

Abstract
Prostate cancer continues to be a major cause of morbidity and mortality in men around the world. The field of prostate cancer research continues to be hindered by the lack of relevant preclinical models to study tumorigenesis and to further development of effective prevention and therapeutic strategies. The Prostate Cancer Foundation held a Prostate Cancer Models Working Group (PCMWG) Summit on August 6th and 7th, 2007 to address these issues. The PCMWG reviewed the state of prostate cancer preclinical models and identified the current limitations of cell line, xenograft and genetically engineered mouse models that have hampered the transition of scientific findings from these models to human clinical trials. In addition the PCMWG identified administrative issues that inhibit the exchange of models and impede greater interactions between academic centers and these centers with industry. The PCMWG identified potential solutions for discovery bottlenecks that include: (1) insufficient number of models with insufficient molecular and biologic diversity to reflect human cancer, (2) a lack of understanding of the molecular events that define tumorigenesis, (3) a lack of tools for studying tumor–host interactions, (4) difficulty in accessing model systems across institutions, and (5) addressing why preclinical studies appear not to be predictive of human clinical trials. It should be possible to apply the knowledge gained molecular and epigenetic studies to develop new cell lines and models that mimic progressive and fatal prostate cancer and ultimately improve interventions.
Keywords: mouse, genetically engineered, cell lines
INTRODUCTION
Prostate cancer continues to be a major cause of morbidity and mortality, accounting for 27,000 deaths in 2007 in the United States alone [1]. While discoveries continue to be made regarding the etiology of this disease, the field of prostate cancer research continues to be hampered by the lack of relevant preclinical models to study tumorigenesis and to develop effective prevention and therapeutic interventions. To address this, the Prostate Cancer Foundation held a Prostate Cancer Models Working Group (PCMWG) Summit on August 6th and 7th, 2007. The charge of the PCMWG was to review the state of the art of prostate cancer preclinical models and identify the current limitations of cell line, xenograft, and genetically engineered mouse (GEM) models that have hampered the transition of scientific findings from these models to human clinical trials. In addition the PCMWG identified administrative issues that inhibit the exchange of models and impede greater interactions between academic centers and these centers with industry.
While an incomplete understanding of the biology of human prostate cancer complicates the development of relevant model systems that mimic the human disease, many genetic changes have been associated with prostate cancer that appear to correlate with microscopic changes in cell structure and gland histology (Fig. 1) [2–15]. Early prostate tumorigenesis appears to be associated with a dysplasia that starts with proliferative inflammatory atrophy (PIA) which may progress to prostatic intraepithelial neoplasia (PIN) which may progress to carcinoma [4]. These early lesions may be initiated by inflammation that occurs with exposure to different infectious agents and/or carcinogens [2–8]. As premalignant lesions progress to primary cancer, to metastatic cancer, and to hormone refractory cancer after hormonal treatment, genetic damage continues to accumulate within the cancer cells [2–15]. The most effective models are, and will be, the ones that can be used to mimic the changes human disease, can be utilized to ask questions that explain observed phenomena of the human condition, and would be predictive for therapeutic efficacy. The field also needs to continually incorporate discoveries in the patient and human tissue studies back into model development and understanding.
Fig. 1.
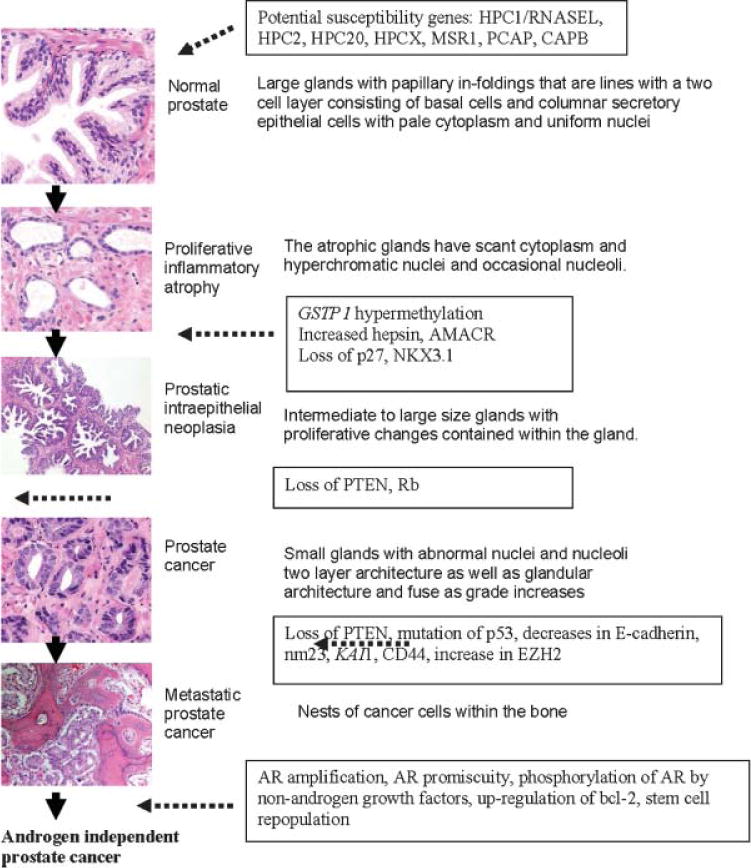
Histologic and molecular changes associated with prostate tumorigenesis. For more information, see Refs. [21–34]. PIA, proliferative inflammatory atrophy; RNASEL, 2′–5′-oligoadenylate-dependent RNase L; AMACR, a-methylacyl-coenzyme A racemase; EZH2, enhancer of zeste homolog2; PcG, polycomb group. Original magnification 100x. Taken from Taichman et al. [1].
