

Introduction
Acute kidney injury (AKI) is a significant cause of morbidity and mortality in hospitalized patients, especially those who are critically ill. The mortality rate in patients with severe AKI requiring renal replacement therapy (RRT) can exceed 50% [1]. Numerous factors contribute to the development of AKI, including reductions in renal blood flow, actions of nephrotoxic drugs, cellular injury/death of proximal tubule epithelial cells, pro-inflammatory responses of renal endothelial cells, influx and activation of inflammatory leukocytes that further reduces renal blood flow through vascular congestion and promotes and extends injury to kidney parenchymal cells [2,3]. The immune response in AKI involves cells of both the innate and adaptive immune systems. Although numerous studies have demonstrated the detrimental role of many different types of immune cells, recent reports have uncovered a protective and possibly therapeutic role of other immune cells in AKI. Studies in animal models of AKI have revealed that innate immune cells, such as neutrophils, macrophages, dendritic cells, natural killer (NK) cells and natural killer T (NKT) cells, and adaptive CD4+ T cells promote renal injury. Indeed, renal inflammation is a common feature of human AKI [4] and detailed analyses of biopsy samples from patients with AKI demonstrated the presence of mononuclear leukocytes (some CD3+ T cells) and neutrophils [5]. In contrast, CD4+FoxP3+ regulatory T cells (Tregs) can protect the kidney from ischemic and nephrotoxic injury in animal models. Understanding the immune mechanisms of renal injury and protection should yield new approaches to prevention and treatment of AKI. This review will summarize current knowledge on the role of the immune system in the pathogenesis of AKI.
Detrimental actions of pro-inflammatory leukocytes
Neutrophils
Polymorphonuclear cells (PMNs or neutrophils) are critical mediators of innate immunity. They respond rapidly (within minutes) to invading pathogens and to sites of tissue damage. Neutrophils clear invading pathogens by phagocytosis or by releasing toxic granules containing proteases and other enzymes and reactive oxygen species (ROS). For this reason, neutrophil degranulation can lead to damage of host cells in the inflamed tissue. In several mouse models of AKI (e.g., ischemia/reperfusion injury and cisplatin-induced kidney injury), neutrophil accumulation in the injured kidney is a consistent and early finding [6-8] and depletion of neutrophils [6] or prevention of neutrophil tracking to the kidney [9] reduces kidney injury. In addition to releasing granules, neutrophils have been shown to produce the pro-inflammatory cytokines, interferon (IFN)-γ and interleukin (IL)-17, and the chemokine CXCL1, in the injured kidney [7,9]. These findings demonstrate the involvement of neutrophils in the pathogenesis of kidney injury in the commonly used murine model of ischemia/reperfusion-induced AKI. Studies in some other species have reported a lack of significant neutrophil accumulation and no benefit after neutrophil depletion; however, these results may be related to differences in experimental models and limitations to methods for neutrophil depletion (see [10] and references therein).
Macrophages
Macrophages are phagocytic cells that arise from monocytes in the blood. Macrophage numbers increase early in the injured kidney (within 1 hour of reperfusion in an ischemia/reperfusion model [11]), and this infiltration is mediated by CCR2 and CX3CR1 signaling pathways [12,13]. These macrophages have a distinct 'inflamed' F4/80lowLy6ChighGR-1+CX3CR1low phenotype [12]. Depletion of macrophages, using liposomal clodronate, prior to kidney ischemia/reperfusion injury, reduced renal injury and adoptive transfer of macrophages reconstituted AKI [14]. Although macrophage infiltration is observed in cisplatin-induced experimental AKI, blockade of macrophage trafficking to the kidney did not prevent renal injury [15]. Analysis of post-ischemic kidney infiltrating macrophages by flow cytometry demonstrated that they are significant producers of many pro-inflammatory cytokines, including IL-6 and tumor necrosis factor (TNF)-α [12]. Another study identified IL-6 expression in renal outer medulla macrophages by in situ hybridization 4 hours after ischemia/reperfusion injury and IL-6 deficient mice are protected from kidney ischemia/reperfusion injury [16]. Recently, different types of macrophages have been described that either promote or inhibit inflammation, M1 and M2 macrophages, respectively [17]. New experimental evidence has demonstrated that once recruited to the post-ischemic kidney, macrophages display an M1 pro-inflammatory phenotype, but several days later those same macrophages change to an M2 phenotype and are vital in the repair process of the kidney [18].
