy in ALS treatment yielded disappointing outcomes (Sorenson et al., 2008). Several technical aspects of these trial designs have been criticized as deficient, including insufficient delivery/accessibility of an intramuscular injection of hrIGF-1 to the affected spinal cord neurons. Below we would like to offer discussion of two additional aspects, which may account for the IGF-1 resistance in these trials: (1) timing of IGF-1 therapy and (2) IGF-1Rβ receptor misregulation.
Targeting upper MNs for more effective ALS therapy?
ALS is an incurable neuromuscular disease manifested by progressive and selective degeneration of MNs in the brain/spinal cord and muscle atrophy. In ALS, both the cortical (upper) and spinal (lower) components of MNs progressively degenerate, which together contributes to the loss of motor functions. The most complex and prototypical projection neurons in cortical layer V, corticospinal MNs (CSMNs; upper MNs), extend their projections to the spinal cord and control voluntary motor functions. CSMN degeneration is believed to occur before that of the lower MNs in ALS patients (Stewart et al., 2006) and SOD1-Tg mice (Ozdinler et al., 2011). However, the cellular and molecular mechanisms of their survival and death are less studied in CSMNs than in lower MNs. Important work from the same research group demonstrated that injured CSMNs can be regenerated by endogenous neurogenesis and that IGF-1 is a specific neurotrophin to these neurons (Chen et al., 2004; Ozdinler et al., 2006). Together with our findings that the IGF-1Rβ level is much higher in the cortex than in the spinal cord (Fig. 5), it appears that IGF-1 is more important to the upper MNs than to the lower MNs. Together these findings show that enrollment of symptomatic ALS patients in clinical trials may invalidate the IGF-1 therapy. Based on our somewhat preliminary data presented in Figure 5A with two pairs of young (P30) SOD1 and control mice showing inversely correlated NEDD4-1 and IGF-1R only in the frontal cortical region, it appears that this mechanism revealed by our in vitro studies is a more dominant event for cortical neurons, including CSMNs, than it is for the lower spinal cord MNs. Additional experiments are required to confirm this critical point.
Should IGF-1Rβ receptor misregulation be considered when designing IGF-1-based therapy?
Despite the significant effort and progress that has been made in investigating the feasibility of using IGF-1 for ALS therapy in both preclinical and clinical studies, the importance of its receptor counterpart has been very underappreciated. The IGF-1Rβ receptor is reportedly regulated at multiple levels involving separate molecular mechanisms. (1) ROS was shown to induce transcriptional upregulation of the receptor gene in certain cell types (Kavurma et al., 2007). (2) In addition to post-translational modification by Tyr-phosphorylation, which directly regulates its receptor kinase activity/signaling, ubiquitination is shown here to regulate receptor protein stability. (3) Similar to previous reports regarding certain cancer types (Larsson et al., 2007; Sehat et al., 2010; Sarfstein et al., 2012), we found nuclear translocation of the receptor in MNs; it is not clear whether these nuclear receptors mediate functional or dysfunctional IGF-1 signaling in the nucleus.
Despite reduced IGF-1 levels, a general upregulation of IGF-1 receptors was observed throughout the spinal cord of primary ALS patients with significant increases observed in the cervical and sacral segments (Adem et al., 1994) in a similar pattern to 125I-IGF-1 binding to the different spinal levels (Doré et al., 1996). This upregulated receptor gene may represent an adaptive stress-response of neurons attempting to survive insults in the early phase of disease onset. However, the insulted MNs still eventually die as the disease progresses. It is plausible that multiple regulatory mechanisms at both the transcriptional and post-translational levels regulate the IGF-1Rβ level/activity, including altered receptor internalization/nuclear translocation during neurodegeneration. Indeed, IGF-1Rβ gene expression is also reportedly downregulated in the glial cells/spinal cords of asymptomatic mutant SOD1 mice (Vargas et al., 2008), suggesting this change occurs before overt neuronal degeneration. Although NEDD4-1 is most abundant in degenerating neurons, the NEDD4-mediated regulation of IGF-1Rβ may also occur in glial cells, the most abundant cell type in spinal cords, which may affect the MN-glia interaction based on the noncell autonomous mechanism in mutant SOD models (Ilieva et al., 2009). Our work demonstrates that NEDD4-1-mediated IGF-1Rβ protein degradation is more dominant in cortical neurons. It may occur in spinal cord MNs as seen in the late stage of disease onset in mutant G93A SOD1 spinal cords. The tipping point for neurodegeneration may result when the ROS insults accumulate to the point where the upregulation of the receptor gene can no longer compensate for IGF-1Rβ protein degradation. Given the increasing evidence of elevated endoplasmic reticulum (ER) stress in ALS disease (Kikuchi et al., 2006; Wu et al., 2006; Saxena et al., 2009; Walker, 2010), it is particularly important to further investigate if ER stress induces IGF-1Rβ misregulation in MNs. Specifically, the interplay of receptor degradation with other regulatory mechanisms (transcriptional upregulation and nuclear translocation) in spinal MNs and other neuronal types (e.g., interneurons and glias) warrants further investigation SOD1-Tg mice.
