

Introduction
Acute lung injury (ALI) is a clinical syndrome characterized by impairment in gas exchange and/or lung mechanics that leads to hypoxemia and increased work of breathing (WOB). When respiratory failure occurs, most patients require mechanical ventilation. This clinical scenario is related to high morbidity and mortality rates.
There have been large amounts of research on the pathogenetic mechanisms of lung injury, which include changes in alveolocapillary permeability, the inflammatory response, extracellular matrix remodeling and abnormal alveolar micromechanics. In spite of this knowledge, no effective therapy, other than treating the initial cause of injury and providing supportive treatment, has been shown to have a significant clinical benefit. Fortunately, the cause of ALI is known in most cases, so specific therapy can be initiated (e.g., antibiotics in sepsis, surgery when appropriate). In other cases, the cause is time-limited, such as in aspiration pneumonitis or polytrauma. However, even in these cases, ALI may persist beyond the initial insult. In this setting, restoration of normal lung structure and function is of paramount importance for survival.
There is increasing evidence of the lung's capacity to repair itself. This process involves an interplay between various cellular and molecular mechanisms, including resolution of edema and inflammation, cell proliferation and tissue remodeling. Moreover, it is possible that some of these mechanisms, activated early in the response to injury, are essential for normal repair later on. With this in mind, timing of a therapeutic intervention becomes a critical issue, as blockade of one mediator may prevent injury when administered early, but also impair the repair phase. Moreover, strategies aimed at promoting repair could represent a new alternative for patients with ALI.
Knowledge of the repair mechanisms could, therefore, be the next step to understanding the lung response to injury. In this review, we will summarize some of these mechanisms and discuss their relevance as potential therapeutic targets in ALI.
An overview of the repair process
The lung response to an injurious stimulus involves transduction of the danger signal into a biochemical response. Depending on the cause, there are many pathways that can be activated. For instance, bacterial antigens may trigger an inflammatory response by activating any of the Toll-like receptors (TLR). Chemical agents induce cell membrane damage and, in some cases, oxidative stress, leading to the activation of a number of intracellular kinases. Mechanical stress, such as positive pressure ventilation, can also precipitate a biological response after a mechanotransduction process [1].
In addition to exogenous causes, an endogenous system detects tissue and cell damage and triggers the physiological response. Alarmins, a subgroup of molecules of a larger set called DAMPs (damage-associated molecular patterns), lead to this system. Structurally different, these endogenous molecules are released in response to tissue damage by dead cells and local inflammatory cells (alveolar macrophages in the case of lung), activating and recruiting immune cells through binding to different receptors, such as TLR, interleukin-1 receptor (IL-1R) and RAGE (receptor of advanced glycation end-products) [2], thereby activating the pro-inflammatory pathway. Irrespective of the cause, these signals converge in a group of transcription factors (e.g., nuclear factor-kappa B [NF-κB], activator protein [AP]-1), which induce the synthesis of new molecules that ultimately mediate the inflammatory response to the aggression. Hallmarks of this response are the increased alveolocapillary permeability, which causes a protein-rich edema, the neutrophil infiltrate (recruited from the circulation in response to chemokines), and the release of a wide variety of mediators, such as cyto- and chemokines, proteases, eicosanoids and growth factors, into the extracellular space. Figure 1 summarizes this process. During this stage, pneumocyte death due to apoptosis (in response to released mediators) and/or necrosis (caused by toxins, proteases...) results in exposure of the basement membrane of the alveolar epithelium.
Figure 1.
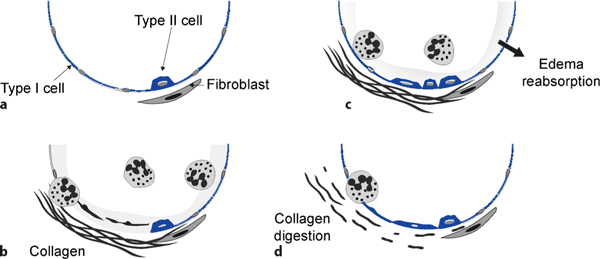
Overview of the injury and repair mechanisms in an alveolus. (a) The normal alveolus is formed by type I and type II alveolar cells. The former cover the majority of the alveolar area, and the latter are reduced in number. (b) After acute lung injury, the inflammatory response results in the recruitment of neutrophils from the circulation, the development of alveolar edema and the deposition of collagen fibers. The necrotic alveolar cells are detached from the basement membrane. (c) During the repair phase, the alveolar fluid is reabsorbed, the inflammatory response attenuated, and type II alveolar cells (among others) proliferate and differentiate into type I pneumocytes. In this phase, collagen fibers may facilitate cellular migration. (d) Finally, digestion of the collagen scar is needed for complete normalization of lung functions.