Natural killer cells
NK cells are similar to lymphocytes of the adaptive immune system but lack a T cell receptor. Their activation is governed by signals received from activating and inhibitory receptors on their cell surface. The ligands for these NK cell receptors are expressed on target cells and are regulated by cell stress such as viral infection. During experimental AKI, the proximal tubule epithelial cells (TECs) upregulate the expression of an NK cell activating ligand, Rae-1, which promotes TEC killing by activating the NKG2D receptor on NK cells [19]. The NK cells utilize perforins to kill the TECs in this model and their numbers in the kidney are elevated as early as 4 h after ischemic insult [19].
Dendritic cells
Dendritic cells classified by the expression of CD11c+ are the most abundant leukocyte subset in the normal mouse kidney [20] suggesting an important role in renal immunity and inflammation. Upon stimulation, dendritic cells transform to a mature phenotype characterized by high levels of class II major histocompatibility complex (MHC class II) and co-stimulatory molecules with low phagocytic capacity. Mature dendritic cells are ideally suited to activate conventional T cells. Dendritic cells migrate to the renal draining lymph nodes after ischemia/reperfusion injury and induce T cell proliferation suggesting that kidney dendritic cells are vital to the adaptive immune response to ischemia/reperfusion injury [21]. Dendritic cells are also important in the innate immune response through several mechanisms. These include releasing pro-inflammatory factors, interacting with NKT cells via the co-stimulatory molecule, CD40, and presenting glycolipid to NKT cells via the CD1d molecule. Dong et al. demonstrated that after ischemia/reperfusion injury, renal dendritic cells produce the pro-inflammatory cytokines/chemokines, TNF, IL-6, monocyte chemotactic protein (MCP)-1 and RANTES (Regulated on Activation, Normal T Expressed and Secreted), and depletion of dendritic cells prior to ischemia/reperfusion injury significantly reduced the kidney levels of TNF produced after ischemia/reperfusion injury [22]. IL-12 and IL-23 are mainly produced from activated dendritic cells and their downstream cytokines, IFN-γ and IL-17, promote macrophage activation and neutrophil recruitment and amplify the immune response following ischemic kidney injury [9]. The use of a genetically-engineered mouse in which the promoter for the mainly dendritic cell-specific surface protein, CD11c, drives expression of the human diphtheria toxin receptor (CD11c-DTR mouse) has facilitated understanding of the role of renal dendritic cells in experimental AKI. Recent studies have demonstrated that dendritic cells can promote or prevent injury to the kidney depending on the stimulus. For example, depletion of dendritic cells prior to ischemia/reperfusion injury reduces subsequent reperfusion injury and renal dysfunction [11]. On the other hand, depletion of dendritic cells prior to cisplatin exposure resulted in worse renal dysfunction and inflammation [8]. These findings suggest that dendritic cells play an important role in orchestrating the immune response during AKI; additional studies are needed to understand what determines whether dendritic cells promote or inhibit kidney inflammation as this information may translate into new therapeutic strategies.
Lymphocytes
T and B cells are the major effector cells of the adaptive immune system. Recognition of antigens, presented by antigen-presenting cells, in the presence of sufficient co-stimulation, causes expansion and activation of T cells with a T-cell receptor specific for that antigen. B cells recognize soluble antigens through cell surface immunoglobulin type receptors. T cell contribution to the pathogenesis of kidney ischemia/reperfusion injury has been established in different mouse models lacking certain types of lymphocytes. In mice which lack CD4 and CD8 T cells (nu/nu mice), kidney injury and dysfunction were significantly reduced compared to wild-type controls after ischemia/reperfusion injury and cisplatin induced injury [23,24]. Reconstitution of nu/nu mice with CD4+ T cells alone but not with CD8+ T cells alone restored kidney injury after ischemia/reperfusion injury [23] and mice lacking CD8 or CD4 T cells alone suffered less kidney dysfunction after cisplatin administration [24]. To investigate the requirement of antigen-dependent CD4 T cell activation in ischemia/reperfusion injury, Satpute et al. reconstituted nu/nu mice with either polyclonal CD4 T cells (with a diverse array of T-cell receptor specificities) or CD4 T cells from the DO11.10 mouse which nearly all express the same T-cell receptor, specific for a chicken ovalbumin peptide [25]. Whereas polyclonal T cells reconstituted injury in the T cell deficient mice, DO11.10 T cells did not, unless their cognate antigen was co-administered with the T cells [25]. These results suggest that antigen specific activation of CD4 T cells is important for early (within 24 h) AKI.
The results of B cell deficiency in mouse models of AKI are not consistent. Mice lacking T and B cells (RAG-1 KO) were protected in some studies [26,27] and in others had similar injury to wild-type mice [28,29]. Deficiency of B cells alone in mu MT mice conferred protection against ischemia/reperfusion injury in one study [30] but resulted in more severe renal dysfunction after ischemia/reperfusion injury in another [31]. Therefore, more studies are needed to determine the role of B cells in AKI.