In summary, we demonstrate for the first time that zinc can induce IGF-1Rβ protein degradation in neurons via NEDD4-mediated ubiquitination and the subsequent UPS mechanism. This may represent a novel molecular mechanism underlying the pathogenesis of MN degeneration in ALS. The interplay between IGF-1Rβ degradation and the upregulation of its gene expression during disease onset warrants further investigation in motor neuronal models. Our work clearly identifies the E3 ligase NEDD4-1 and IGF-1Rβ as two new molecular targets for ALS research. A fuller understanding of misregulated IGF-1Rβ during disease onset will open new avenues for treatment options and trial designs.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS054880 and R01 AG030197 (F.-F.L.); R01 AG021173, R01NS046673, and R01 AG030197 (H.X.); and R01 AG022074 (E.M.); Alzheimer's Association Award IIRG-11-204030 (F.-F.L.); and a Zenith Award (H.X.). We thank Dr. Rengang Wang for technical advices, Dr. Daniela Rotin for providing pRc-CMV-T7-NEDD4-1-WT and pRc-CMV-T7-NEDD4-1-CS plasmids, and Drs. Dong-Xian Zhang and Paul J. Taylor for constructive discussions.
The authors declare no competing financial interests.
References
- Adem A, Ekblom J, Gillberg PG, Jossan SS, Höög A, Winblad B, Aquilonius SM, Wang LH, Sara V. Insulin-like growth factor-1 receptors in human spinal cord: changes in amyotrophic lateral sclerosis. J Neural Transm Gen Sect. 1994;97:73–84. doi: 10.1007/BF01277964. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Burnett BG, Muñoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Yakar S, Gewolb IH, Karas M, Okubo Y, LeRoith D. Insulin-like growth factor-I receptor signal transduction: at the interface between physiology and cell biology. Comp Biochem Physiol B Biochem Mol Biol. 1998;121:19–26. doi: 10.1016/s0305-0491(98)10106-2. [DOI] [PubMed] [Google Scholar]
- Cao XR, Lill NL, Boase N, Shi PP, Croucher DR, Shan H, Qu J, Sweezer EM, Place T, Kirby PA, Daly RJ, Kumar S, Yang B. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci Signal. 2008;1:ra5. doi: 10.1126/scisignal.1160940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Magavi SS, Macklis JD. Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A. 2004;101:16357–16362. doi: 10.1073/pnas.0406795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Chen Q, Cheng XW, Lu TJ, Liu HX, Jia JM, Zhang C, Xu L, Xiong ZQ. Zn2+ mediates ischemia-induced impairment of the ubiquitin-proteasome system in the rat hippocampus. J Neurochem. 2009;111:1094–1103. doi: 10.1111/j.1471-4159.2009.06401.x. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Lees GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol Dis. 1997;4:137–169. doi: 10.1006/nbdi.1997.0163. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- Doré S, Krieger C, Kar S, Quirion R. Distribution and levels of insulin-like growth factor (IGF-I and IGF-II) and insulin receptor binding sites in the spinal cords of amyotrophic lateral sclerosis (ALS) patients. Brain Res Mol Brain Res. 1996;41:128–133. doi: 10.1016/0169-328x(96)00081-2. [DOI] [PubMed] [Google Scholar]
- Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 2010;65:341–357. doi: 10.1016/j.neuron.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr Alzheimer Res. 2009;6:213–223. doi: 10.2174/156720509788486527. [DOI] [PubMed] [Google Scholar]
- Friedlich AL, Lee JY, van Groen T, Cherny RA, Volitakis I, Cole TB, Palmiter RD, Koh JY, Bush AI. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer's disease. J Neurosci. 2004;24:3453–3459. doi: 10.1523/JNEUROSCI.0297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Xu H. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci. 2003;26:404–406. doi: 10.1016/S0166-2236(03)00163-2. [DOI] [PubMed] [Google Scholar]
- Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A. 2003;100:8247–8252. doi: 10.1073/pnas.1431613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kavurma MM, Figg N, Bennett MR, Mercer J, Khachigian LM, Littlewood TD. Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem J. 2007;407:79–87. doi: 10.1042/BJ20070380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe H, Neeb A, Dimova K, Young SM, Jr, Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang DE, Umikawa M, Kariya K, Goebbels S, Nave KA, Rosenmund C, Jahn O, Rhee J, Brose N. Regulation of Rap2A by the ubiquitin ligase NEDD4-1 controls neurite development. Neuron. 2010;65:358–372. doi: 10.1016/j.neuron.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guégan C, Nagai M, Xu Z, Sosunov AA, Mckhann GM, 2nd, Przedborski S. Spinial cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim TY, Hwang JJ, Lee JY, Shin JH, Gwag BJ, Koh JY. Accumulation of labile zinc in neurons and astrocytes in the spinal cords of G93A SOD-1 transgenic mice. Neurobiol Dis. 2009;34:221–229. doi: 10.1016/j.nbd.