Bottleneck: Prostate Cancer Research Has Generated an Insufficient Number of Preclinical Models With Insufficient Molecular and Biological Diversity
Currently, preclinical models of prostate cancer in major use include those derived from rat, canine, mouse, and human sources. The Dunning rat model, although not as widely used presently as it has been in the past, continues to be the only spontaneously derived prostate cancer model with multiple sublines that metastasize quickly and reproducibly to multiple organs (Fig. 2) [16–20]. The canine ACE-1 grows as a cell line and as a xenograft and was derived from a primary adenocarcinoma. When implanted in bone, it demonstrates an osteoblastic pattern [21]. Most laboratories are now utilizing human cell lines and/or xenografts or genetically engineered models (GEM) in mice for preclinical prostate studies.
Fig. 2.
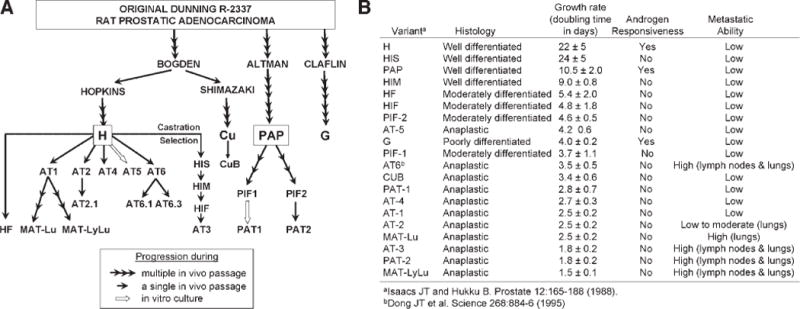
The development of the Dunning rat prostate adenocarcinoma cell lines. A: Schematic of the development of the Dunning sub lines. B: Characteristics of the Dunning sublines. [Personal communication, J. Isaacs].
There have been several excellent reviews of the available human prostate cancer cells [22–26]. Table I includes the top 10 most cited in the literature by PUBMED search. By far, the classic three cell lines, PC-3, LNCaP, and DU145, are the most widely used, each with thousands of studies published. Each of the other human cell lines have less than 200 citations. Despite the use of all of these models, it is clear that a basic understanding and characterization of existing models and how their properties compare to the human disease is lacking and needed for growth in the field. It is critical that researchers understand the properties of cell lines on a general level, but also to use the correct cell line/model for the scientific problem that they are studying. Thus, while the androgen receptor (AR) status of a cell line is generally known, the growth factor receptor status and signal transduction pathways are also important to delineate prior to initiating studies. The PC-3 and LNCaP cell lines each have multiple sublines associated with them, however, in general, these are not fully genetically and phenotypically characterized, nor is there a method for standardization (Figs. 3 and 4) [22]. It is possible that many of the deficiencies that exist in the preclinical models could be addressed simply through better genetic and phenotypic characterization of the models already in existence. For example, each cell line should have published chromosomal analyses, state of growth factor and signal transduction pathways, and sensitivities to chemotherapies, hormonal therapies, and radiation. Each cell line should be characterized as to how it grows in vitro, and in vivo after being implanted/injected within orthotopic, subcutaneous, lymphatic, intra-osseous, intravenous, and intra-cardiac sites. However, as biological models evolve after serial passages in vitro and in vivo, caution should be taken in the interpretation of published studies and characterization of key features may be advisable in each lab prior to the use of the model of interest. Each publication with a prostate cancer model needs to standardize the models used with existent molecular/phenotypic data.
TABLE I.