Natural killer T cells
NKT cells are a unique subset of T lymphocytes with surface receptors and functional properties shared with conventional T cells and NK cells. Invariant or Type I NKT cells express a conserved T-cell receptor (Vα14/Jα18 and Vβ8.2,Vβ2 or Vβ7) together with the NK cell marker, NK1.1. In contrast to conventional T cells, this invariant NKT cell T-cell receptor does not recognize peptide antigens presented by MHC-class I or II; it recognizes glycolipids in the context of the class I-like molecule, CD1d. One of the most important functions of NKT cells is their ability to rapidly produce large amounts of cytokines, including Th1-type (IFN-γ, TNF) and Th2-type (IL-4, IL-13) at the same time. The rapid response by NKT cells following activation can amplify and regulate the function of dendritic cells, Tregs, NK and B cells, as well as conventional T cells. The number of IFN-γ producing type I NKT cells in the kidney is significantly increased by 3 hours of reperfusion after ischemic insult [7] and NKT cells were observed in kidneys of patients with acute tubular necrosis (ATN) [32]. Blockade of NKT cell activation with the anti-CD1d mAb, NKT cell depletion with an anti-NK1.1 monoclonal antibody in wild-type mice, or use of type I NKT cell-deficient mice (Jα18-/-) inhibited the accumulation of IFN-γ producing neutrophils after IRI and prevented AKI [7]. Type II NKT cells are not restricted to an invariant T-cell receptor but still recognize glycolipids, such as sulfatide. In a recent study by Yang et al. sulfatide-activated Type II NKT cells migrated to the injured kidney and protected mice from ischemia/reperfusion injury [32]. Taken together these studies suggest that different types of NKT cells may play divergent roles in AKI.
Protective actions of regulatory T cells
Tregs make up an indispensible counter-balance to the pro-inflammatory cells of the immune system. This aspect is illustrated in patients with immunodysregulation, polyendocrinopathy, enteropathy, x-linked (IPEX) syndrome and scurfy mice which both have devastating autoimmunity caused by a lack of functional Tregs [33]. Numerous types of Tregs have been described in the literature, but the most abundant are CD4+ T cells, which express CD25, and the Treg-specific transcription factor, FoxP3. FoxP3 suppresses the expression of pro-inflammatory genes and promotes the anti-inflammatory phenotype of Tregs [34]. The main mechanisms of suppression employed by Tregs include production of anti-inflammatory cytokines, such as IL-10 and transforming-growth factor (TGF)-β, generation of extracellular adenosine, direct contact-mediated inhibition of dendtritic cells through cell surface molecules such as lymphocyte activation gene (LAG)-3 and cytotoxic T-lymphocyte antigen (CTLA)-4 and several others [35,36]. Based on the important contribution of the pro-inflammatory immune cells to AKI discussed above we hypothesized that Tregs would serve to protect the kidney from inflammation and injury. To test this hypothesis, Tregs were partially depleted from naïve wild-type mice prior to a short ischemic insult that was not severe enough to induce kidney dysfunction in control mice [37]. Reduction in numbers of Tregs predisposed mice to tubular necrosis, renal inflammation and loss of function [37]. In other studies, Treg depletion prior to more severe ischemia also worsened renal injury measured at 72 h of reperfusion [38]. These results suggest that enhancement of Treg numbers or function may be an effective preventative therapy for AKI. Indeed, adoptive transfer of isolated Tregs, by i.v. injection prior to ischemia/reperfusion injury [37,39] or cisplatin administration [40] protected mice from kidney injury. This protection was associated with a reduction in infiltrating innate immune cells and pro-inflammatory gene expression in the kidney [37,39,40]. In the ischemia/reperfusion injury model, IL-10-deficient Tregs were unable to offer any protection [37], suggesting that IL-10 production is an important mechanism for Treg-mediated protection from kidney injury. Two other methods of protecting the kidney, ischemic preconditioning [39,41] and use of FTY720 [42], have been shown to be partially dependent on the presence of Tregs.
In addition to prevention of injury, Tregs also promote the recovery of kidney function after ischemia/reperfusion injury [43]. This effect was demonstrated by depleting Tregs after ischemic injury, which caused reduced renal repair and increased mortality; on the other hand, adoptive transfer of Tregs 24 h after ischemic insult accelerated the recovery of renal function in mice [43]. The effects on kidney recovery did not appear to be mediated through effects on infiltrating innate immune cells, but were associated with increased tubular epithelial cell proliferation and reduced cytokine production from infiltrating T cells [43]. This study is important since it is difficult to detect renal injury in a timely manner and this suggests that manipulations to enhance Treg numbers or function can have beneficial effects after injury has occurred.