One of the key steps in the tissue response is the deposition of collagen fibers at the sites of injury. Similarly to what happens in skin wounds, lung fibroblasts release procollagen peptides into the extracellular space, in order to create a scar. This is probably an attempt to keep the lung structure as intact as possible. So collagen deposition must not be viewed as a late response to abnormal healing, but as an early phenomenon. Some experimental studies corroborate this early onset of collagen deposition [3]. Moreover, patients show an increase in procollagen levels in the first 48 hours after meeting ALI criteria [4].
Tissue repair involves a variety of mechanisms including edema reabsorption, resolution of inflammation and cell proliferation in order to repopulate the alveolar epithelium (Figure 1c). Lung edema clearance is a crucial step. It has been documented that mild alveolar injury results in increased alveolar fluid reabsorption. However, in severe cases, the injured pneumocytes cannot sustain the active transport of ions and water across the epithelium. Therefore, cell integrity is essential for edema clearance. The molecular mechanisms of ion and water transport in lung exceed the scope of this article, and have been reviewed elsewhere [5]. Regulation of the inflammatory response is a complex mechanism that requires interplay between several immune mediators [6]. Some anti-inflammatory cytokines, IL-10 being the most studied, are released even during a pro-inflammatory response as a negative feedback mechanism. When the pro-inflammatory pathways are downregulated (i.e., after cessation of the stimulus), these anti-inflammatory mediators decrease cytokine expression. Apoptosis of inflammatory cells (mainly neutrophils) has also been documented when pro-survival signals, such as granulocyte-colony stimulating factor (G-CSF), disappear. Alveolar macrophages also play a role in this phase engulfing death cells.
Finally, the regeneration of the alveolar structure requires the proliferation and differentiation of some progenitors into type I pneumocytes (Figure 2). Different growth factors (e.g. epidermic [EGF], keratinocyte [KGF] or hepatic growth factor [HGF]), acting through tyrosinkinase receptors, promote cell proliferation. The cell lines implicated in this step are a matter of research, stimulated by the growing interest in stem cells and regenerative medicine [7]. Endogenous progenitor cells include both resident stem cells and bone marrow-derived cells. Regarding the first, type II pneumocytes proliferate after injury and can originate type I cells. This has been demonstrated after pneumonectomy, hyperoxia or repeated bleomycin instillation in mice. Moreover, bone marrow-derived stem cells could also participate in alveolar repair, although data on the engraftment and differentiation of these cells are more focused on their therapeutic use than to clarify their role in the normal repair process. In addition, other cell types may also play a role in alveolar regeneration. Lung mesenchymal cells are activated after ALI and, in addition to collagen synthesis, they may secrete growth factors and even modulate the immune response by secreting anti-inflammatory cytokines [8].
Figure 2.
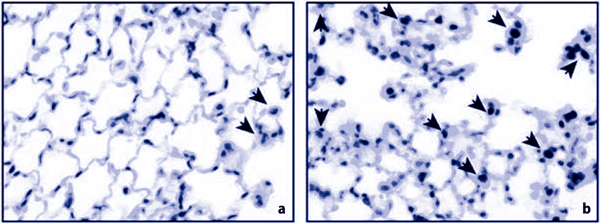
Cell proliferation in control mice (a) and during the repair phase after ventilator-induced lung injury . (b) Some cells (arrows) show positive staining for Ki-67, a marker of cell proliferation.
