

Abstract
Objective
Pediatric ischemic stroke is a poorly understood, yet clinically important, problem. The sole approved treatment for acute stroke is tissue type plasminogen activator (tPA). However, tPA vasoactivity aggravates hypoxia/ischemia (H/I)-induced impairment of cerebrovasodilation in response to hypercapnia and hypotension in newborn pigs. Mitogen activated protein kinase (MAPK, a family of 3 kinases, ERK, p38 and JNK) is upregulated after H/I. Coupling of tPA to RBC prevented H/I induced impairment of dilation and suppressed ERK MAPK activation. This study investigated the differential roles of MAPK isoforms in the effects of RBC-tPA on cerebrovasodilation in a translationally relevant injury model, photothrombosis.
Design
Prospective, randomized animal study.
Setting
University laboratory.
Subjects
Newborn (1–5 day old) pigs.
Interventions
Cerebral blood flow (CBF) and pial artery diameter were determined before and after photothrombotic injury (PTI, laser 532 nm and erythrosine B) was produced in piglets equipped with a closed cranial window. CSF ERK, p38, and JNK MAPK were determined by ELISA.
Measurements and Main Results
tPA and RBC-tPA alleviated reduction of CBF after PTI. Cerebrovasodilation was blunted by PTI, reversed to vasoconstriction by tPA, but dilation was maintained by RBC-tPA. CSF JNK and p38 MAPK but not ERK MAPK were elevated by PTI, an effect potentiated by tPA. RBC-tPA blocked JNK, but potentiated p38 MAPK upregulation after PTI. A JNK MAPK antagonist prevented, a p38 MAPK antagonist potentiated, while an ERK MAPK antagonist had no effect on dilator impairment after PTI.
Conclusions
These data indicate that in addition to restoring perfusion, RBC-tPA prevents impairment of cerebrovasodilation after PTI through blockade of JNK and potentiation of p38 MAPK. These data suggest tPA coupling to RBC offers a novel approach to increase benefit/risk ratio of thrombolytic therapy to treat CNS ischemic disorders.
Keywords: plasminogen activators, cerebral hemodynamics, signal transduction, pediatric, stroke
Introduction
Pediatric stroke may occur in as many as 1 in 4000 births (1), with 30% being the result of thrombosis (2). The thrombolytic agent tissue-type plasminogen activator (tPA) remains the only approved treatment for acute stroke, but its use in children has been limited and its benefit remains unclear (3,4). Indeed, the brief therapeutic window of tPA and the high incidence of post-treatment complications, including intracranial hemorrhage (ICH), have constrained the actual clinical use of tPA to approximately 3–8% of all patients eligible for such therapy (5).
In basic science studies, tPA has been shown to increase the volume of injured tissue after stroke, as exemplified in tPA null mice, and exacerbate excitotoxic neuronal death by enhancing signaling through the N-methyl-D-Aspartate glutamate receptor via activation of matrix metalloproteinases (MMPs) (6–8). MMPs are upregulated after brain injury, in part, by activating mitogen activated protein kinase (MAPK) (8), a family of at least 3 kinases (extracellular signal-related kinase - ERK -, p38, and c-Jun-N-terminal kinase – JNK). Our recent studies show that urokinase plasminogen activator (uPA) contributes to impaired stimulus-induced cerebrovascular dilation following cerebral hypoxia/ischemia in the newborn pig through upregulation of ERK MAPK (9).
Contemporaneous studies from our group demonstrate that anchoring tPA on red blood cells (RBC) endows the resultant complex, RBC-tPA, with dramatically prolonged circulation time (many hours vs minutes for tPA), while spatially constraining it to the intravascular space (10–12). In rodent models of cerebrovascular thrombosis and traumatic brain injury, treatment with this RBC-tPA complex provided effective thromboprophylaxis, rapid reperfusion, neuroprotection, and reduction in mortality all without causing ICH (13,14). RBC-tPA also prevents impairment of cerebral vasodilatory responses and tissue injury through inhibition of ERK MAPK upregulation in a piglet model of cerebral hypoxia/ischemia (15). These studies suggest that RBC carriage may offer a unique opportunity to increase the benefit risk ratio of tPA within the CNS. However, our mechanistic studies to date have not considered the possibility that a shift in the MAPK isoform profile may ultimately link RBC-tPA to improved cerebral hemodynamics following CNS injury.
The present study was designed to investigate the differential roles of MAPK isoforms in the effects of RBC-tPA on cerebrovasodilation in a translationally relevant CNS injury model, photothrombosis.