Prostate Cancer Cell Lines
| Species | Model | Source | Androgen receptor status | Other properties |
|---|---|---|---|---|
| Canine | Ace-1 | Cell line from primary adenocarcinoma | AR− | CyK8, 18+, vimentin+ |
| Rat | Dunning | Spontaneous from primary adenocarcinoma | Multiple sublines | Multiple sublines |
| Human (top 12 by citation index) | PC-3 | Cell line from bone metastasis | − | PSA−, negative for p53 |
| LNCaP | Cell line from lymph node metastasis | Mutated AR | PSA+, CK8, 18+; WT p53 | |
| DU145 | Cell line from dural metastasis | − | PSA−, PAP+, CK 7, 8,18, 19+, mutated p53 | |
| CWR22rv1 | Gleason 9 TURP | + | PSA+, 22Rv1 cells express CK-8, 18+, mutated p53 | |
| LuCaP 23 | Lymph node metastasis | + | PSA, PAP+ | |
| LuCaP 35 | Xenograft from lymph node metastasis | +, WT AR | PSA+ | |
| MDA PCa 2a | Cell line from bone metastasis | Mutated AR | PSA+; WT p53, CK 5, 8, 18+ | |
| VCaP | Cell line from bone metastasis | +, WT AR | PSA+, PAP+, PSMA+CK 8, 18 +, mutated p53 | |
| LAPC-4 | Cell line from lymph node metastasis | + | PSA+, PSCA+, PSMA+, CK 8, 18+ Mutated p53 | |
| PC-82 | Primary prostate, cribiform pattern | + | PAP+ | |
| PreC | Non-immortalized prostate epithelial cells | AR− | PSA−, p63+, CK 8, 18+ | |
| RWPE | Immortalized prostate epithelial cells | − | PSA+, CK 8, 18+ |
Fig. 3.
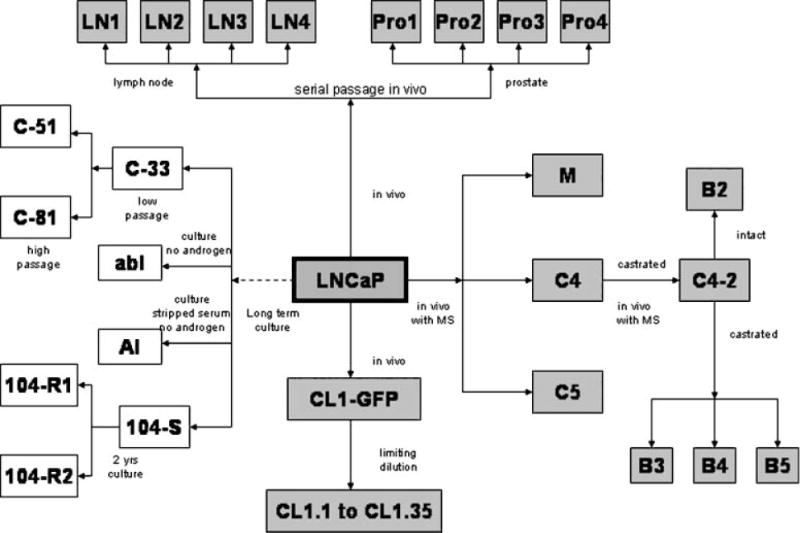
The development of the LNCaP prostate adenocarcinoma cell sublines. Taken from Sobel and Sadar [22].
Fig. 4.
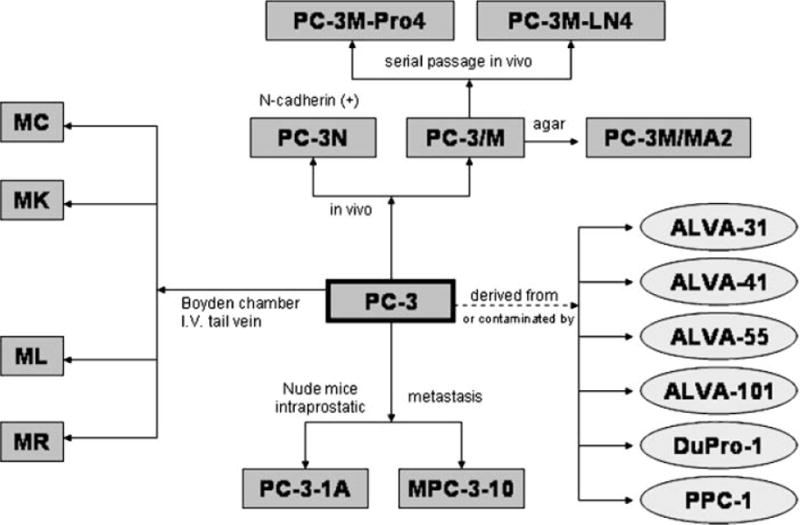
The development of the PC-3 prostate adenocarcinoma cell lines. Taken from Sobel and Sadar [22].
Summary of potential solutions
Create a repository of early passage (within 5–10 passages) of well-characterized cell lines and xenografts at the genetic and biochemical levels. This should include chromosomal analysis as well as gene expression data from in vitro and xenograft data. Data should include growth factor receptor expression as well as the common signal transduction pathways. Gene expression data on a minimal subset of biomarker genes (e.g. AR, PSA, PAP, K8, K18, Vimentin, NSE) on the existing cell lines and xenografts could be deposited in a public domain website, which can be updated by researchers. This information could be helpful to gain a better understanding on genetic drifts of existing models and to provide a context of research findings from different groups.
