

Abstract
Bacteria are recognized as important drivers of biogeochemical processes in all aquatic ecosystems. Temporal and geographical patterns in ocean bacterial communities have been observed in many studies, but the temporal and spatial patterns in the bacterial communities from the South China Sea remained unexplored. To determine the spatiotemporal patterns, we generated 16S rRNA datasets for 15 samples collected from the five regularly distributed sites of the South China Sea in three seasons (spring, summer, winter). A total of 491 representative sequences were analyzed by MOTHUR, yielding 282 operational taxonomic units (OTUs) grouped at 97% stringency. Significant temporal variations of bacterial diversity were observed. Richness and diversity indices indicated that summer samples were the most diverse. The main bacterial group in spring and summer samples was Alphaproteobacteria, followed by Cyanobacteria and Gammaproteobacteria, whereas Cyanobacteria dominated the winter samples. Spatial patterns in the samples were observed that samples collected from the coastal (D151, D221) waters and offshore (D157, D1512, D224) waters clustered separately, the coastal samples harbored more diverse bacterial communities. However, the temporal pattern of the coastal site D151 was contrary to that of the coastal site D221. The LIBSHUFF statistics revealed noticeable differences among the spring, summer and winter libraries collected at five sites. The UPGMA tree showed there were temporal and spatial heterogeneity of bacterial community composition in coastal waters of the South China Sea. The water salinity (P=0.001) contributed significantly to the bacteria-environment relationship. Our results revealed that bacterial community structures were influenced by environmental factors and community-level changes in 16S-based diversity were better explained by spatial patterns than by temporal patterns.
Introduction
Bacteria are recognized as important agents in nutrient cycles and considered to be releasing inorganic matter through the decomposition of organic matter, thereby recycling nutrients to the phytoplankton [1–7]. Studies of bacteria have disclosed that marine bacterial populations are complex, widespread and often consisting of unidentified or uncultivated members [8–11]. Advances in molecular techniques and ecological genomics have greatly improved our understanding of the processes mediated by bacteria in the marine environments, including marine sediments, oligotrophic open sea, coastal temperate [12]. Although large populations of bacteria are well documented in coastal waters, their diversity and spatiotemporal variations remain largely unexplored.
As one of the most variable marine habitats, coastal waters are generally characterized by a high biodiversity and high primary production because these waters contain significant bacterial and nutritional inputs from terrestrial sources [13]. Numerous environmental factors have been suggested to influence the bacterial diversity (e.g. salinity, temperature and nutrients) [14]. Because of environmental heterogeneity, seasonal currents, anthropogenic effects, the coastal waters could harbor high bacterial diversity in response to geochemical and eutrophication gradients [15]. Long-term studies in the coastal ocean showed the robust seasonal patterns in species richness [16]. Marine bacteria demonstrate seasonal patterns in diversity with, generally, higher diversity during the winter than the summer in pelagic ecosystems [17]. It is possible that the ability of bacteria respond to seasonal variations could allow bacteria to respond to changing environmental conditions. Recently, the spatiotemporal profiles from ten samples of microbes in the coastal sediment of the South China Sea (SCS) were examined, showing that the microbial community structure was correlated with spatiotemporal variation [18].
The SCS is one of the largest marginal seas in the tropical Pacific Ocean, covers an area approximately 3,500,000 km2. It has a remarkable amount of biological diversity, including over 30% of the world’s coral reefs and many valuable fisheries [19]. With the rapid development of the tourism and the sudden increase in the population in the past 10 years, the coastal regions of the SCS are facing many ecological problems. However, to date, studies that describe the structure and composition of the bacterial communities in the coastal waters of the SCS are still limited due to the highly variable physical and biogeochemical conditions.
In this study, we aimed at exploring the composition of bacterial communities in the coastal waters of the SCS during different seasons using 16S rRNA gene sequences. Our primary focus was to find temporal and spatial patterns of bacterial communities and to gain an overall understanding of the bacterial diversity in this marine system. The results considerably extend our knowledge of the variations of bacterial communities responding to spatiotemporal variations. To our knowledge, this is the first comparison of the bacterial communities among different seasons in the coastal waters of the SCS by means of 16S rDNA sequences analysis.
