

Abstract
Quinazolin-4-one 1 was identified as an inhibitor of the HIF-1α transcriptional factor from a high-throughput screen. HIF-1α up-regulation is common in many cancer cells. In this paper, we describe an efficient one-pot sequential reaction for the synthesis of quinazolin-4-one 1 analogues. The structure-activity relationship (SAR) study led to the 5-fold more potent analogue, 16.
Keywords: hypoxia-inducible factor-1α, quinazolin-4-ones, parallel synthesis
Hypoxia-inducible factor (HIF-1) is a dimeric transcription factor consisting of an oxygen regulated α-component and a constitutively expressed β-component. At normal oxygen levels, HIF-1α is degraded via the pVHL-mediated ubiquitin-proteosomal pathway. Under hypoxic conditions, HIF-1α rapidly accumulates and dimerizes with HIF-1β. This heterodimer binds to the DNA hypoxia-response element (HRE) and activates a diverse array of target genes.1 This pathway is particularly relevant to the cancer field because oxygen levels in tumors are commonly lower than in the surrounding tissues. Hypoxic cells are resistant to radiation damage and their distances from blood vessels reduce the potency of anti-cancer drugs. Hypoxia additionally promotes the up-regulation of genes involved in drug resistance. HIF-1 is directly responsible for the induction of numerous genes that are present at higher levels in cancer cells, in particular VEGF. The overexpression of HIF-1 has been related to the aggressiveness and vascularity of tumors, and mortality rate in patients. Despite the introduced difficulties in treating hypoxic tumors, the hypoxic environment found in tumor cells can be exploited for targeted therapy. One strategy to achieve this involves the identification of HIF-1 inhibitors as potential anti-cancer drugs.2 We recently reported a high-throughput cell-based HIF-1 mediated β-lactamase reporter gene assay. Upon screening a library of 73,000 compounds (PubChem AID:915 (http://pubchem.ncbi.nlm.nih.gov)), several compounds were identified as novel inhibitors of the HIF-1 signaling pathway.3 Among these hits, quinazolin-4-one 1 (NCGC00056044) showed good drug-like properties and was selected for further exploration.
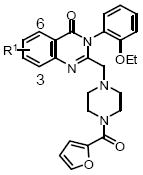
Three areas were selected for structure-activity relationship (SAR) studies: (1) substitution in area A; (2) piperazine region B; and (3) phenyl substitution in area C (Figure 1).
Figure 1.
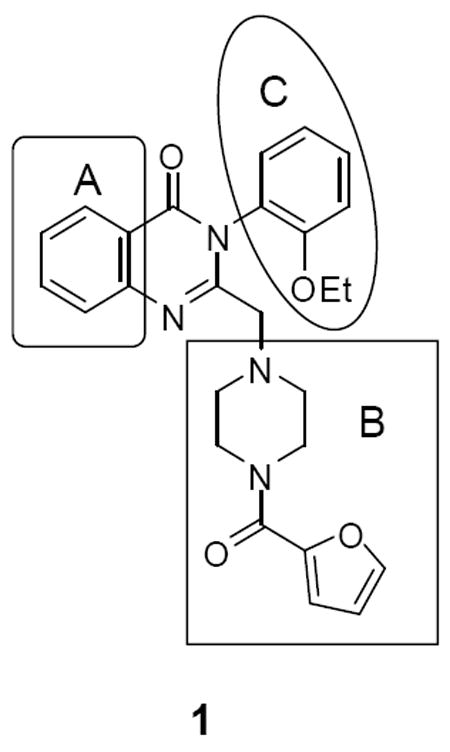
Optimization plan for NCGC00056044 (1)
To facilitate our compound synthesis for the SAR study, we modified a reported method4 to remove the need for intermediate purification. In addition, a microwave reactor was used to accelerate the synthesis. Acylation of anthranilic acid 2 with chloroacetyl chloride gave 3, which was treated with aniline 4 to afford chloride 5 (Scheme 1). The chloride was reacted with amine 6 to give compounds 1, 7-36. All three steps were conducted in one-pot without the need for intermediate isolation. This protocol was carried out in a parallel fashion to prepare the analogues which were purified via HPLC.5
Scheme 1.