The previously formed collagen scar can facilitate cell attachment to keep the alveolar structure. Again, collagen synthesis should be viewed as part of the normal healing process. However, excessive collagen deposition may impair gas exchange and lung mechanics. So, in order to restore normal respiratory function, the previously formed scar must be processed and removed (Figure 1d). This can be carried out by matrix metalloproteinases (MMPs), a family of enzymes that can digest virtually all types of extracellular fibers [9]. One of the most important sources of MMPs is inflammatory cells (neutrophils and macrophages contain significant amounts of MMP-8 and MMP-9). Therefore, it can be hypothesized that the inflammatory response is important for adequate lung repair, and that MMPs are one of the links between these two phenomena. The underlying mechanisms that regulate this step in ALI are unknown, but knowledge of these mechanisms could help clarify why some patients develop a severe fibrotic response, which can cause long-term disabilities.
The special case of repair after ventilator-induced lung injury
Ventilation with high tidal volumes or transpulmonary pressures may cause severe injury to the lungs. In experimental models, there is a clear causality relationship between ventilatory settings and so-called ventilator-induced lung injury (VILI). Although this relationship is less clear in patients, especially those with previous ALI, the development of ventilatory strategies aimed to avoid further lung injury has been shown to decrease mortality. Therefore, although experimental models of VILI cannot be directly extrapolated to critically ill patients, they highlight the mechanisms of injury and repair involved.
A few studies have focused on repair after VILI, giving some insights into this process. The first was published by Nin et al. in 2008 [10]. These authors submitted Sprague-Dawley rats to injurious ventilation for one hour, reestablishing spontaneous breathing and letting them recover. Histological studies showed a significant reduction in capillary congestion, interstitial edema, type-I pneumocyte necrosis and hyaline membrane formation after 24 hours of recovery, reaching normality after 72 hours. Inflammatory markers showed a similar pattern. Aortic vascular and pulmonary microvessel responses to acetylcholine and norepinephrine were impaired and returned to normal at 168 and 72 hours respectively. This study demonstrated that VILI can revert rapidly after spontaneous breathing is reestablished.
In another study, Gonzalez-Lopez et al. [11] submitted CD1 mice to a combination of two ventilator strategies. One group was ventilated for 90 minutes with high pressures and another group was ventilated with the same strategy followed by up to 4 hours of protective ventilation. Histological score, pulmonary edema, alveolar permeability and tumor necrosis factor (TNF)-α increased during injury and returned to baseline values during repair. Later comparisons of the repair phase showed that survivors had higher pneumocyte proliferation and leukocyte infiltration and lower alveolar permeability and collagen content than non-survivors. MMP-2 levels were also increased in survivors. This MMP improves wound healing in ex vivo models using mice and human alveolar epithelial cell lines. Taken together, these studies suggest that an appropriate inflammatory response and tissue remodeling are key events during repair.
The role of inflammation in tissue damage and repair
Inflammation is necessary for the development of a proper response to the insult, but it can damage the tissue where it takes place. Inflammation leads initially to an alteration in homeostasis, during which tissue partially sacrifices cellular and extracellular matrix integrity and tissue functionality for the benefit of a quick response. A good example of this compromise is the early recruitment of immune cells to the site of injury. The first cells arriving are neutrophils [12]. Representing 70% of total circulating leukocytes, these cells are rapid responders to chemokines, appearing in the lungs a few minutes after initial injury. But this recruitment has a price for lung integrity; after migrating through endothelial cells and before arriving in the alveolar space, leukocytes find the basement membrane, a highly organized extracellular matrix mainly composed of four families of glycoproteins (laminin, collagen type IV isoforms, nidogen and heparan sulfate proteoglycans). The basement membrane is actively involved in leukocyte recruitment by its potential to bind and release cytokines and chemotactic factors. The only way neutrophils can cross this barrier is by proteolytic degradation [13], contributing to further damage of lung structure. Once in the lung, the release of a variety of alarmins, free oxygen radicals, leukotrienes, proteases and other pro-inflammatory molecules maintain the inflammatory state, thereby contributing to the ALI phase.