Materials and Methods
Closed cranial window technique and cerebral photothrombosis
Newborn pigs (1–5 days, 1.0–1.6 Kg) of either sex were studied. All protocols were approved by the University of Pennsylvania IACUC. Animals were sedated with isoflurane (1–2 MAC). Anesthesia was maintained with a-chloralose (30–50 mg/kg. supplemented with 5 mg/kg/h i.v.). Catheters were inserted into femoral arteries and veins, while the trachea cannulated for ventilation with room air. The closed cranial window technique was used to measure pial artery diameter and collect CSF for ELISA analysis (9). The cranial window was placed on the side ipsilateral to the injury site.
Induction of photothrombosis was based on that described for the newborn pig (16), but in our studies, we used the area of the closed cranial window to expose two to three main and 1–3 smaller arteries supplying the MCA territory. Arterial occlusion was achieved by photothrombosis, in which a stable thrombus consisting of aggregating platelets, fibrin and other blood components is formed in response to endothelial peroxidative damage. The photochemical reaction occurs due to interaction of iv photosensitizing dye erythrosine B (20 mg/kg iv) and the focused beam of a solid state laser operated at 532 nm, powers of 200–250 mW, average intensity of 60–75 W/cm2, and durations of up to 3–5 minutes using a Snake Creek minilaser (Hallstead, PA).
Hypotension was induced by the rapid withdrawal of either 5–8 or 10–15 ml blood/Kg to induce moderate or severe hypotension (decreases in mean arterial blood pressure of 25 and 45%, respectively). Such drops in blood pressure were maintained constant for 10 min by titration of additional blood withdrawal or blood reinfusion. Two levels of hypercapnia (low and high) were induced via inhalation of graded levels of a 10% CO2-21% O2-balance N2 gas mixture for 10 min to produce levels of pCO2 of 50–60 mm Hg for the low exposure and 70–80 mm Hg for the high exposure.
Protocol
Two types of pial vessels, small arteries (resting diameter, 120–160 μm) and arterioles (resting diameter, 50–70 μm) were examined to determine whether segmental differences in the effects of photothrombosis could be identified. Fifteen experimental groups were studied (Table 1). Pre-treatment time was 30min prior to insult, while post-treatment time was 2h post insult (Fig. 1, timeline). The vehicle for all agents was 0.9% saline, except for U 0126, SB 203580, and SP 600125 which used dimethyl sulfoxide (100 μl) diluted with 9.9 ml 0.9% saline. In sham control and photothrombotic animals, responses to hypercapnia, hypotension, and isoproterenol (10−8, 10−6 M) were obtained initially and then again 1 and 2.5h later in the presence of vehicle (Fig. 1). In drug treated photothrombotic animals, drugs were administered either 30 min before or 2h after injury, and the insult protocol followed (Fig. 1). Pial artery reactivity was determined in pial small arteries and arterioles close to the area of injury (peri-ischemic area) using the closed cranial window technique (9).
Table 1.
| Group Name | Agent Used | Comparisons Made |
|---|---|---|
| 1. sham control | vehicle | CBF, ELISA, pial reactivity |
| 2. photothrombosis | vehicle pre-treated | compared between groups |
| 3. photothrombosis | tPA (2mg/kg iv), pre-treated | |
| 4. photothrombosis | RBC-tPA (0.1 mg/kg iv), pre-treated | |
| 5. photothrombosis | ERK-antagonist U 0126 (1mg/kg iv), pre-treated | |
| 6. photothrombosis | p38-antagonist SB 203580 (1 mg/kg iv), pre-treated | |
| 7. photothrombosis | JNK-antagonist SP 600125 (1 mg/kg iv), pre-treated | |
| 8. photothrombosis | JNK-antagonist, D-JNKI1 (1 mg/kg iv), pretreated | |
| 9. photothrombosis | vehicle post-treated | |
| 10. photothrombosis | tPA (2mg/kg iv), post-treated | |
| 11. photothrombosis | RBC-tPA (0.1 mg/kg iv), post-treated | |
| 12. photothrombosis | U 0126 (1mg/kg iv), post-treated | |
| 13. photothrombosis | SB 203580 (1 mg/kg iv), post-treated | |
| 14. photothrombosis | SP 600125 (1 mg/kg iv), post-treated | |
| 15. photothrombosis | D-JNKI1 (1 mg/kg iv), post-treated | |
n=5 for each group
Figure 1.
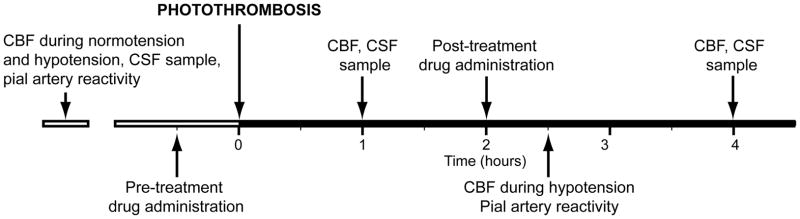
Timeline for experimental protocol.