Characterization of how each cell line/xenograft grows after intra-cardiac and intravenous injection as well as after implantation at subcutaneous, orthotopic, lymphatic, and osseous sites. The lack of cell lines/xenografts which spontaneously metastasize in a reproducible manner continues to be a major problem for the field. How each of the cell lines behaves when injected at various primary and metastatic sites and in hosts with various genetic backgrounds has not been well explored. How each of the cell lines may undergo genetic drifts when passaged in culture or implant in mice under various hormonal conditions or cell culture media should be better defined.
Characterization and standard reporting of how each cell line/xenograft responds to radiation, chemotherapies, and targeted therapies. The therapeutic dose response curves for each cell line in vitro and in vivo should be readily accessible so investigators can compare their data with published standards. This would allow comparison of agents and demonstrate that controls were comparable across different laboratories and between different experiments.
Placement of all data in a public domain website so that it is accessible to the field. All of the data generated on the cell lines should be placed in a website that is continually updated. Expression array data could be maintained in an “Oncomine” format [27]. Since there is no centralized and standardized quality assurance of cell lines to make sure genetic drift has not occurred, investigators could determine if the model that they are using has “drifted” from those previously published by placing appropriate control data on the website.
Generation of new cell lines. It remains unclear why prostate cancer cells are so difficult to isolate and successfully transfer to tissue culture as compared to other disease sites. Multiple investigators have utilized varying methods, including co-culture, to increase the number of available cell lines. Immortalization with hTERT or other methods may be needed to increase the number of human models, especially those derived from primary tumors. The NCI 60 panel has too few prostate cancer cell lines and the prostate cancer cell lines it contains are biologically suboptimal. As new cell lines are generated, they should be added to the NCI panel.
Deficiencies in anatomic, functional, and metabolic imaging complicate monitoring of progression in xenograft models. Knowledge on the limitations of different imaging modalities and relevant application to current models is lacking. The understanding of cancer progression and therapeutics can benefit greatly from novel imaging tracers or innovative imaging reporter systems that can reveal the activities of specific molecular pathway in the living subjects.
Bottleneck: The Molecular Events That Define Prostate Tumorigenesis Remain Unclear
GEM models are helping define the molecular events of prostate tumorigenesis (Fig. 5, Table II) [28–31]. GEM models break into two broad areas, those generated by overexpression of an oncogene with a prostate specific promoter and those with targeted deletion of specific genes. The transgenic mouse model of prostate (TRAMP) model is currently the most widely used, and best characterized of all of these models (http://thegreenberglab.fhcrc.org/) [32–34]. The autochthonous TRAMP utilizes a transgene specifically overexpressed in the epithelial cells of the prostate which results in the development of prostate cancers in a manner that mimics the histopathology of human PCa development (http://thegreenberglab.fhcrc.org/). The mice exhibit progressive stages of prostate cancer ranging from mild to severe prostatic hyperplasia with cribriform structures and focal adenocarcinoma and seminal vesicle invasion to occasional metastatic spread to the lymph nodes, lung, and bone. Since the development of TRAMP, several other transgenic models have become available [28–31].
Fig. 5.
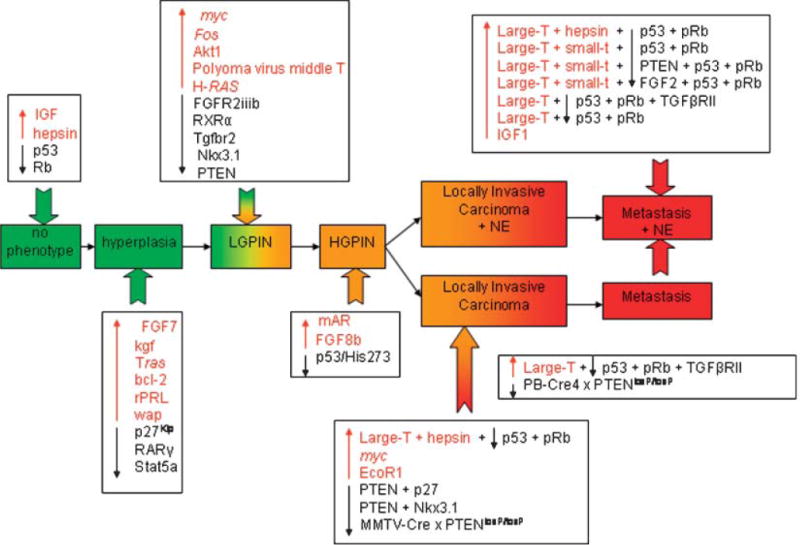
Diagrammatic summary of the onset and progression of prostate cancer resulting from one, two, or multiple genetic disruptions. Taken from Kasper [31].