The role of proteases, especially of MMPs, in the pathogenesis of ALI has been controversial. Extracellular matrix processing releases some bioactive molecules. For example, type I collagen degradation generates an acetylated tripeptide with similar chemotactic activity to IL-8 [14]; moreover, MMPs process many immune mediators, like pro- and anti-inflammatory cytokines and chemokines, altering their activity (e.g., IL-1β, transforming growth factor [TGF]β and lipopolysaccharide-induced CXC chemokine [LIX] activation and macrophage inflammatory protein [MIP]-1α inactivation) and bioavailability (TNF-α and TGFβ release from cell surface and extracellular matrix, respectively) [15]. Their ability to regulate the inflammatory mediators and degrade collagen fibers also makes MMPs key elements in the later stages of inflammation, when resolution and repair of the injured tissue are of paramount importance. In a model of liver injury, it has been demonstrated that neutropenic animals develop more severe fibrosis, probably due to the lack of MMPs (released by neutrophils) in the repair phase [16]. A similar dependence between inflammation and collagenolysis in the lung has not been demonstrated, but these findings warrant more research.
Research on models of lung injury using knock-out mice for single MMPs, such as MMP-2, MMP-3, MMP-7, MMP-8 and MMP-9, has shed some light on this issue. MMP-2 [11], MMP-7 [17] and MMP-9 [18] have been shown to be involved in alveolar epithelial repair in experimental models of wound healing. Furthermore, a model of VILI [19] demonstrated that MMP-9 function is worth preserving, given that mice deficient in this protease had increased levels of lung injury. On the other hand, MMP-3 may be detrimental, as mice lacking this MMP were protected against lung injury caused by administration of nonspecific IgG [20] or bleomycin [21]. Regarding MMP-8, this protease has shown different effects depending on the experimental model. It has a detrimental role in models of VILI [22] and lung fibrosis, due to IL-10 cleavage [23]. However, this enzyme may be beneficial after lipopolysaccharide (LPS) or hyperoxia exposition [24]. Moreover, it may have a role in resolution stages of ALI, as it seems to be involved in neutrophil apoptosis through modulation of IL-4 levels [25].
Therefore, this complex family serves not only to regulate the influx and clearance of leukocytes and the inflammatory process itself, but also the removal of excess deposits of collagen fibers released by fibroblasts during ALI. It is becoming clearer that pharmacotherapy should be aimed at blocking specific MMPs during the early stages of ALI, in order to avoid destruction of the basement membrane and extracellular matrix caused by their proteolytic action. Although this could have an initial benefit it may turn detrimental to the repair process if treatment is continued.
Signs of tissue repair in patients
The assessment of lung repair in patients could be of great interest because of its prognostic relevance in ALI patients. However, measurement of any mediator involved in tissue repair has one fundamental limitation: If a patient shows high levels of a given marker, it could be due to ongoing repair and, therefore, associated with a good prognosis. However, the same high levels could also be due to massive injury triggering a full-blown response. In this case, the outcome may not be so good.
Collagen levels illustrate this two-sided interpretation of biomarkers. As mentioned before, collagen deposition is an early event during ALI. Increased levels of procollagen in pulmonary edema fluid may have a prognostic significance in patients with ALI. For instance, Chesnutt et al. [26] reported that a procollagen concentration above 1.75 U/ml had a positive predictive value for death of 0.75. However, a recent article by Quesnel et al. [27] showed that patients with ALI/acute respiratory distress syndrome (ARDS) with fibroblasts in their alveolar fluid had increased levels of type I procollagen, a decreased pro-inflammatory response (lower neutrophil count, decreased IL-8 levels), and improved prognosis, suggesting a switch from inflammation and tissue destruction to alveolar repair.
Research on the prognostic value of MMP-9 has yielded similar conflicting results. Abundance of this protease in bronchoalveolar lavage (BAL) fluid has been related to a worse outcome. However, MMP-9 has shown a protective role in experimental models of lung injury [19,28], and a clinical study demonstrated that this enzyme could have therapeutic value (see below). Finally, the prognostic value of growth factors in ALI was addressed years ago by Verghese et al. [29]. Lower levels of HGF and KGF were found in survivors. Although these mediators promote cell proliferation, differentiation and, ultimately, alveolar repair, their presence reflects a worse outcome. But, as the authors of the article discussed, the increased levels in non-survivors may be a marker of more severe injury.
Collectively, published results suggest that measurement of a single biomarker in patients can be misleading, as it can reflect both the severity of the injury and the subsequent healing process. An accurate prediction may require complex reasoning that takes into account the initial conditions but also the host's biological response. Therefore, an approach based on multiple markers could offer an alternative to monitor the course of this disease in patients.