Determination of CBF
CBF was measured in the cerebral cortex using radioactively labeled microspheres (17). Briefly, a known amount of radioactivity in 15-μm microspheres (300,000–800,000 spheres) was injected into the cardiac left ventricle and the injection line flushed with 1 ml of saline. Blood flow can be calculated as Q = C X R X CR−1, where Q is brain blood flow (in ml/min), C is counts per minute (cpm) in the tissue sample determined by a gamma counter, R is the rate of withdrawal of reference blood sample from the femoral artery (1.03 ml/min), and CR is the total counts in the reference blood sample. CBF was determined in the cerebral cortex both ipsilateral (peri-infarct area) and contralateral to the injury site.
Preparation of RBC-tPA
RBCs were isolated by centrifugation from fresh anti-coagulated (heparin, 1000U/Kg) animal blood. Biotinylated tPA was coupled to biotinylated RBC via streptavidin, producing RBC-tPA complexes possessing 5×104 tPA molecules per RBC, as described previously (10,11).
ELISA
Commercially available ELISA Kits were used to quantity CSF ERK, p38, and JNK MAPK (Assay Designs, EMD) concentration. Phosphorylated MAPK isoform enzyme values were normalized to total form and then expressed as percent of the control condition.
Statistical analysis
Pial artery diameter, CBF, CSF ERK, p38, and JNK MAPK values were analyzed using ANOVA for repeated measures. If the value was significant, the data were then analyzed by Fishers protected least significant difference test. An α level of p<0.05 was considered significant in all statistical tests. A sample size calculation determined that with an n of 5, statistical determination could be made with p<0.05 and power of 0.87. Values are represented as mean ± SEM of the absolute value or as percentage changes from control value.
Results
tPA and RBC-tPA cause comparable increase in CBF after cerebral photothrombosis
Photothrombosis reduced CBF in the peri-infact area but not in the cerebral cortex located contralateral to the injury site (Fig. 2). Animals were treated with tPA (2 mg/kg iv) or RBC-tPA (0.1 mg/kg iv) 30 min prior to photothrombosis (pre-treatment) or 2h post insult (post-treatment) and the effect on CBF was measured. In the peri-ischemic area, post-treatment with tPA increased CBF within 5 min from 32 ± 3 to 77 ± 6 ml/min. 100g while RBC-tPA elevated CBF from 30 ± 4 to 73 ± 10 ml/min. 100g (not shown in Fig. 2). Similar increases in CBF to that observed with post-injury administration were produced by tPA and RBC-tPA when these agents were delivered as a pre-treatment. In the photothrombotic vehicle treated group, CBF was unchanged (32 ± 4 and 34 ± 5 ml/min. 100g). Effects of treatment on pial artery diameter mirrored those observed for CBF above (123 ± 11 to 149 ± 13 μm for tPA, 126 ± 13 to 155 ± 14 μm for RBC-tPA, and 124 ± 12 to 125 ± 15 μm for vehicle treatment. Dilation for RBC tPA was prolonged (15–20 min) compared to tPA (5 min). RBC carriage enhanced the potency of tPA induced reperfusion approximately 10- fold based on the dose administered. However, the reperfusion produced by tPA was not of long duration, since CBF in this animal treatment group showed significant hypoperfusion at 4h post injury, whereas the perfusion afforded by RBC-tPA persisted (Fig. 2). Further, only RBC-tPA improved reperfusion at 1h post injury when administered pre-treatment.
Figure 2.

Effect of photothrombosis (PTI) on CBF (ml/min.100g) during normotension at 1 and 4h post insult and hypotension at 2.5 hr post insult in the ipsilateral and contralateral cortex in the absence and presence of tPA (2mg/kg iv), RBC-tPA (0.1 mg/kg iv), U 0126, SB 203580, SP 600125, or D-JNKI1 (all 1 mg/kg iv), n=5. Drugs are given either 30 min prior to or 2h after photothrombosis, designated as pre and post treatment (30 min prior or 2h post insult). *p<0.05 versus corresponding 0 time value +p<0.05 versus corresponding PTI alone value #p<0.05 versus corresponding PTI alone normotension value.