TABLE II.
A Summary of Murine Models of Prostate Cancer
| Hyperplasia/PIN only | Invasive carcinoma without metastasis | Metastatic carcinoma |
|---|---|---|
| Nkx3.1−/− | ARR2PB-c Myc | TRAMP |
| PSACre; NKx3.1loxP/loxP | PB-c-Myc | C3(1)-Tag |
| PB-AR | Nkx3.1−/− and Pten+/− | PSP94-Tag |
| MPAKT | LADY (LPB-Tag) | gp91-phox-Tag |
| C(3)1-c-Myc | Ptenhy/− | |
| PB-Ras | LADY (LPB-Tag, line 12T-10) | |
| PBCre4; RXRαloxP/loxP | PBCre4; PTENloxP/loxP | |
| PBCre; RbloxP/loxP | PB-AR-E231G | |
| PB-FGF2 | ||
| ARR2PBi-FGFR1 | ||
| AR2PBi-caFGFR1 | ||
| ARR2PB-SKP2 | ||
| PB-rPRL Pten+/− | ||
| ARR2Pb-ETV1 |
Adapted from Abdulkadir and Kim [29].
AR, androgen receptor; ARR2PBi, androgen-responsive regions 2 probasin bigenic promoter; caFGFR, constitutively active fibroblast growth receptor mutant; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; gp, glycoprotein; hy, hypomorph; PB, probasin promoter; PBCre, probasin promoter cAMP responseelement; phox, phox homology; PIN, prostatic intraepithelial neoplasia; PSA, prostate-specific antigen; Pten, phosphatase and tensin homolog; rPRL, rat prolactin; RXR, retinold X receptor; SKP, S-phase kinase-associated protein; TRAMP, transgenic mouse model of prostate.
Several murine models generated by disruption or overexpression of genes in the prostate develop premalignant as well as malignant lesions. The best characterized of these models is the loss of the tumor suppressor gene phosphatase and tensin homologue (PTEN) [35–37]. The lipid phosphatase PTEN, acts as a tumor suppressor gene by acting as a negative regulator of the phosphatidylinositol 3-kinase/AKT pathway. Approximately 70% of primary prostate cancers exhibit a loss of at least one PTEN allele and loss of both alleles is associated with advanced disease [35, 36]. In mice, loss of one allele of PTEN is associated with the development of high-grade PIN and conditional loss of PTEN results in invasive prostate cancer that metastasizes to lymph nodes and lung in some animals [38]. Combining PTEN loss with other genetic abnormalities observed in human prostate cancers has led to several models of disease [39–42]. Mice nullizygous for PTEN and p53 in the prostate develop aggressive prostate cancer [39]. NKX3.1 encodes a homeodomain transcription factor located on a region of human chromosome 8p21 that is frequently deleted in prostatic intraepithelial neoplasia (PIN) as well as prostate cancer (reviewed [40–43]). Loss of NKX3.1 function in mice leads to PIN [43]. Nkx3.1;Pten compound mutant mice develop androgen-independent prostate cancer that retains wild-type AR expression and function [40–42]. These mice, as well as others, have given the field powerful new tools to understand prostate cancer biology. However, biology, however, their usefulness in the areas of prevention and therapeutic discovery remain largely unexplored, in large part because of the time it takes for these models to generate cancers and the expense of generation and maintenance, but also because of uncertainty about their predictive value for these interventions in humans. Moreover, GEM models also preclude testing of human-specific diagnostic or therapeutic reagents (e.g., antibodies).
Summary of potential solutions
Increase the number and sophistication of GEM models to better mimic human disease progression. While the current GEM models have given increasing insight into the molecular events that contribute to prostate tumorigenesis, additional models that target new oncogenic events, that modify the tumor ecology (e.g., stroma, cytokines), and that combine mutations will add significantly to the knowledge base.
Better define the biology of the mouse prostate as compared to the human prostate. The mouse prostate differs significantly from the human. The human prostate is a discrete, encapsulated organ composed of central (25%), transitional (5%), and peripheral zones (70%). The mouse prostate is not a discrete unit and is composed of four-paired lobes; anterior, dorsal, lateral, and ventral without a discrete capsule. Adding to these differences in microscopic and gross anatomy is the fact that the mouse prostate does not spontaneously develop prostate cancer and does not make PSA. These differences in mouse and human prostates should be well characterized at the genetic and molecular levels so as to make interpretation of results derived from models as meaningful as possible.
Develop more cell lines from GEM models to allow more rapid study of prostate cancer cell lines in immunocompetent hosts. One of the drawbacks to GEM models is that they generally take months to develop and therefore, their use for testing therapeutic interventions has been limited. GEM models, do however, allow the testing of agents in immunocompetent hosts. One way to speed agent testing in these models is to develop more cell lines from the GEM models to allow autochthonous xenograft testing.