Therapeutic strategies aimed at lung repair
Since identification of the pathways involved in lung injury, most literature has been focused on the use of therapies aimed at reducing ALI by truncating the inflammatory response. Unfortunately, none of these strategies has been successfully applied in the clinical practice. Recently, a significant number of studies have centered their efforts on enhancing the repair process using different approaches, ranging from the use of bio-compatible materials or cells to the therapeutic use of mediators aimed at promoting cell proliferation, migration and differentiation. Figure 3 summarizes the different therapeutic targets aimed at favoring lung repair.
Figure 3.
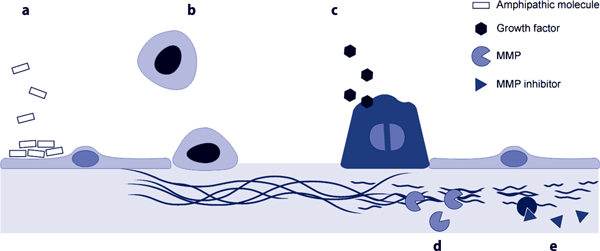
Therapeutic approaches to promote lung repair. (a) Direct repair of the plasma membrane could be achieved using amphipathic compounds that seal injured membranes. (b) Exogenous stem cells to repair the denuded areas. (c) Administration of growth factors to stimulate the proliferation of endogenous stem cells. (d) Stimulation or (e) selective blockade of different matrix metalloproteinases (MMPs) to promote collagen processing or avoid the adverse effects of these enzymes.
In many cases, these studies are still at the in vitro stages, such as the use of vimentin to improve wound repair [30], or just suggest a possible beneficial role (e.g., connexins [31], adrenomedullin [32] or a possible modulation of the transcription factors, FoxM1 and Runx3 [33,34]) and need further research to prove their viability in vivo. Improvement of plasma membrane repair is a possible direct treatment. A recent study in an ex vivo model using an amphiphilic macromolecule (Poloxamer 188) with sealing properties showed signs of membrane repair in alveolar resident cells and an improvement in conventional measures of lung injury [35].
Enhancement of the epithelial repopulation is a promising therapeutic target that could be achieved in different ways. In recent years, therapy using stem cells is gaining considerable interest. It has been demonstrated that these cells are active players in lung repair [36]. In spite of doubts about their safety and the best administration route to improve their engraftment, use of stem cells in animal models has been demonstrated to attenuate damage and fibrosis in lungs challenged with endotoxin [37]. In a recent study, Curley et al. [38] submitted a group of rats to VILI followed by an intravenous injection of mesenchymal stem cells; these animals showed less lung injury and increased levels of the anti-inflammatory and anti-fibrotic cytokine, IL-10, than did rats who did not receive the stem cells. The specific mechanisms by which stem cells perform their functions in tissue repair are still under study, but the release of several growth factors and the suppression of pro-inflammatory cytokines seem to be involved.
An alternative approach is the therapeutic use of exogenous growth factors to induce the proliferation of endogenous stem cells. Among these factors, EGF, KGF and HGF have been the most studied. All of them are mitogens in type II pneumocytes, and act synergistically to mediate their maturation and increase surfactant synthesis [8]. EGF had beneficial effects in an animal model of ALI [39], and inhibition of EGF receptor had a detrimental effect during airway epithelium repair [40]. KGF has been linked to upregulation of anti-inflammatory cytokines and modulation of epithelial cell migration [41]. In the same way, HGF attenuates inflammation and showed antifibrotic effects in a murine model of bleomycin-induced fibrosis [42]. Vascular endothelial growth factor (VEGF) could also have a therapeutic effect by its ability to repair damaged endothelium, therefore helping in clearance of lung edema, but animal models have shown disappointing results [43].
Finally, manipulation of tissue remodeling could improve the outcome of ALI patients by favoring re-epithelization or avoiding fibrosis. The use of steroids in ALI has yielded conflicting results, depending on the time of application. These discrepancies can be explained if one takes into account the beneficial effects of inflammation during the repair phase discussed earlier. However, no study has specifically addressed the effects of steroids during lung repair.