RBC-tPA prevents, whereas tPA aggravates, photothrombosis induced impairment of hypotensive and hypercapnic cerebrovasodilation
Hypotension, hypercapnia, and isoproterenol elicited reproducible dilation of pial small arteries and arterioles. Dilation of small pial arteries in response to hypotension and hypercapnia was blunted after photothrombosis, and further reversed to vasoconstriction in pigs treated before (30 min) or after (2h) insult with tPA (2 mg/kg iv) (Fig. 3,4). In contrast, dilation was maintained at levels nearly equivalent to pre-photothombosis values in animals given RBC-tPA (0.1 mg/kg iv) pre- or post-insult (Fig. 3,4). CBF was decreased during hypotension in vehicle photothrombotic animals (Fig. 2), indicating disturbed autoregulation. Pre and post-treatment with tPA further decreased CBF during hypotension (Fig. 2). In contrast, RBC-tPA pre- and post-treatment prevented the fall in CBF observed during hypotension (Fig. 2), indicating preservation of cerebral autoregulation. Thus, RBC-tPA and tPA caused opposite effects on pial artery vasoreactivity impairment of hypercapnia and hypotension. Vasodilation of small pial arteries in response to isoproterenol was unaffected by photothrombosis, tPA, and RBC-tPA (Fig. 5). Similar observations were made in pial arterioles.
Figure 3.
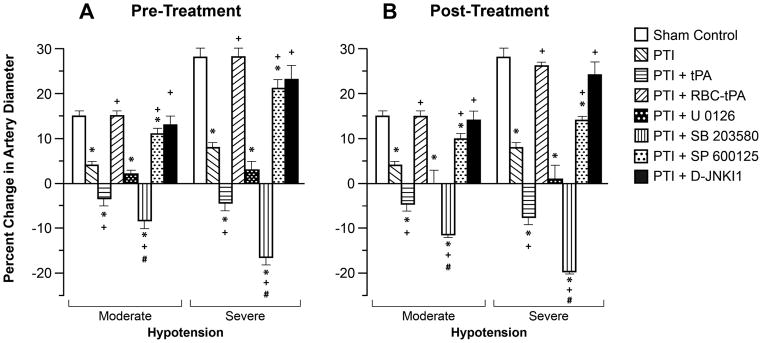
Influence of hypotension (moderate, severe) on pial artery diameter in newborn pigs before (control), after photothrombotic injury (PTI), or treated with tPA (2 mg/kg iv), RBC-tPA (0.1 mg/kg iv), U 0126, SB 203580, SP 600125 or D-JNKI1 (all 1 mg/kg iv), n=5 A: pretreatment 30 before PTI B: post treatment 2h after PTI *P<0.05 versus corresponding control value. Measurements were made at 2.5 h post PTI. +P<0.05 versus corresponding non treated PTI value #p<0.05 versus corresponding PTI +tPA value.
Figure 4.
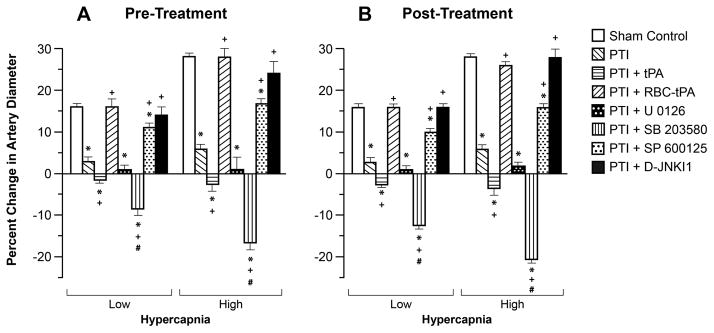
Influence of hypercapnia (Low, High) on pial artery diameter in newborn pigs before (control), after photothrombotic injury (PTI), or treated with tPA (2 mg/kg iv), RBC-tPA (0.1 mg/kg iv), U 0126, SB 203580, SP 600125 or D-JNKI1 (all 1 mg/kg iv), n=5 A: pretreatment 30 before PTI B: post treatment 2h after PTI. Measurements were made at 2.5 h post PTI. *p<0.05 versus corresponding control value +p<0.05 versus corresponding non treated PTI value #p<0.05 versus corresponding PTI +tPA value.
Figure 5.
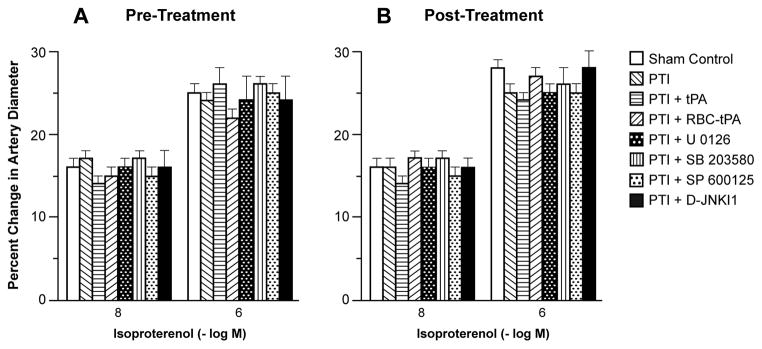
Influence of isoproterenol (10−8, 10−6 M) on pial artery diameter in newborn pigs before (control), after photothrombotic injury (PTI), or treated with tPA (2 mg/kg iv), RBC-tPA (0.1 mg/kg iv), U 0126, SB 203580, SP 600125 or D-JNKI1 (all 1 mg/kg iv), n=5 A: pretreatment 30 before PTI B: post treatment 2h after PTI. Measurements were made at 2.5 h post PTI.