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Koh JY. Zinc and disease of the brain. Mol Neurobiol. 2001;24:99–106. doi: 10.1385/MN:24:1-3:099. [DOI] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Wang B, Pan W, Xu H, Jiang X, Liao FF. Functional interaction of phosphatase and tensin homologue (PTEN) with the E3 ligase NEDD4-1 during neuronal response to zinc. J Biol Chem. 2010a;285:9847–9857. doi: 10.1074/jbc.M109.091637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, Liao FF. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010b;5:49. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signaling in cancer. Br. J Cancer. 2007;96(Suppl):R2–R6. [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoith D, Werner H, Neuenschwander S, Kalebic T, Helman LJ. The role of the insulin-like growth factor-I receptor in cancer. Ann N Y Acad Sci. 1995;766:402–408. doi: 10.1111/j.1749-6632.1995.tb26689.x. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Liu Y, Oppenheim RW, Sugiura Y, Lin W. Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev Biol. 2009;330:153–166. doi: 10.1016/j.ydbio.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdinler PH, Macklis JD. IGF-1 specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH, Jr, Macklis JD. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. J Neurosci. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, Hensley K, Thongboonkerd V, Merchant ML, Lynn BC, Pierce WM, Klein JB, Calabrese V, Butterfield DA. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice–a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39:453–462. doi: 10.1016/j.freeradbiomed.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Sang Q, Kim MH, Kumar S, Bye N, Morganti-Kossman MC, Gunnersen J, Fuller S, Howitt J, Hyde L, Beissbarth T, Scott HS, Silke J, Tan SS. Nedd4-WW domain-binding protein 5 (Ndfil1) is associated with neuronal survival after acute cortical brain injury. J Neurosci. 2006;26:7234–7244. doi: 10.1523/JNEUROSCI.1398-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfstein R, Pasmanik-Chor M, Yeheskel A, Edry L, Shomron N, Warman N, Wertheimer E, Maor S, Shochat L, Werner H. Insulin-like growth factor-I receptor (IGF-1R) translocates to nucleus and autoregulates IGF-1R gene expression in breast cancer cell. J Biol Chem. 2012;287:2766–2776. doi: 10.1074/jbc.M111.281782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motorneuron subtype selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Sehat B, Andersson S, Girnita L, Larsson O. Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis. Cancer Res. 2008;68:5669–5677. doi: 10.1158/0008-5472.CAN-07-6364. [DOI] [PubMed] [Google Scholar]
- Sehat B, Tofigh A, Lin Y, Trocmé E, Liljedahl U, Lagergren J, Larsson O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci Signal. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- Smith AP, Lee NM. Role of zinc in ALS. Amyotroph Lateral Scler. 2007;8:131–143. doi: 10.1080/17482960701249241. [DOI] [PubMed] [Google Scholar]
- Sorenson EJ, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71:1770–1775. doi: 10.1212/01.wnl.0000335970.78664.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart HG, Andersen PM, Eisen A, Weber M. Corticomotoneuronal dysfunction in ALS patients with different SOD1 mutations. Clin Neurophysiol. 2006;117:1850–1861. doi: 10.1016/j.clinph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL. Ubiquitin ligase Nedd4 promotes {alpha}-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A. 2011;108:17004–17009. doi: 10.1073/pnas.1109356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo JL, Carro E, Garcia-Galloway E, Torres-Aleman I. Role of insulin-like growth factor I signaling in neurodegenerative diseases. J. Mol Med (Berl) 2004;82:156–162. doi: 10.1007/s00109-003-0499-7. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Dewil M, Robberecht W, Van Den Bosch L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:147–159. doi: 10.1159/000089620. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Díaz-Amarilla PJ, Beckman JS, Barbeito L. Transcriptional profile of primary astrocytes expressing ALS-linked mutant SOD1. J Neurosci Res. 2008;86:3515–3525. doi: 10.1002/jnr.21797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchione A, Marchese A, Henry P, Rotin D, Morrione A. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol Cell Biol. 2003;23:3363–3372. doi: 10.1128/MCB.23.9.3363-3372.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK. Protein disulfide isomerase and the endoplasmic reticulum in Amyotrophic lateral sclerosis. J Neurosci. 2010;30:3865–3867. doi: 10.1523/JNEUROSCI.0408-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of the Alzheimer amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17:68–77. doi: 10.1038/cdd.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Oberley LW, Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]