Bottleneck: A Lack of Tools for Studying Tumor – Host Interactions
Prostate cancer can no longer be considered a disease of a single cell type but rather must be viewed as a complex system of epithelial cells that exhibit dysregulated growth within the framework of a microenvironment of multiple cells supporting that growth as well as the macroenvironment of the host with a unique genotype and immune system. Collectively, these components constitute an ecosystem that selects for the biological characteristics of the tumor. Further research is needed to better define these interactions, many of which are potential targets for therapy. While no model fully mimics human host biology and the human tumor microenvironment, in vivo models can be utilized to study specific components of tumor initiation and progression, including the interactions of prostate cancer cells with stroma and with bone bearing human xenografts. Development of future mouse models with high frequencies of spontaneous prostate cancer bone metastasis will prove to be highly attractive.
Summary of potential solutions
Development of better mouse models that allow studying host immune response to prostate cancer. Continual development of more relevant models of tumor–immune system interactions is desperately needed.
Generation of promoters that can regulate gene expression in stroma and other sites of interaction, such as bone. This is critical for generating relevant animal models both utilizing xenografts as well as GEM models.
Generation of more stromal cell lines. These lines should include fibroblasts and endothelial cells from prostate as well as common metastatic sites, including bone, liver, and lymph node. Other lines would include osteoclasts, osteoblasts, and nerve cells. The interaction of prostate cancer cells with the hematopoetic progenitor cells of the bone marrow should be explored.
Characterization of xenografts that are generated with stromal cells as well as tumor cells. Data suggest that tumor cells react differently to therapy when they are in contact with different substrata as well as in contact with different cell types. Relevant models should be generated that include combining tumor cells with as many normal cell types as possible. It is well recognized that spontaneous metastasis of xenografts is a deficient property of prostate cancer models and that this complicates attempts to study the process of metastasis in vivo. It is possible that the creation of more complex tumor—microenvironment models will facilitate spontaneous metastasis in vivo.
Bottleneck: Access to Model Systems Across Institutions
Collaboration between different academic laboratories as well as between academia and industry is complicated by multiple factors. Models held by the ATCC as well as different mouse distribution centers often have demands that exceed their capacity for timely response. Collaboration between laboratories, whether academic or industrial, is further complicated by the process of materials transfer agreements. These agreements also make it hard to freely test proprietary compounds in models.
Summary of potential solutions
Create a virtual systematic database for reagents, methods, characteristics, and controls for cell lines, xenograft models, and GEM models. This type of database would allow informed experiment design and model use and would increase the quality of research in the field.
Create a central organizational structure for MTAs between individual laboratories as well as between investigators and industry. This may help facilitate the movement of materials between universities as well as simply licensing.
Bottleneck: Preclinical Studies Appear Not to be Predictive of Human Clinical Trials
The conundrum of why preclinical models have not been more predictive of results in human studies is the result of inadequate models, inappropriate use of the models that are available, and subsequent design of clinical trials that do not mirror the preclinical model testing.
Summary of potential solutions
Design of appropriate preclinical studies that utilize the appropriate agent doses, pharmacokinetic and pharmacodynamic parameters to take into account the differences in metabolism between mouse and human. Preclinical studies should be done that mirror what can be done in patients. The lack of clinical feedback in the design of many preclinical studies complicates the translation of findings to first in man trials.
Design human clinical trials that mimic the preclinical trials in animals. While it is true many preclinical studies of drug efficacy are designed without appropriate thought of how the agents can be given in humans, it is also true that the design of clinical trials rarely takes into account how the preclinical testing was accomplished.
CONCLUSION
The genetically engineered, xenograft and cell culture models of prostate cancer have contributed to multiple important discoveries in the field. No single model accurately represents all the molecular or cellular features of prostate cancer as it develops from normal prostate and progresses to metastatic, hormone refractory disease. Given the vast amount of information gained through genomic, proteomic, and epigenetic studies it should be possible to apply this knowledge to develop a new cell lines and models that mimic progressive and fatal prostate cancer and ultimately improve interventions.
Acknowledgments
The PCMWG meeting was sponsored by the Prostate Cancer Foundation. KJP receives support as an American Cancer Society Clinical Research Professor, NIH P01CA093900, SPORE 2P50 CA69568, U19 CA113317 and the Prostate Cancer Foundation. LWKC receives support from DOD PC040260, PC060866 and NIH P01CA098912. NMG receives support from NIH U01 CA84296. WCH receives support from the Prostate Cancer Foundation. MJW receives support from the Prostate Cancer Foundation, NIH R01 Ca105402, P01 CA104106. LW receives support from the Prostate Cancer Foundation, NIH R01 CA101904.