RBC-tPA blunts, whereas tPA augments, photothrombosis induced phosphorylation of JNK MAPK
We next investigated the mechanism by which RBC-tPA preserved vasodilation beginning with its effect on phosphorylation of CSF JNK MAPK after injury. The activation (phosphorylation) state of the JNK MAPK isoform was determined by expressing the data as a percent of control (total). Photothrombosis induced a marked phosphorylation of JNK MAPK within 1h post injury (Fig. 6). Exogenous tPA administered 30 min prior to, or 2h after, photothrombosis potentiated the phosphorylation of JNK MAPK (Fig. 6). In contrast, administration of RBC-tPA pre- or post-injury blunted insult-induced phosphorylation of CSF JNK MAPK. Notably, RBC-tPA not only blocked the potentiation of CSF JNK MAPK release observed with tPA, but almost completely restored the values to those measured under sham control conditions (Fig. 6). SP 600125 and D-JNKI1 (1mg/kg iv), purported JNK MAPK antagonists, blocked JNK MAPK phosophorylation, (Fig. 6), while having no effect on p38 MAPK (Fig. 7) or ERK MAPK (data not shown). Both SP 600125 and D-JNKI1 pre-and post-treatment prevented reductions in CBF post injury both in the absence and presence of hypotension, thereby preserving autoregulation (Fig. 2). Similarly, SP 600125 and D-JNKI1 prevented impairment of hypotensive and hypercapnic pial artery dilation post insult (Fig. 3,4), while having no effect on isoproterenol-induced vasodilation (Fig. 5).
Figure 6.
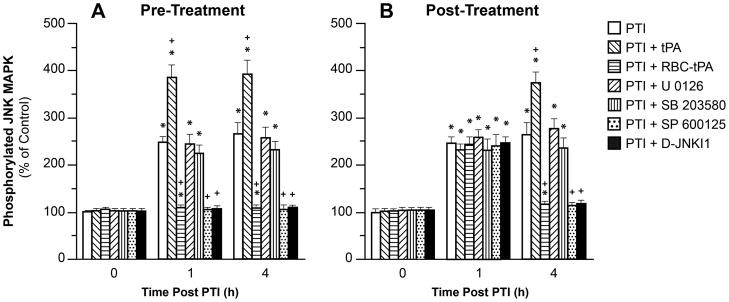
Phosphorylation of JNK MAPK in CSF prior to photothrombosis (PTI) (0 min) and as a function of time (hour) after PTI in vehicle, or treated with tPA (2 mg/kg iv), RBC-tPA (0.1 mg/Kg iv), U 0126, SB 203580, SP 600125 or D-JNKI1 (all 1 mg/kg iv), n=5. Data expressed as percent of control by ELISA determination of phospho MAPK and total MAPK isoforms and subsequent normalization to total form. A: pretreatment 30 min before H/I B: post treatment 2h after H/I. *p<0.05 versus corresponding 0 time value +p<0.05 versus corresponding PTI non treated value.
Figure 7.
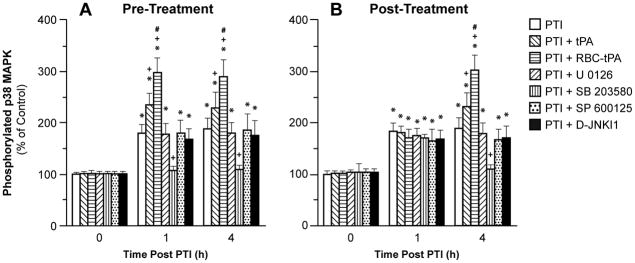
Phosphorylation of p38 MAPK in CSF prior to photothrombosis (PTI) (0 min) and as a function of time (hour) after PTI in vehicle, or treated with tPA (2 mg/kg iv), RBC-tPA (0.1 mg/Kg iv), U 0126, SB 203580, SP 600125 or D-JNKI1 (all 1 mg/kg iv), n=5. Data expressed as percent of control by ELISA determination of phospho MAPK and total MAPK isoforms and subsequent normalization to total form. A: pretreatment 30 min before H/I B: post treatment 2h after H/I. *p<0.05 versus corresponding 0 time value +p<0.05 versus corresponding PTI non treated value #p<0.05 versus corresponding PTI + tPA value.