MMPs are alternative targets to promote repair. Non-selective MMP inhibitors are available, but the lack of specificity could limit their benefits and none of them has been tested in ALI. Selective blockade or stimulation (depending on the role of the MMP and timing) could be a more promising approach. One of the first examples that has arrived in human trials is the use of beta-2 adrenergic receptor agonists. Intravenous administration of salbutamol decreased the duration and severity of lung injury by reducing lung edema in patients with ARDS [44]. This finding was associated with an upregulation of MMP-9 [18] and, therefore, with better alveolar epithelial repair [45]. Nevertheless, the route of administration could be relevant, as a recent trial has concluded that patients with ALI treated with inhaled salbutamol show no significant improvement in clinical outcomes [46]. On the other hand, blockade of MMP-8 has shown beneficial effects in experimental models of lung injury [22], including decreased lung fibrosis after bleomycin administration [23]. However, no clinical study aimed at modulating this protease has yet been proposed.
Conclusion
The lungs have a substantial potential for recovery after ALI. Of note, the mechanisms that can cause tissue disruption in the early phase also contribute to its repair later on, inflammation and matrix remodeling being paradigmatic examples. Therefore, therapies that disrupt these pathways, such as MMP inhibition, may have a prophylactic value, but their application at a later phase could be detrimental. Knowledge of the mediators involved in tissue repair could lead to new therapeutic strategies being applied after the initial insult has been controlled. Growth factors, exogenous stem cells (including type II pneumocytes) or drugs that promote matrix remodeling could be new alternatives to improve the prognosis of patients with ALI.
Abbreviations
ALI: acute lung injury; AP: activator protein; ARDS: acute respiratory distress syndrome; BAL: bronchoalveolar lavage; DAMPs: damage-associated molecular patterns; EGF: epidermic growth factor; G-CSF: granulocyte-colony stimulating factor; HGF: hepatic growth factor; IL-1R: interleukin-1 receptor; KGF: keratinocyte growth factor; LIX: lipopolysaccharide-induced CXC chemokine; LPS: lipopolysaccharide; MIP: macrophage inflammatory protein; MMPs: matrix metalloproteinases; NF-κB: nuclear factor-kappa B; RAGE: receptor of advanced glycation end-products; TGF: transforming growth factor; TLR: Toll-like receptors; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor; VILI: ventilator induced lung injury; WOB: work of breathing.
Competing interests
The authors declare that they have no competing interests.
Note
This article is one of eleven reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2012 (Springer Verlag, DOI: 10.1007/978-3-642-25716-2) and co-published as a series in Critical Care. Other articles in the series can be found online at http://ccforum.com/series/annualupdate2012. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Contributor Information
Adrián González-López, Email: guillermo.muniz@sespa.princast.es.
Guillermo M Albaiceta, Email: guillermo.muniz@sespa.princast.es.
Acknowledgements
Supported by Instituto de Salud Carlos III (Fondo de Investigación Sanitaria PI 10/00606) and Universidad de Oviedo (UNOV-09-BECDOC), Spain.
References
- Ventrice EA, Marti-Sistac O, Gonzalvo R, Villagra A, Lopez-Aguilar J, Blanch L. [Molecular and biophysical mechanisms and modulation of ventilator-induced lung injury] Med Intensiva. 2007;16:73–82. doi: 10.1016/S0210-5691(07)74779-4. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;16:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- de Carvalho ME, Dolhnikoff M, Meireles SI, Reis LF, Martins MA, Deheinzelin D. Effects of overinflation on procollagen type III expression in experimental acute lung injury. Crit Care. 2007;16:R23. doi: 10.1186/cc5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong L, Thickett DR, Mansell JP. et al. Changes in collagen turnover in early acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;16:1910–1915. doi: 10.1164/ajrccm.160.6.9811084. [DOI] [PubMed] [Google Scholar]
- Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;16:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;16:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- Kubo H. Molecular basis of lung tissue regeneration. Gen Thorac Cardiovasc Surg. 2011;16:231–244. doi: 10.1007/s11748-010-0757-x. [DOI] [PubMed] [Google Scholar]
- Lindsay CD. Novel therapeutic strategies for acute lung injury induced by lung damaging agents: the potential role of growth factors as treatment options. Hum Exp Toxicol. 2011;16:701–724. doi: 10.1177/0960327110376982. [DOI] [PubMed] [Google Scholar]
- Davey A, McAuley DF, O'Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J. 2011;16:959–970. doi: 10.1183/09031936.00032111. [DOI] [PubMed] [Google Scholar]
- Nin N, Lorente JA, de Paula M. et al. Rats surviving injurious mechanical ventilation show reversible pulmonary, vascular and inflammatory changes. Intensive Care Med. 2008;16:948–956. doi: 10.1007/s00134-007-0959-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lopez A, Astudillo A, Garcia-Prieto E. et al. Inflammation and matrix remodeling during repair of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;16:L500–L509. doi: 10.1152/ajplung.00010.2011. [DOI] [PubMed] [Google Scholar]
- Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;16:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- Korpos E, Wu C, Sorokin L. Multiple roles of the extracellular matrix in inflammation. Curr Pharm Des. 2009;16:1349–1357. doi: 10.2174/138161209787846685. [DOI] [PubMed] [Google Scholar]
- Lin M, Jackson P, Tester AM. et al. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide Pro-Gly-Pro. Am J Pathol. 2008;16:144–153. doi: 10.2353/ajpath.2008.080081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;16:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- Harty MW, Muratore CS, Papa EF. et al. Neutrophil depletion blocks early collagen degradation in repairing cholestatic rat livers. Am J Pathol. 2010;16:1271–1281. doi: 10.2353/ajpath.2010.090527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC. Matrix metalloproteinases in lung repair. Eur Respir J. 2003;16(Suppl 44):36s–38s. doi: 10.1183/09031936.03.00001203. [DOI] [PubMed] [Google Scholar]
- O'Kane CM, McKeown SW, Perkins GD. et al. Salbutamol up-regulates matrix metalloproteinase-9 in the alveolar space in the acute respiratory distress syndrome. Crit Care Med. 2009;16:2242–2249. doi: 10.1097/CCM.0b013e3181a5506c. [DOI] [PubMed] [Google Scholar]
- Albaiceta GM, Gutierrez-Fernandez A, Parra D. et al. Lack of matrix metalloproteinase-9 worsens ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;16:L535–L543. doi: 10.1152/ajplung.00334.2007. [DOI] [PubMed] [Google Scholar]
- Nerusu KC, Warner RL, Bhagavathula N, McClintock SD, Johnson KJ, Varani J. Matrix metalloproteinase-3 (stromelysin-1) in acute inflammatory tissue injury. Exp Mol Pathol. 2007;16:169–176. doi: 10.1016/j.yexmp.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita CM, Dolgonos L, Zemans RL. et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol. 2011;16:1733–1745. doi: 10.1016/j.ajpath.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaiceta GM, Gutierrez-Fernandez A, Garcia-Prieto E. et al. Absence or inhibition of matrix metalloproteinase-8 decreases ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2010;16:555–563. doi: 10.1165/rcmb.2009-0034OC. [DOI] [PubMed] [Google Scholar]
- Garcia-Prieto E, Gonzalez-Lopez A, Cabrera S. et al. Resistance to bleomycin-induced lung fibrosis in MMP-8 deficient mice is mediated by interleukin-10. PLoS One. 2010;16:e13242. doi: 10.1371/journal.pone.0013242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol. 2010;16:1575–1588. doi: 10.4049/jimmunol.0900290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueders MM, Balbin M, Rocks N. et al. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;16:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med. 1997;16:840–845. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- Quesnel C, Nardelli L, Piednoir P. et al. Alveolar fibroblasts in acute lung injury: biological behaviour and clinical relevance. Eur Respir J. 2010;16:1312–1321. doi: 10.1183/09031936.00074709. [DOI] [PubMed] [Google Scholar]