Recently, a clinical trial was conducted to determine the safety and feasibility of ex vivo expansion of human Tregs for adoptive transfer into patients [44]. The authors demonstrated that it is possible to expand Tregs in vitro and then infuse up to 3 million Tregs/kg to patients, with no adverse effects observed [44]. Although this trial was conducted in the study of a different disease model, it demonstrates that Treg cells themselves may be a novel therapeutic strategy in humans for many diseases, including AKI.
Conclusion
The immune response to kidney damage during AKI is an important contributor to the prolonged lack of renal function and progression of kidney injury. This response is complex, involving numerous pro-inflammatory leukocytes which employ diverse effector mechanisms inside the kidney (Figure 1). The response also involves an anti-inflammatory arm that protects the kidney from subthreshold insults, which is mediated by Tregs and possibly dendritic cells in some conditions (Figure 1). In the future, it may be possible to prevent AKI or treat existing AKI by inhibiting the pro-inflammatory immune response to kidney injury and/or promoting the anti-inflammatory response through drugs or maneuvers that enhance Treg action or by direct administration of Tregs themselves.
Figure 1.
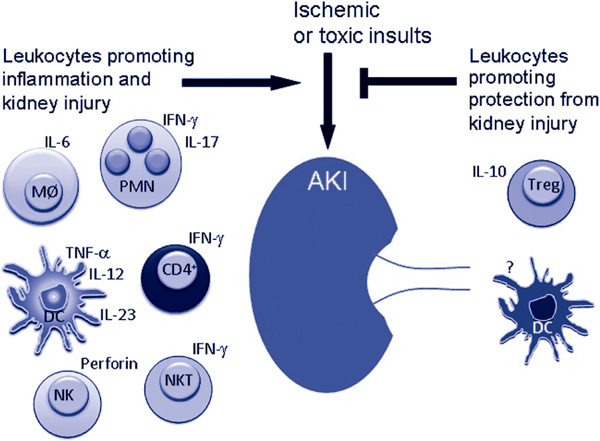
Toxic or ischemic insults initiate inflammation in the kidney which consists of interferon (IFN)-γ- and interleukin (IL)-17-producing neutrophils (PMN), IL-6-producing macrophages (MØ), dendritic cells (DC) which produce tumor necrosis factor (TNF)-α, IL-12 and IL-23, IFN-γ-producing CD4+ T cells, invariant type I natural killer T (NKT) cells and natural killer (NK) cells which kill renal tubule epithelial cells in a perforin-dependent manner. On the other hand toxic and ischemic injuries are prevented by regulatory T cells (Tregs) which produce IL-10, and in cisplatin-induced nephrotoxicity DCs protect the kidney by an unknown mechanism.
Abbreviations
AKI: acute kidney injury; ATN: acute tubular necrosis; CTLA: cytotoxic T-lymphocyte antigen; DC: dendritic cells; IFN: interferon; IL: interleukin; IPEX: immunodysregulation: polyendocrinopathy: enteropathy: x-linked; LAG: lymphocyte activation gene; MØ: IL-6-producing macrophages; MCP: monocyte chemotactic protein; NK: natural killer; NKT: natural killer T; PMNs: polymorphonuclear cells or neutrophils; RANTES: Regulated on Activation: Normal T Expressed and Secreted; ROS: reactive oxygen species; RRT: renal replacement therapy; TECs: tubule epithelial cells; TGF: transforming-growth factor; TNF: tumor necrosis factor; Tregs: regulatory T cells.
Competing interests
The authors declare that they have no competing interests.
Note
This article is one of eleven reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2012 (Springer Verlag, DOI: 10.1007/978-3-642-25716-2) and co-published as a series in Critical Care. Other articles in the series can be found online at http://ccforum.com/series/annualupdate2012. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Contributor Information
Gilbert R Kinsey, Email: grk4n@virginia.edu.
Mark D Okusa, Email: grk4n@virginia.edu.