Photothrombosis increases p38 MAPK, which is potentiated by tPA and RBC-tPA
Photothrombosis elevated CSF p38 MAPK, which was potentiated slightly by tPA, and more potently by RBC-tPA (Fig. 7). SB 203580 (1 mg/kg iv), a p38 MAPK antagonist, blocked injury induced elevation of CSF p38 MAPK (Fig. 7), while having no effect on JNK MAPK (Fig. 6) or ERK MAPK (data not shown). Pre- and post-treatment with SB 203580 potentiated reductions in CBF after photothrombosis (Fig. 2) as well as impairment of hypotensive and hypercapnic pial artery dilation (Fig. 3,4), while having no effect on isoproterenol-induced pial artery dilation (Fig. 5).
Photothrombosis has no effect on ERK MAPK
CSF ERK MAPK was unchanged by photothrombosis. Pre- and post-treatment with the ERK MAPK antagonist U 0126 (1 mg/kg iv) had no influence on CBF, isoproterenol dilation, or impairment of hypotensive and hypercapnic pial artery dilation (Fig. 2–5).
MAPK antagonists have no effect on CBF in absence of photothrombosis
U 0126, SB 203580, SP 600125 and D-JNKI1 all had no effect on CBF in either the ipsilateral or contralateral cortex in the absence of photothrombosis (eg 62 ± 8 and 66 ± 10 ml/min. 100g before and after U 0126 in the contralateral cortex).
Blood Chemistry
Blood chemistry values were collected before and after all experiments. There were no statistically significant differences among groups. Low levels of hypercapnia raised pCO2 to 58 ± 8 and high levels of hypercapnia raised pCO2 to 78 ± 9 mm Hg. Oxygen levels were kept constant during periods of hypercapnia.
Discussion
The data in this paper show that coupling of tPA to carrier RBCs potentiates its ability to improve post-thrombotic reperfusion when given post-injury and endows it with ability to improve reperfusion when given prior to injury, features lacking in free tPA. Further, RBC-tPA, not free tPA, protects against impaired cerebrovasodilation to hypercapnia and hypotension caused by thrombosis, in a setting when tPA is given clinically, eg thrombotic stroke.
In prior studies, we observed that pial artery dilation in response to these two stimuli was blunted following global cerebral hypoxia/ischemia, reversed to pial artery vasoconstriction with administration of tPA prior to the injury, and the vasoconstrictor blocked and the vasodilation allowed to occur with the PAI-1 inhibitor, EEIIMD and RBC-tPA (15,18). These data indicate that coupling tPA to RBC enhances its reperfusing potency and pro-vasodilation effect in the piglet CNS exposed to focal and global ischemia, likely due to prolongation of the half-life in the circulation and retention in the intravascular compartment (15). RBC carriage enhanced the potency of tPA induced vasodilation approximately 10- fold based on the dose administered.
The protective effect on stimulus-induced vasodilation provided by RBC-tPA appears selective for several reasons. First, RBC without tPA did not cause vasodilation nor did it prevent impairment in dilator responses after global cerebral hypoxia/ischemia (15). Second, photothrombosis does not exert a non-specific global impairment since CBF was unchanged contralateral to the injury site and responses to isoproterenol were unchanged post insult. Third, RBC-tPA did not alter the dilation in animals given isoproterenol, excluding a non-specific enhancement of cerebrovasodilator responsiveness. The observation that RBC-tPA preserves cerebrovasodilation whether given pre- or post-photothrombosis suggests its utility both as a treatment for cerebral ischemia and a means to prevent delayed injury in settings where the risk of recurrent ischemic occlusion is high.
Blockade of JNK MAPK and potentiation of p38 MAPK upregulation preserved vasodilation in animals given RBC-tPA. These data suggest that JNK MAPK upregulation is deleterious while p38 MAPK is protective in the setting of focal thrombotic ischemia. Interestingly, CSF ERK MAPK was unchanged and U 0126 was not protective after photothrombosis, in contrast to global cerebral hypoxia/ischemia (15). These data indicate that global and focal ischemia utilize distinct signaling pathways to impair cerebral hemodynamics. Because JNK MAPK antagonists blocked JNK upregulation post injury while having no effect on p38 or ERK, these data, conducted as part of a cross-selectivity paradigm, support the efficacy and specificity of these agents as probes for the role of JNK MAPK in hemodynamic effects of photothrombosis. These data extend previous findings which indicate that upregulation of JNK MAPK contributes to neuronal cell death in a rodent model of focal middle cerebral artery occlusion (19), but are the first to investigate the role of this MAPK isoform in impaired cerebral hemodynamics. Potential cellular sources for MAPK detected in CSF include neurons, glia, vascular smooth muscle and endothelial cells. The experimental design of these studies, however, does not permit us to identify the cellular site of origin.