Materials and Methods
Ethics Statement
No specific permits were required for the described field studies. The South China Sea Institute of Oceanology and Chinese Academy of Sciences issued the permission for each location. The location is not privately owned. The field studies did not involve endangered or protected species.
Sampling Sites and sample collection
Five sampling sites (D151, D157, D1512, D221, D224) were selected from the Hainan Island coastal area of the SCS (Figure 1). Water samples were collected from the five sites during summer (May) of 2006, spring (January) and winter (October) of 2007 by the Department of Guangzhou Marine Geological Survey. In total, we obtained 15 seawater samples. The characters used in the sample names indicate sampling seasons (SP, spring; S, summer; W, winter).
Figure 1.
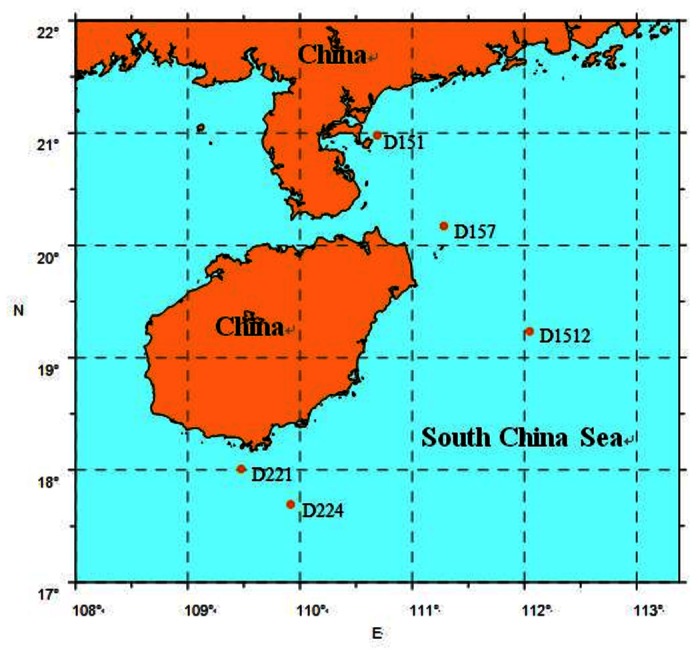
Map showing the sampling stations in the South China Sea.
For each sample, 10 L of seawater were collected from surface (0-5 m) and pre-filtered through a 0.8-µm filter to separate free-living microorganisms. The filtrates were passed through a 0.2-µm filter. Then the 0.2-µm filter was immediately stored in sterile bags and frozen at -20°C before being processed. DNA samples were extracted under sterile conditions and to avoid cross contamination the manipulation of each sample was performed separately.
Environmental parameters
Chemical data were determined in triplicates by standard oceanographic methods. Ambient water temperature and salinity was determined at the moment of sample collection using a hand-held refractometer (Leica). Other parameters were measured in the laboratory. Total organic carbon (TOC), total nitrogen (TN) was determined in accordance with the methods described by Grasshoff et al [20].
DNA extraction and 16S rRNA gene amplification by PCR
Total genomic DNA was extracted from the filters using the bacteria genomic extraction kit (TianGen, Beijing) according to the manufacturer’s instructions. Genomic DNA of each sample was extracted induplicate. The 16S rRNA genes were amplified from the DNA templates by PCR with the universal primers Uni515F (5’ –GTGYCAGCMGCCGCGCGGTAA-3’) and Uni1406R (5’-TGACGGGCGGTGTGTRCAA-3’) [18]. PCR was performed in 25 µL reaction mixtures (1 × PCR buffer, 0.5mM dNTP, 1.25 U LA Taq polymerase [Takara], 1 ng of each primer and 100 ng of DNA template). To screen for potential contamination of PCR reagents, a negative PCR control using H2O instead of a DNA template was used. PCR amplification began with a 5 min denaturing step at 94 °C; followed by 26 cycles of 94°C 45 s, 50°C 45 s, 72°C 2 min; The final cycle was an extension at 72°C for 10 min. PCR products with expected sizes were excised from agarose gel after electrophoresis. The products were purified using EZNA Cycle-Pure Kits (Omega). The purity of the PCR amplifications was assessed by 1% (w/v) agarose gel electrophoresis.