References
- Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol. 2011;16:209–217. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey GR, Okusa MD. Pathogenesis of acute kidney injury: foundation for clinical practice. Am J Kidney Dis. 2011;16:291–301. doi: 10.1053/j.ajkd.2011.02.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;16:5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solez K, Morel-Maroger L, Sraer JD. The morphology of "acute tubular necrosis" in man: analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine (Baltimore) 1979;16:362–376. [PubMed] [Google Scholar]
- Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;16:486–491. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- Kelly KJ, Williams WW Jr, Colvin RB. et al. Intercellular adhesion molecule-1- deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;16:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang L, Sung SS. et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;16:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- Tadagavadi RK, Reeves WB. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol. 2010;16:53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang L, Vergis AL. et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;16:331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolisetty S, Agarwal A. Neutrophils in acute kidney injury: not neutral any more. Kidney Int. 2009;16:674–676. doi: 10.1038/ki.2008.689. [DOI] [PubMed] [Google Scholar]
- Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;16:268–277. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang L, Sung S. et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;16:1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DJ, Dursun B, He Z. et al. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. 2008;16:F264–F271. doi: 10.1152/ajprenal.00204.2007. [DOI] [PubMed] [Google Scholar]
- Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;16:F722–F731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- Lu LH, Oh DJ, Dursun B. et al. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J Pharmacol Exp Ther. 2008;16:1111–1117. doi: 10.1124/jpet.107.130161. [DOI] [PubMed] [Google Scholar]
- Kielar ML, John R, Bennett M. et al. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol. 2005;16:3315–3325. doi: 10.1681/ASN.2003090757. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;16:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Lee S, Huen S, Nishio H. et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;16:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZX, Wang S, Huang X. et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol. 2008;16:7489–7498. doi: 10.4049/jimmunol.181.11.7489. [DOI] [PubMed] [Google Scholar]
- Soos TJ, Sims TN, Barisoni L. et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;16:591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005;16:1096–1108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;16:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- Burne MJ, Daniels F, El Ghandour A. et al. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. 2001;16:1283–1290. doi: 10.1172/JCI12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Chien C, Burne-Taney M. et al. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J Am Soc Nephrol. 2006;16:765–774. doi: 10.1681/ASN.2005010102. [DOI] [PubMed] [Google Scholar]
- Satpute SR, Park JM, Jang HR. et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. 2009;16:984–992. doi: 10.4049/jimmunol.0801928. [DOI] [PubMed] [Google Scholar]
- Bajwa A, Jo SK, Ye H. et al. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. J Am Soc Nephrol. 2010;16:955–965. doi: 10.1681/ASN.2009060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol. 2006;16:3108–3114. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- Burne-Taney MJ, Yokota-Ikeda N, Rabb H. Effects of combined T- and B-cell deficiency on murine ischemia reperfusion injury. Am J Transplant. 2005;16:1186–1193. doi: 10.1111/j.1600-6143.2005.00815.x. [DOI] [PubMed] [Google Scholar]
- Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol. 2002;16:F352–F357. doi: 10.1152/ajprenal.00160.2001. [DOI] [PubMed] [Google Scholar]
- Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H. B cell deficiency confers protection from renal ischemia reperfusion injury. J Immunol. 2003;16:3210–3215. doi: 10.4049/jimmunol.171.6.3210. [DOI] [PubMed] [Google Scholar]
- Renner B, Strassheim D, Amura CR. et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. 2010;16:4393–4000. doi: 10.4049/jimmunol.0903239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Lee JP, Jang HR. et al. Sulfatide-reactive natural killer T cells abrogate ischemia-reperfusion injury. J Am Soc Nephrol. 2011;16:1305–3514. doi: 10.1681/ASN.2010080815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005;16(Suppl):56–62. doi: 10.1016/j.jaut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;16:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol. 2009;16:1105–1111. doi: 10.1093/intimm/dxp095. [DOI] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;16:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey GR, Sharma R, Huang L. et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;16:1744–1753. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro RMM, Camara NOS, Rodrigues MM. et al. A role for regulatory T cells in renal acute kidney injury. Transplant Immunol. 2009;16:50–55. doi: 10.1016/j.trim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 2010;16:771–780. doi: 10.1038/ki.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Nho D, Chung HS, Shin MK, Kim SH, Bae H. CD4+CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int. 2010;16:1100–1109. doi: 10.1038/ki.2010.139. [DOI] [PubMed] [Google Scholar]
- Cho WY, Choi HM, Lee SY, Kim MG, Kim HK, Jo SK. The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 2010;16:981–992. doi: 10.1038/ki.2010.266. [DOI] [PubMed] [Google Scholar]
- Kim MG, Lee SY, Ko YS. et al. CD4+ CD25+ regulatory T cells partially mediate the beneficial effects of FTY720, a sphingosine-1-phosphate analogue, during ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2011;16:111–124. doi: 10.1093/ndt/gfq480. [DOI] [PubMed] [Google Scholar]
- Gandolfo MT, Jang HR, Bagnasco SM. et al. Foxp3(+) regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;16:717–729. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- Brunstein CG, Miller JS, Cao Q. et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;16:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]