Many studies of cerebral ischemia have been performed in rodent models. Piglets offer a unique advantage in elucidating pathways involved in CNS ischemic injury by virtue of having a gyrencepahalic brain that contains substantial white matter similar to humans, which is more sensitive to ischemic damage than grey matter (20). On the basis of interspecies extrapolation of brain growth curves (21), the age of the newborn pig used in these studies roughly approximated the newborn-infant time period in the human. Although the incidence of cerebral ischemic events in the pediatric population is relatively low compared to the adult (4), the magnitude of the condition is amplified by the potential long-term loss of quality of life years for children with CNS ischemic disorders. Many individual children, however, are receiving tPA based on the assumption that studies in adults are generalizable to children. The data from this study provide additional evidence that both safety and efficacy of tPA must be evaluated systematically in children before being widely adopted in clinical care. A potential limitation of the present study is the non consideration that safety and efficacy could equally be different in neonates and older children, indicating the need for scientific investigation using different maturational ages of pigs and/or other species.
Other studies of the delivery of plasminiogen activators by carrier RBCs, first published in 2003 (10) provide a strong logical framework for explanation of the obtained results and evaluation of the translational outlook of this novel drug delivery strategy. A unique protective effect of RBC-tPA observed in this study most likely arose from both prolonged duration RBC-tPA that may facilitate thrombolytic reperfusion and spatial constraint may contribute to the unique protective effects of RBC-tPA. Our present studies show that anchoring tPA on RBCs does not affect its enzymatic activity (11). However data on biodistribution of radiolabeled RBC-tPA in animals (10) and in vitro studies of accessibility of RBC anchored tPA to vascular cells (22) indicate that anchoring tPA on RBCs does constrain it to remain with the vasculature and inhibiting its ability to interact with vascular receptors. These factors provide a longer time for its vascular effect to be observed with lesser side effects. In particular, it is conceivable that differences in MAPK isoform upregulation between tPA and RBC-tPA do relate to the latter being spatially constrained and therefore unable to diffuse into the brain tissue from the vasculature. We think that different MAPK isoforms are located within different cell types, eg vascular smooth muscle, neurons, glia, and endothelial cells. From the translational standpoint, it is encouraging that most recent studies from our group indicate that recombinant proteins fusing mutant plasminogen activators with single chain fragments of antibodies safely anchoring to RBC allow to form RBC-PA complexes in the bloodstream by a simple IV injection of the fusion (23,24). This new approach will support wider medical use of RBC carried plasminogen activators without the need for ex vivo coupling of tPA to isolate RBCs and reinfusion, as utilized in this study.
In summary, our results indicate that RBC-tPA prevents impairment of cerebrovasodilation after photothrombosis through blockade of JNK and potentiation of p38 MAPK-dependent mechanisms. These data suggest that RBC coupling to tPA may offer a novel approach to increase the benefit/risk ratio of thrombolytic therapy to treat CNS ischemic disorders.
Acknowledgments
Sources of Financial Support: This research was supported by grants from the National Institutes of Health, NS53410 and HD57355 (WMA), HL76406, CA83121, HL76206, HL07971, and HL81864 (DBC), HL77760 and HL82545 (AARH), HL66442 and HL090697 (VRM), the University of Pennsylvania Research Foundation (WMA), the University of Pennsylvania Institute for Translational Medicine and Therapeutics (DBC), and the Israeli Science Foundation (AARH).