Construction of 16S rRNA gene clone libraries and amplified ribosomal DNA restriction analysis (ARDRA)
PCR products were cloned into the PMD20-T vectors as described by the manufacturer (Invitrogen, CA), and then transformed into chemically competent E. coli DH5α. Positive colonies for the blue-white colony screen used for the vector were picked. Approximately 100 white clones for each library were randomly selected for the 16S rRNA-ARDRA assay. The inserted 16S rDNA sequences were amplified by PCR with M13+/- primers. The 25 µL PCR reaction mixture contained 2.5 µL 10 × PCR buffer, 0.5 µL dNTP (2.5 mM), 1.25 U LA Taq polymerase, 0.5 µL each primer (10 µM) and 2 µL recombinant plasmid from each white clone. PCR amplification began with a 5 min denaturing at 94°C, and followed by 30 cycles of 94°C 30 s, 55°C 30 s, and 72°C 90 s; The final cycle was an extension at 72°C for 10 min. The resulting PCR products were further used for ARDRA analysis. All clones with both the same HaeⅢ and the same TaqⅠrestriction patterns were assigned to one ARDRA pattern [21].
Sequencing and Phylogenetic Analyses
Representative clone of each ARDRA pattern was sequenced (Sangon Biological Engineering Technology and Services, China). All sequences obtained were edited to exclude the vector and the primer sequences, and checked for chimerical structures using the CHECK-CHIMERA program on the Ribosomal Database Project [22]. All sequences that were free of chimeras were compared with those in the GenBank database (http://www.ncbi.nlm.nih.gov/BLAST/) and the Ribosomal Database Project II (http://rdp.cme.msu.edu) using the Basic Local Alignment Search Tool (BLAST) to identify known sequences with a high degree of similarity. Meanwhile, the sequences were clustered as OTUs at an overlap identity cutoff of 97% by MOTHUR software [23]. One representative sequence from each dominant OTU was manually complied and aligned with their closest neighbors from GenBank database using Clustal X [24]. Phylogenetic tree including the representative sequences and their closest neighbors was constructed by neighbor-joining algorithm based on Jukes-Cantor-corrected distances in MEGA 4.0 [25]. The branches of the resultant tree were evaluated by bootstrap analysis based on 1000 replicates.
Statistical methods for community analysis
Based on the result of ARDRA analysis, the coverage of the constructed 16S rRNA gene libraries were calculated using C = [1-(n/N)] according to Good (1953) [26], where n is the number of sequence types that occur only once in the library and N is the total number of clones examined. All the sequenced clones from the libraries were used for further analysis. Rarefaction analysis was conducted using MOTHUR program, Chaol richness estimates and Shannon-Weaver diversity index were calculated to further assess bacterial diversity and richness. The LIBSHUFF analysis was performed for pair-wise comparisons in each library to determine the significance of differences between clone libraries using the LIBSHUFF function available in MOTHUR, and P value was estimated by 10, 000 random permutations of sequences between libraries.
Bacterial community similarity analyses were conducted by the unweighted pair group method with arithmetic (UPGMA) algorithm according to the Yue & Clayton theta structural diversity measure using the MOTHUR software at OTU definition of 0.03 [23], which measures the molecular evolutionary distances of the sequences and can compare the evolutionary relationships among microbial communities that exist in different environments.
Correlations between bacterial communities and environmental factors were analyzed using the redundancy analysis (RDA) with the R package vegan [27]. The dominant OTUs were used as species input, and the environmental variables entered into the RDA were normalized (z-score transformation) [28]. Automatic forward selection with significant tests of Monte Carlo permutations were used to build the optimal models of bacteria-environment relationship (999 permutations) [27].