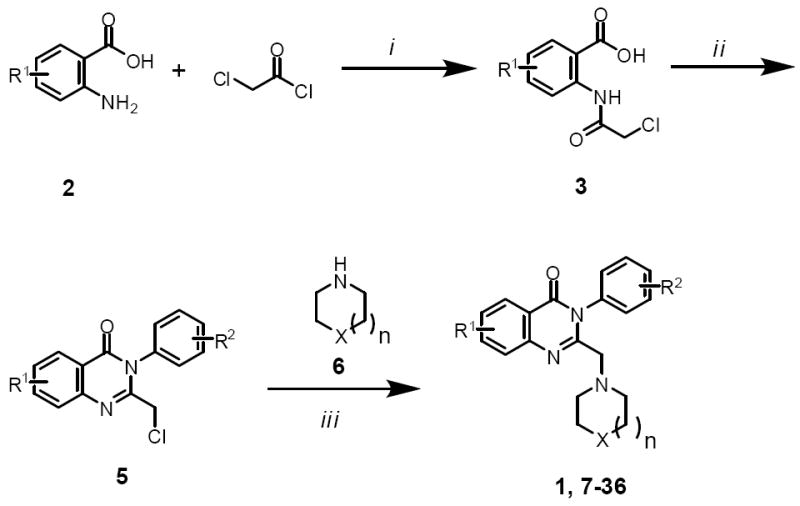
Reagents and conditions: (i) iPrNEt2, ACN, r.t.; (ii) ArNH2 (4), POCl3, MW 150 °C, 15 min; (iii) K2CO3, EtOH, MW 150 °C, 5 min; then amine 6, MW 150 °C, 10 min.
Compound 39 was prepared as described in Scheme 2. Reaction of 37 with 2-furoyl chloride, followed by a hydrolysis reaction yielded acid 38. The desired 39 was obtained via a microwave assisted one-pot three-component reaction of 38, acid 2a, and 2-ethoxyaniline.6
Scheme 2.
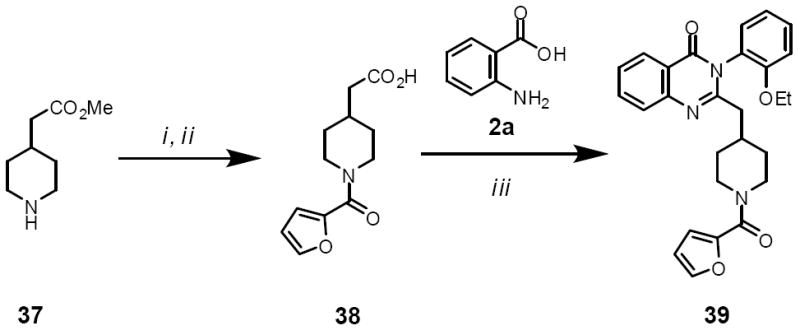
Reagents and conditions: (i) iPrNEt2, DCM, 2-furoyl chloride; (ii) LiOH; (iii) 2-ethoxyaniline, 2a, pyridine, MW 230 °C, 10 min.
Scheme 3 describes the synthesis of the area C analogue 42. Nitro-reduction of 40 gave 41. Alkylation of the aniline nitrogen in 41 using ethyl iodide followed by a Boc-deprotection gave 42.
Scheme 3.
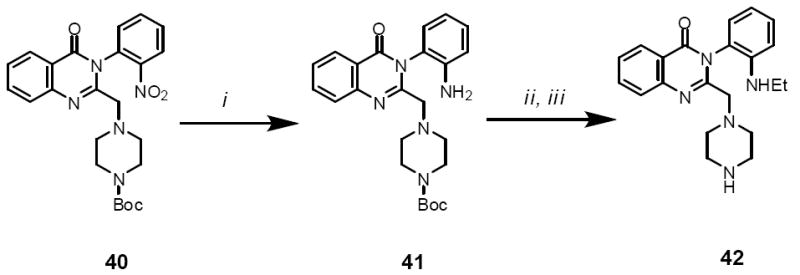
Reagents and conditions: (i) Na2S2O4; MW 100 °C, 10 min (ii) EtI, DMF, iPr2NEt, K2CO3, MW 150 °C, 15 min; (iii) DCM, TFA, r.t. 2 h
All analogues were evaluated in a cell-based HIF-1 mediated β-lactamase reporter gene assay under hypoxic conditions.7 Area A showed little tolerance for substitution (Table 1). The C-6 methoxy (7), C-5 iodo (9), and C-4 and C-5 dimethoxy (10) substitutions were inactive. Compound 8 with a methyl group at C-6 was active, but it was 3-fold less potent than the original hit (1). Considering these results, our efforts focused on the optimization of areas B and C (Figure 1).