References
- 1.Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–158. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- 2.DeVeber G, Andrew M. Cerebral sinovenous thrombosis in children. N Engl J Med. 2001;345:417–423. doi: 10.1056/NEJM200108093450604. [DOI] [PubMed] [Google Scholar]
- 3.Benedict SL, Ni OK, Schloesser P, et al. Intra-arterial thrombolysis in a 2-year-old with cardioembolic stroke. J Child Neurol. 2007;22:225–227. doi: 10.1177/0883073807300296. [DOI] [PubMed] [Google Scholar]
- 4.Janjua N, Nasar A, Lynch JK, et al. Thrombolysis for ischemic stroke in children. Data from the nationwide inpatient sample. Stroke. 2007;38:1850–1854. doi: 10.1161/STROKEAHA.106.473983. [DOI] [PubMed] [Google Scholar]
- 5.Lapchak PA. Hemorrhagic transformation following ischemic stroke: significance, causes, and relationship to therapy and treatment. Curr Neurol Neurosci Rep. 2002;2:1–6. doi: 10.1007/s11910-002-0051-0. [DOI] [PubMed] [Google Scholar]
- 6.Wang YF, Tsirka SE, Strickland S, et al. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 7.Nicole O, Docagne F, Ali C, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nature Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Mori T, Jung JC, et al. Secretion of matrix metalloproteinase-2–9 after mechanical trauma injury in rat cortical cultures and involvement of MAP kinase. J Neurotruama. 2002;19:615–625. doi: 10.1089/089771502753754082. [DOI] [PubMed] [Google Scholar]
- 9.Armstead WM, Cines DB, Bdeir K, et al. uPA impairs cerebrovasodilation after hypoxia-ischemia through LRP and ERK MAPK. Brain Res. 2008;1231:121–131. doi: 10.1016/j.brainres.2008.06.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murciano JC, Medinilla S, Eslin D, et al. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat Biotechnol. 2003;21:891–896. doi: 10.1038/nbt846. [DOI] [PubMed] [Google Scholar]
- 11.Ganguly K, Krasik T, Medinilla S, et al. Blood clearance and activity of erythrocyte-coupled fibrinolytics. J Pharm Exp Ther. 2005;312:1106–1113. doi: 10.1124/jpet.104.075770. [DOI] [PubMed] [Google Scholar]
- 12.Zaitsev S, Danielyan K, Murciano JC, et al. CR-1-directed targeting of tPA to circulating erythrocytes for prophylactic fibrinolysis. Blood. 2006;108:1895–1902. doi: 10.1182/blood-2005-11-012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danielyan K, Ganguly K, Ding B, et al. Cerebrovascular thromboprophylaxis by erythrocyte coupled tPA. Circulation. 2008;118:1442–1449. doi: 10.1161/CIRCULATIONAHA.107.750257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein SC, Ganguly K, Belfeld CM, et al. Erthrocyte bound tissue plasminogen activator (tPA) is neuroprotective in experimental traumatic brain injury. J Neurotrauma. 2009;26:1585–1592. doi: 10.1089/neu.2008.0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstead WM, Ganguly K, Kiessling JW, et al. RBC-coupled tPA prevents impairment of cerebral vasodilatory responses and tissue injury in pediatric cerebral hypoxia/ischemia through inhibition of ERK MAPK. J Cereb Blood Flow Metab. 2009;29:1463–1474. doi: 10.1038/jcbfm.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuluz JW, Prado R, He D, et al. New pediatric model of ischemic stroke in infant piglets by photothrombosis. Stroke. 2007;38:1932–1937. doi: 10.1161/STROKEAHA.106.475244. [DOI] [PubMed] [Google Scholar]
- 17.Leffler CW, Busija DW, Mirro R, et al. Effects of ischemia on brain blood flow and oxygen consumption of newborn pigs. Am J Physiol. 1989;257:H1917–1926. doi: 10.1152/ajpheart.1989.257.6.H1917. [DOI] [PubMed] [Google Scholar]
- 18.Armstead WM, Cines DB, Higazi AA. Plasminogen activators contribute to impairment of hypercapnic and hypotensive cerebrovasodilation after cerebral hypoxia/ischemia in the newborn pig. Stroke. 2005;36:2265–2269. doi: 10.1161/01.STR.0000181078.74698.b0. [DOI] [PubMed] [Google Scholar]
- 19.Borsello T, Clarke PGH, Hirt L, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 20.Shaver EG, Duhaime AC, Curtis M, et al. Experimental acute subdural hematoma in infant piglets. Pediatric Neurosurgery. 1996;125:123–129. doi: 10.1159/000121109. [DOI] [PubMed] [Google Scholar]
- 21.Dobbing J. The later growth of the brain and its vulnerability. Pediatrics. 1974;53:2–6. [PubMed] [Google Scholar]
- 22.Murciano JC, Higazi AA, Cines DB, Muzykantov VR. Soluble urokinase receptor conjugated to carrier red blood cells binds latent pro-urokinase and alters its functional profile. J of Controlled Release. 2009;139:190–196. doi: 10.1016/j.jconrel.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaitsev S, Spitzer D, Murciano JC, et al. Targeting of a mutant plasminogen activator to circulating red blood cells for prophylactic fibrinolysis. J Pharm Exp Ther. 2010;332:1022–1031. doi: 10.1124/jpet.109.159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaitsev S, Spitzer D, Murciano JC, et al. Sustained thromboprophylaxis mediated by an RBC-targeted pro-urokinase zymogen activated at the site of clot formation. Blood. 2010;115:5241–5248. doi: 10.1182/blood-2010-01-261610. [DOI] [PMC free article] [PubMed] [Google Scholar]