Footnotes
Grant sponsor: American Cancer Society; Grant numbers: NIH P01CA093900, SPORE 2P50 CA69568, U19 CA113317; Grant sponsor: Prostate Cancer Foundation; Grant sponsor: DOD; Grant numbers: PC040260, PC060866; Grant sponsor: NIH; Grant numbers: P01CA098912, U01 CA84296, R01 Ca105402, P01 CA104106, R01 CA101904.
References
- 1.Taichman RS, Loberg RD, Mehra R, Pienta KJ. The evolving biology and treatment of prostate cancer. J Clin Invest. 2007;117(9):2351–2361. doi: 10.1172/JCI31791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majumder PK, Sellers WR. Aktregulated pathways in prostate cancer. Oncogene. 2005;24:7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 3.Jong JT. Prevalent mutations in prostate cancer. J Cell Biochem. 2006;97:433–447. doi: 10.1002/jcb.20696. [DOI] [PubMed] [Google Scholar]
- 4.Cansino Alcaide JR, Martinez-Pineiro L. Molecular biology in prostate cancer. Clin Transl Oncol. 2006;8:148–152. doi: 10.1007/s12094-006-0004-1. [DOI] [PubMed] [Google Scholar]
- 5.Rennert H, Zeigler-Johnson CM, Addya K, Finley MJ, Walker AH, Spangler E, Leonard DG, Wein A, Malkowicz SB, Rebbeck TR. Association of susceptibility alleles in ELAC2/HPC2, RNASEL/HPC1, and MSR1 with prostate cancer severity in European American and African American men. Cancer Epidemiol Biomarkers Prev. 2005;14:949–957. doi: 10.1158/1055-9965.EPI-04-0637. [DOI] [PubMed] [Google Scholar]
- 6.Mazzucchelli R, Barbisan F, Tarquini LM, Galosi AB, Stramazzotti D. Molecular mechanisms in prostate cancer. A review. Anal Quant Cytol Histol. 2004;26:127–133. [PubMed] [Google Scholar]
- 7.Prowatke I, Devens F, Brenner A, Gröne EG, Mertens D, Gröne HJ, Lichter P, Joos S. Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br J Cancer. 2007;96:82–88. doi: 10.1038/sj.bjc.6603490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berezovska OP, Glinskii AB, Yang Z, Li XM, Hoffman RM, Glinsky GV. Essential role for activation of the Polycomb group (PcG) protein chromatin silencing pathway in metastatic prostate cancer. Cell Cycle. 2005;5:1886–1901. doi: 10.4161/cc.5.16.3222. [DOI] [PubMed] [Google Scholar]
- 9.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–1671. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 10.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–1490. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]
- 11.Harnden P, Shelley MD, Coles B, Staffurth J, Mason MD. Should the Gleason grading system for prostate cancer be modified to account for high-grade tertiary components? A systematic review and meta-analysis. Lancet Oncol. 2007;8:411–419. doi: 10.1016/S1470-2045(07)70136-5. [DOI] [PubMed] [Google Scholar]
- 12.Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM, Shah RB. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–544. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- 13.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Keufer R, Lee C, Montie JM, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 14.Cerveira N, Ribeiro FR, Peixoto A, Costa V, Henrique R, Jerónimo C, Teixeira MR. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–832. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosquera JM, Perner S, Demichelis F, Kim R, Hofer MD, Mertz KD, Paris PL, Simko J, Collins C, Bismar TA, Chinnaiyan AM, Rubin MA. Morphological features of TMPRSS2-ERG gene fusion prostate cancer. J Pathol. 2007;212:91–101. doi: 10.1002/path.2154. [DOI] [PubMed] [Google Scholar]
- 16.Rinker-Schaeffer CW, Wharam JF, Simons J, Isaacs JT. Development of a high-efficiency method for gene marking of Dunning prostate cancer cell lines with the enzyme beta-galactosidase. Prostate. 1996;29:60–64. doi: 10.1002/(SICI)1097-0045(199607)29:1<60::AID-PROS9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 17.Isaacs JT. Relationship between tumor size and curability of prostatic cancer by combined chemo-hormonal therapy in rats. Cancer Res. 1989;49:6290–6294. [PubMed] [Google Scholar]
- 18.Cooke DB, Quarmby VE, Mickey DD, Isaacs JT, French FS. Oncogene expression in prostate cancer: Dunning R3327 rat dorsal prostatic adenocarcinoma system. Prostate. 1988;13:263–272. doi: 10.1002/pros.2990130402. [DOI] [PubMed] [Google Scholar]
- 19.Isaacs JT. Hormonally responsive versus unresponsive progression of prostatic cancer to antiandrogen therapy as studied with the Dunning R-3327-AT and -G rat adenocarcinomas. Cancer Res. 1982;42(12):5010–5014. [PubMed] [Google Scholar]