Table 1.
Modification at the R1 position*
| Structure | Entry | Compd | R1 | 1% O2 IC50 (uM) |
|---|---|---|---|---|
|
1 | 1 | H | 0.43 |
| 2 | 7 | 6-Methoxy | inactive | |
| 3 | 8 | 6-Methyl | 1.2 | |
| 4 | 9 | 5-Iodo | inactive | |
| 5 | 10 | 4,5-Dimethoxy | inactive |
Values of IC50 are the mean of three independent experiments.
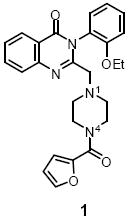
Modification of piperazine region B is shown in Table 2. Acetylation of N-4 (11) resulted in similar activity to the hit compound (1), but capping the piperazine nitrogen with a benzamide (12) or ethyl carbamate (13) resulted in a loss of activity. N-4 methylation (14) or benzylation (15) resulted in a 2-fold and 64-fold loss of activity respectively. Ultimately, the most active compound was the unsubstituted N-4 analogue (16), which was about 5-fold more potent than 1. N-4 was critical for activity because when it was replaced with either a carbon (19) or oxygen (18), activity was lost. In fact, both piperazine nitrogens were important because replacement of N-1 with a carbon (39) also resulted in a 40-fold loss of activity. Finally, the piperazine ring was expanded to homopiperazine (17) and there was a slight loss in activity relative to 16, but this analogue was still more potent than 1.
Table 2.
SAR study for the piperazine region*
| Entry | Compd | X | 1% O2 IC50 (uM) | |
|---|---|---|---|---|
|
1 | 1 |
|
0.43 |
| 2 | 11 |
|
0.47 | |
| 3 | 12 |
|
1.7 | |
| 4 | 13 |
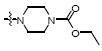
|
9.4 | |
| 5 | 14 |
|
0.81 | |
| 6 | 15 |
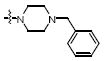
|
27.5 | |

|
7 | 39 |
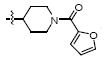
|
27.6 |
| 8 | 16 |
|
0.09 | |
| 9 | 17 |
|
0.16 | |
| 10 | 18 |
|
27.6 | |
| 11 | 19 |
|
inactive |
Values of IC50 are the mean of three independent experiments.
The modification of area C was explored in table 3. The first set of compounds was based on piperazine scaffold A (Table 3, entries 1-12) and there was almost no tolerance for substitution. The only moderately successful analogue was 2-OMe (29), but even this was 8-fold less active than 1. Scaffold B presented a greater opportunity for SAR analysis (entries 13-20). Large alkoxy groups, such as benzyloxy (33), or isobutyloxy (34) at C-2 resulted in significant loss of activity in comparison with ethoxy (16). Moving the methoxy group from the 2 to 4 position resulted in a complete loss of activity (29 vs. 36). A dramatic substitution effect was observed at the 5 position. Replacement of the nitro group (32) with a CF3 (31) resulted in more than a 20-fold improvement in potency. Finally, by comparing 35, 16, and 42, the ethoxy group appeared to be better than ethoxythio or ethylamine at the C-2 position.
Table 3.