- Yoon HK, Cho HY, Kleeberger SR. Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ Health Perspect. 2007;16:1557–1563. doi: 10.1289/ehp.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese GM, McCormick-Shannon K, Mason RJ, Matthay MA. Hepatocyte growth factor and keratinocyte growth factor in the pulmonary edema fluid of patients with acute lung injury. Biologic and clinical significance. Am J Respir Crit Care Med. 1998;16:386–394. doi: 10.1164/ajrccm.158.2.9711111. [DOI] [PubMed] [Google Scholar]
- Rogel MR, Soni PN, Troken JR, Sitikov A, Trejo HE, Ridge KM. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. in press . [DOI] [PMC free article] [PubMed]
- Losa D, Chanson M, Crespin S. Connexins as therapeutic targets in lung disease. Expert Opin Ther Targets. 2011;16:989–1002. doi: 10.1517/14728222.2011.584875. [DOI] [PubMed] [Google Scholar]
- Vadivel A, Abozaid S, van Haaften T. et al. Adrenomedullin promotes lung angiogenesis, alveolar development, and repair. Am J Respir Cell Mol Biol. 2010;16:152–160. doi: 10.1165/rcmb.2009-0004OC. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sadikot RT, Adami GR. et al. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med. 2011;16:1473–1484. doi: 10.1084/jem.20102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Kwon HJ, Bae SC, Jung HS. Lung tissue regeneration after induced injury in Runx3 KO mice. Cell Tissue Res. 2010;16:465–470. doi: 10.1007/s00441-010-1011-7. [DOI] [PubMed] [Google Scholar]
- Plataki M, Lee YD, Rasmussen DL, Hubmayr RD. Poloxamer 188 facilitates the repair of alveolus resident cells in ventilator injured lungs. Am J Respir Crit Care Med. 2011;16:939–947. doi: 10.1164/rccm.201104-0647OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangreco A, Arwert EN, Rosewell IR, Snyder J, Watt FM, Stripp BR. Stem cells are dispensable for lung homeostasis but restore airways after injury. Proc Natl Acad Sci USA. 2009;16:9286–9291. doi: 10.1073/pnas.0900668106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KY, Shih HC, How CK. et al. IV delivery of induced pluripotent stem cells attenuates endotoxin-induced acute lung injury in mice. Chest. 2011;16:1243–1253. doi: 10.1378/chest.11-0539. [DOI] [PubMed] [Google Scholar]
- Curley GF, Contreras M, Higgins BD, O'Toole DP, Laffey JG. The role of mesenchymal stem cells during repair from ventilator induced lung injury. J Respir Crit Care Med. in press .
- Plopper CG, St George JA, Read LC. et al. Acceleration of alveolar type II cell differentiation in fetal rhesus monkey lung by administration of EGF. Am J Physiol. 1992;16:L313–L321. doi: 10.1152/ajplung.1992.262.3.L313. [DOI] [PubMed] [Google Scholar]
- Harada C, Kawaguchi T, Ogata-Suetsugu S. et al. EGFR tyrosine kinase inhibition worsens acute lung injury in mice with repairing airway epithelium. Am J Respir Crit Care Med. 2011;16:743–751. doi: 10.1164/rccm.201002-0188OC. [DOI] [PubMed] [Google Scholar]
- Panoskaltsis-Mortari A, Ingbar DH, Jung P. et al. KGF pretreatment decreases B7 and granzyme B expression and hastens repair in lungs of mice after allogeneic BMT. Am J Physiol Lung Cell Mol Physiol. 2000;16:L988–L999. doi: 10.1152/ajplung.2000.278.5.L988. [DOI] [PubMed] [Google Scholar]
- Dohi M, Hasegawa T, Yamamoto K, Marshall BC. Hepatocyte growth factor attenuates collagen accumulation in a murine model of pulmonary fibrosis. Am J Respir Crit Care Med. 2000;16:2302–2307. doi: 10.1164/ajrccm.162.6.9908097. [DOI] [PubMed] [Google Scholar]
- Medford AR, Millar AB. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): paradox or paradigm? Thorax. 2006;16:621–626. doi: 10.1136/thx.2005.040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins GD, McAuley DF, Thickett DR, Gao F. The beta-agonist lung injury trial (BALTI): a randomized placebo-controlled clinical trial. Am J Respir Crit Care Med. 2006;16:281–287. doi: 10.1164/rccm.200508-1302OC. [DOI] [PubMed] [Google Scholar]
- Perkins GD, Gao F, Thickett DR. In vivo and in vitro effects of salbutamol on alveolar epithelial repair in acute lung injury. Thorax. 2008;16:215–220. doi: 10.1136/thx.2007.080382. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Brower RG, Carson S. et al. Randomized, placebo-controlled clinical trial of an aerosolized beta-2 agonist for treatment of acute lung injury. Am J Respir Crit Care Med. 2011;16:561–568. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]