- 20.Isaacs JT, Heston WD, Weissman RM, Coffey DS. Animal models of the hormone-sensitive and -insensitive prostatic adenocarcinomas, Dunning R-3327-H, R-3327-HI, and R-3327-AT. Cancer Res. 1978;38:4353–4359. [PubMed] [Google Scholar]
- 21.LeRoy BE, Thudi NK, Nadella MV, Toribio RE, Tannehill-Gregg SH, van Bokhoven A, Davis D, Corn S, Rosol TJ. New bone formation and osteolysis by a metastatic, highly invasive canine prostate carcinoma xenograft. Prostate. 2006;66:1213–1222. doi: 10.1002/pros.20408. [DOI] [PubMed] [Google Scholar]
- 22.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: A compendium of old and new lines–part 1. J Urol. 2005;173:342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- 23.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: A compendium of old and new lines–part 2. J Urol. 2005;173:360–372. doi: 10.1097/01.ju.0000149989.01263.dc. [DOI] [PubMed] [Google Scholar]
- 24.Sobel RE, Wang Y, Sadar MD. Molecular analysis and characterization of PrEC, commercially available prostate epithelial cells. In Vitro Cell Dev Biol Anim. 2006;42:33–39. doi: 10.1007/s11626-006-0009-7. [DOI] [PubMed] [Google Scholar]
- 25.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 26.van Bokhoven A, Caires A, Maria MD, Schulte AP, Lucia MS, Nordeen SK, Miller GJ, Varella-Garcia M. Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. Prostate. 2003;57:226–244. doi: 10.1002/pros.10291. [DOI] [PubMed] [Google Scholar]
- 27.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3. 0:Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simin K, Hill R, Song Y, Zhang Q, Bash R, Cardiff RD, Yin C, Xiao A, McCarthy K, van Dyke T. Deciphering cancer complexities in genetically engineered mice. Cold Spring Harb Symp Quant Biol. 2005;70:283–290. doi: 10.1101/sqb.2005.70.038. [DOI] [PubMed] [Google Scholar]
- 29.Abdulkadir SA, Kim J. Genetically engineered murine models of prostate cancer: Insights into mechanisms of tumorigenesis and potential utility. Future Oncol. 2005;1:351–360. doi: 10.1517/14796694.1.3.351. [DOI] [PubMed] [Google Scholar]
- 30.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, Humphrey PA, Sundberg JP, Rozengurt N, Barrios R, Ward JM, Cardiff RD. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 31.Kasper S. Survey of genetically engineered mouse models for prostate cancer: Analyzing the molecular basis of prostate cancer development, progression, and metastasis. J Cell Biochem. 2005;94:279–297. doi: 10.1002/jcb.20339. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ, Maddison LA, Foster BA, Greenberg NM. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55:219–237. doi: 10.1002/pros.10215. [DOI] [PubMed] [Google Scholar]
- 33.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer Prostatic Dis. 1999;2(2):70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 34.Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ, Angelopoulou R, Rosen JM, Greenberg NM. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996;56:4096–4102. [PubMed] [Google Scholar]
- 35.Shen MM, Abate-Shen C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res. 2007;67:6535–6538. doi: 10.1158/0008-5472.CAN-07-1271. [DOI] [PubMed] [Google Scholar]
- 36.Carver BS, Pandolfi PP. Mouse modeling in oncologic pre-clinical and translational research. Clin Cancer Res. 2006;12:5305–5311. doi: 10.1158/1078-0432.CCR-06-0482. [DOI] [PubMed] [Google Scholar]
- 37.Liao CP, Zhong C, Saribekyan G, Bading J, Park R, Conti PS, Moats R, Berns A, Shi W, Zhou Z, Nikitin AY, Roy-Burman P. Mouse models of prostate adenocarcinoma with the capacity to monitor spontaneous carcinogenesis by bioluminescence or fluorescence. Cancer Res. 2007;67:7525–7533. doi: 10.1158/0008-5472.CAN-07-0668. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banach-Petrosky W, Jessen WJ, Ouyang X, Gao H, Rao J, Quinn J, Aronow BJ, Abate-Shen C. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Res. 2007;67:9089–9096. doi: 10.1158/0008-5472.CAN-07-2887. [DOI] [PubMed] [Google Scholar]
- 41.Gao H, Ouyang X, Banach-Petrosky WA, Shen MM, Abate-Shen C. Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Res. 2006;66:7929–7933. doi: 10.1158/0008-5472.CAN-06-1637. [DOI] [PubMed] [Google Scholar]
- 42.Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci USA. 2006;103:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogal AP, van der Meer R, Crooke PS, Abdulkadir SA. Haploinsufficient prostate tumor suppression by Nkx3.1: A role for chromatin accessibility in dosage-sensitive gene regulation. J Biol Chem. 2007;282:25790–25800. doi: 10.1074/jbc.M702438200. [DOI] [PubMed] [Google Scholar]