SAR study for R2 position*
| Scaffold | Entry | Comp | Scaffold | R2 | 1% O2 IC50 (uM) |
|---|---|---|---|---|---|
| 1 | 20 | A | 4-F | inactive | |
| 2 | 21 | A | 2-tBu | inactive | |
| 3 | 22 | A | 2-NO2 | inactive | |

|
4 | 1 | A | 2-EtO | 0.43 |
| 5 | 23 | A | H | inactive | |
| 6 | 24 | A | 2-Cl | inactive | |
| 7 | 25 | A | 2-Me | inactive | |
| 8 | 26 | A | 2-Benzyloxy | inactive | |
| 9 | 27 | A | 2-F | inactive | |
| 10 | 28 | A | 2-PhO | inactive | |

|
11 | 29 | A | 2-MeO | 3.3 |
| 12 | 30 | A | 2-CF3 | inactive | |
| 13 | 31 | B | 2-MeO, 5-CF3 | 0.2 | |
| 14 | 32 | B | 2-MeO, 5-NO2 | 4.2 | |
| 15 | 42 | B | 2-EtNH | 4.1 | |
| 16 | 33 | B | 2-Benzyloxy | 22.9 | |
| 17 | 34 | B | 2-isobutoxy | 6.2 | |
| 18 | 35 | B | 2-EtS | 3.5 | |
| 19 | 36 | B | 4-MeO | inactive | |
| 20 | 16 | B | 2-EtO | 0.09 |
Values of IC50 are the mean of three independent experiments.
To confirm HIF-1α inhibition activity, compounds 18 and 16 were evaluated in a Western blot analysis.8 At 1 μM, 16 completely suppressed HIF-1α accumulation while 18 had no effect on the protein accumulation (Figure 2). This result is in agreement with the compounds’ activities observed in the cell-based assay. However, compound 18 at 10 μM also inhibited HIF-1α protein accumulation. Stockwell and coworkers reported that these quinazolin-4-ones caused rapid death of human tumor cells (BJ-TERT/LT/ST/RASV12 cells) via RAS-RAF-MEK dependent signaling.9 Because Ras, a well known oncogene, has been shown to stimulate HIF-1α expression via the Raf/Mek/ERK pathway,10 it is possible that the activity of these quinazolin-4-ones against HIF-1α accumulation is via the RAS signaling pathway.
Figure 2.
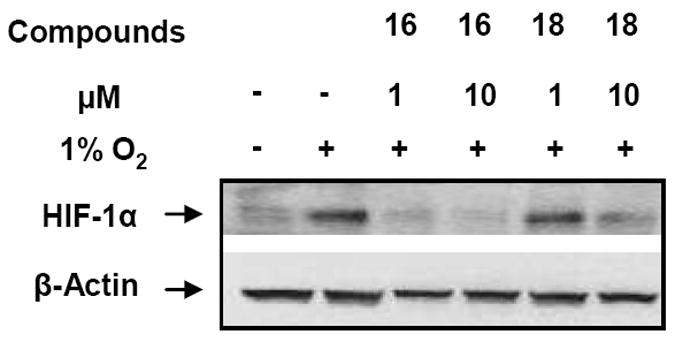
Effect of compounds 16 and 18 on the accumulation of the HIF-1α protein under hypoxia conditions
In conclusion, we have identified a series of novel quinazolin-4-one HIF-1α inhibitors. A library synthesis and SAR studies revealed analogue 16 as the new lead, which was almost 5-fold more potent than the hit (1). The inhibition of HIF-1α was further confirmed in Western blot analysis. Detailed mechanistic studies and evaluation of these compounds as anti-cancer agents in rare types of cancer are currently under investigation and will be reported in due course.
Supplementary Material
Acknowledgments
We thank Paul Shinn and Danielle Van Leer for compound management, William Leister and Jeremy Smith for analytical chemistry. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Weidemann A, Johnson RS. Cell Death Differ. 2008;15:621. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 2.(a) Yewalkar N, Deore V, Padgaonkar A, Manohar S, Sahu B, Kumar P, Jalota-Badhwar A, Joshi K, Sharma S, Kumar S. Bioorg Med Chem Lett. 2010;20:6426. doi: 10.1016/j.bmcl.2010.09.083. [DOI] [PubMed] [Google Scholar]; (b) Park S-Y, Jang W-J, Yi E-Y, Jang J-Y, Jung Y, Jeong J-W, Kim Y-J. J Pineal Res. 2010;48:178. doi: 10.1111/j.1600-079x.2009.00742.x. [DOI] [PubMed] [Google Scholar]; (c) Shimizu K, Maruyama M, Yasui Y, Minegishi H, Ban HS, Nakamura H. Bioorg Med Chem Lett. 2010;20:1453. doi: 10.1016/j.bmcl.2009.12.037. [DOI] [PubMed] [Google Scholar]; (d) Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, Van Meir EG. Clin Cancer Res. 2009;15:6128. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kasper AC, Moon EJ, Hu X, Park Y, Wooten CM, Kim H, Yang W, Dewhirst MW, Hong J. Bioorg Med Chem Lett. 2009;19:3783. doi: 10.1016/j.bmcl.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Won M-S, Im N, Park S, Boovanahalli SK, Jin Y, Jin X, Chung K-S, Kang M, Lee K, Park S-K, Kim HM, Kwon BM, Lee JJ, Lee K. Biochem Biophys Res Commun. 2009;385:16. doi: 10.1016/j.bbrc.2009.05.022. [DOI] [PubMed] [Google Scholar]; (g) Uno M, Ban HS, Nakamura H. Bioorg Med Chem Lett. 2009;19:3166. doi: 10.1016/j.bmcl.2009.04.122. [DOI] [PubMed] [Google Scholar]; (h) Diaz-Gonzalez JA, Russell J, Rouzaut A, Gil-Bazo I, Montuenga L. Cancer Biol Ther. 2005;4:1055. doi: 10.4161/cbt.4.10.2195. [DOI] [PubMed] [Google Scholar]; (i) Harada H, Hiraoka M. Curr Signal Transduction Ther. 2010;5:188. [Google Scholar]; (j) Mooring SR, Wang BH. Sci China:Chem. 2011:24. [Google Scholar]; (k) Li Y, Ye D. Curr Cancer Drug Tar. 2010;10:782. doi: 10.2174/156800910793605857. [DOI] [PubMed] [Google Scholar]; (l) Semenza G. Drug Discovery Today. 2007:19. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]; (m) DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. Cell Metab. 2008:11. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]; (n) Wang X-H, Cavell BE, Alwi SS, Packham G. Biochem Pharmacol. 2009;78:261. doi: 10.1016/j.bcp.2009.04.010. [DOI] [PubMed] [Google Scholar]; (h) Garcia-Maceira P, Mateo J. Oncogene. 2009;28:313. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]; (o) Li SH, Shin DH, Chun Y-S, Lee MK, Kim M-S, Park J-W. Mol Cancer Ther. 2008;7:3729. doi: 10.1158/1535-7163.MCT-08-0074. [DOI] [PubMed] [Google Scholar]; (p) Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, Albaek C, Schroder H, Orum H. Mol Cancer Ther. 2008;7:3598. doi: 10.1158/1535-7163.MCT-08-0510. [DOI] [PubMed] [Google Scholar]; (q) Ban HS, Yoshikazu U, Nakamura H. Expert Opin Ther Patents. 2011:131. doi: 10.1517/13543776.2011.547477. [DOI] [PubMed] [Google Scholar]
- 3.Xia M, Bi K, Huang R, Cho M-H, Sakamuru S, Miller SC, Li H, Sun Y, Printen J, Austin CP, Inglese J. Mol Cancer. 2009;8:117. doi: 10.1186/1476-4598-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becklin RR, Chepanoske CL, Pelter JM, Qi L, Robbins PB, Sahasrabudhe SR, Selliah R, Simmons K, Stockwell BR, Venkat RG, Von Rechenberg M, Zhen E. PCT Int Appl. 2006 WO2006081331. [Google Scholar]
- 5.General procedure for synthesis of compounds 1, 7-36. To a solution of anthranilic acid 2 (0.40 mmol) and Hünig’s base (0.60 mmol) in anhydrous acetonitrile (1.0 mL) was added a solution of chloroacetyl chloride (0.44 mmol) in acetonitrile (0.5 mL) at 4 °C. After stirring at r.t. for 2 h, POCl3 (112 μL, 1.2 mmol, 3.0 equiv) was added followed by a solution of aniline (0.60 mmol) in acetonitrile (0.50 mL). The resulting mixture was heated in a microwave reactor at 150 °C for 15 min, cooled to r.t. and treated with K2CO3 (300 mg) and EtOH (1.0 mL). The mixture was heated at 150 °C for 5 min. To this mixture, a solution of amine (0.44 mmol) in EtOH (0.5 mL) was added. The mixture was heated in microwave for 10 min at 150 °C and the solid was filtered. The filtrate was concentrated under reduced pressure and the residue was purified by preparative HPLC. Example: 3-(2-Ethoxyphenyl)-2-(piperazin-1-ylmethyl)quinazolin-4(3H)-one (16) 1H NMR (400 MHz, CDCl3) δ 1.22 (t, J=6.9 Hz, 3H), 2.40-2.53 (m, 2H), 2.60-2.72 (m, 2H), 2.91-3.09 (m, 4H), 3.27 (d, J=14.3 Hz, 1H), 3.34 (d, J=14.3 Hz, 1H), 3.96-4.17 (m, 2H), 6.99-7.15 (m, 2H), 7.22 (dd, J=7.6, 1.4 Hz, 1H), 7.38-7.55 (m, 2H), 7.70-7.83 (m, 2H), 8.30 (d, J=7.6 Hz, 1H); LCMS: Rt = 4.5 min, 98%; HRMS (ESI): m/z calcd for C21H24N4O2 [M+1]+ 365.1983, found 365.1982.
- 6.Liu JF, Lee JY, Dalton AM, Bi G, Yu LB, Baldino CM, McElory E, Brown E. Tetrahedron Lett. 2005:1241. [Google Scholar]
- 7.HRE β-lactamase reporter gene assay: HRE-bla ME-180 cells were dispensed at 2500 cells/6 μl/well in 1536-well black wall/clear bottom plates (Greiner Bio-One North America, Monroe, NC) using a Flying Reagent Dispenser (Aurora Discovery, CA). 23 nL of the compound were transferred to the assay plate using a pin tool (Kalypsys, San Diego, CA) resulting in a 261-fold dilution. The assay plates were incubated at 37 °C in hypoxia chamber (1% oxygen) for 17 h. After 1 μL of LiveBLAzer™ B/G FRET substrate (Invitrogen, Carlsbad, CA) mixture was added into the plates, the assay plates were incubated at room temperature for 2.5 h, and the fluorescence intensity (405 nm excitation, and 460 and 530 nm emissions) was measured by an Envision plate reader (Perkin Elmer, Shelton CT).
- 8.For western blot analysis, 1 × 106 HRE-bla ME-180 cells per well were cultured in a 6-well plate. After 6 h, the cells were treated for 17 h with DMSO in a normal atmosphere (negative control), or with 1 or 10 μM solutions of 16 or 18 in DMSO at 1% oxygen. After the treatment, the cells were lysed in lysis buffer (Invitrogen) containing 1% of protease inhibitor (Sigma) for 10 min on ice. After the samples were centrifuged for 10 min at 14000 rpm and 4 °C, supernatants were collected and subjected to SDS-PAGE analysis on a 10% Tris-Glycine gel (Invitrogen). Same amount of protein (25 ug) from each sample was loaded into the gel. Proteins were transferred to a PVDF membrane (Invitrogen) and the membrane was blotted with primary antibodies against HIF-1α at a 1:250 dilution (Santa Cruz Biotechnologies, Santa Cruz, CA) or β-Actin at 1:5000 dilution (Sigma) as a loading control. HRP conjugated secondary antibody (1:1000 dilution; anti-goat anti-rabbit or anti-mouse, Santa Cruz, CA) was used together with the Western Blot Luminol reagent kit (Santa Cruz Biotechnology, CA) to develop the membrane that was read with the ChemiDoc XRS system (Bio-Rad, Hercules, CA).
- 9.(a) Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. Nature. 2007;447:864. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang WS, Stockwell BR. Chem Biol. 2008:234. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Lim J, Lee E, You H, Lee JW, Park J, Chun J. Oncogene. 2004:9427. doi: 10.1038/sj.onc.1208003. [DOI] [PubMed] [Google Scholar]; (b) Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Cancer Res. 2005:999. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.