Nucleotide sequence accession numbers
The nucleotide sequences determined in this study have been deposited at GenBank under accession numbers: EU181973-EU182214.
Results
Environmental parameters of the study sites
Abiotic parameters for each sampling site are shown in Table 1. Temperature varied from 15.1 to 28.5. The low salinity found at D151 site and D221 site is explained by the input of freshwater from terrestrial sources (P<0.01). The water geochemical parameters varied greatly between samples. For further analysis, the D151 site and D221 site were considered as coastal environments, and the D157 site, D1512 site and D224 site as offshore environments. Analysis of geochemical content showed the highest TOC and TN concentration at the coastal site D151, D221, which indicated a high input of organic carbon and the associated particles from terrestrial environment.
Table 1. Description and geochemical characteristics of the sampling stations.
| Sample | Location(E, N) | Temp (°C) | Salinity (‰) | TOCa (mg/L) | TNb (mg/L) |
|---|---|---|---|---|---|
| D151SP | 110°43′07", 20°54′22″ | 17.2 | 21.2 | 1.39 | 0.46 |
| D151S | 110°41′32″, 20°58′30″ | 26.2 | 16.9 | 2.05 | 0.65 |
| D151W | 110°41′45″, 20°57′44″ | 19.7 | 19.7 | 1.46 | 0.51 |
| D157SP | 111°17′03", 20°09’56" | 16.5 | 27.9 | 1.52 | 0.55 |
| D157S | 111°17′13″, 20°10′03" | 23.7 | 25.6 | 1.87 | 0.67 |
| D157W | 111°17′06", 20°10′13″ | 16.5 | 28.3 | 1.41 | 0.46 |
| D1512SP | 112°00’36", 19°13′15″ | 19.7 | 29.8 | 1.62 | 0.38 |
| D1512S | 112°03’06", 19°13′48″ | 27.2 | 27.9 | 1.70 | 0.60 |
| D1512W | 112°00’35", 19°13′11″ | 15.1 | 29.7 | 1.42 | 0.52 |
| D221SP | 109°29′28″, 17°59′55″ | 20.6 | 23.4 | 1.29 | 0.41 |
| D221S | 109°28′51″, 18°00’15" | 26.8 | 18.2 | 1.53 | 0.62 |
| D221W | 109°29′22″, 17°59′57″ | 25.6 | 20.8 | 1.47 | 0.53 |
| D224SP | 109°55′16″, 17°41′28″ | 19.8 | 24.7 | 1.49 | 0.51 |
| D224S | 109°55′18″, 17°41′21″ | 28.5 | 25.0 | 1.56 | 0.59 |
| D224W | 109°54′55″, 17°41′35″ | 24.9 | 22.6 | 1.39 | 0.60 |
a total organic carbon;
b total nitrogen
Environmental parameters varied greatly between samples collected in different seasons. The concentration of TOC in the station D151, for instance, was 1.39 in spring, 2.05 in summer and 1.46 in winter (P<0.01) (Table 1), indicating complicated biogeochemical processes and hydrodynamic conditions between seasons.
Coverage and diversity of clone libraries
A total of 15 bacterial clone libraries were constructed for the five sample sites. Approximately 90-100 clones from each clone library were used for the ARDRA analysis. The coverage of the respective libraries ranged from 72% to 91% (Table 2), indicating the clone numbers screened in each library can exhibit the diversity of the sample site.
Table 2. Analyses of the 15 bacterial clone libraries in the South China Sea.
| Sample | No. of ARDRA patterns | No. of sequenced clones | No. of OTUs | Coverage (%) | H′a | Chao1 |
|---|---|---|---|---|---|---|
| D151SP | 24 | 39 | 26 | 75.5% | 3.26 | 351 |
| D151S | 21 | 42 | 23 | 78.4% | 3.13 | 276 |
| D151W | 19 | 37 | 31 | 80.0% | 3.43 | 496 |
| D157SP | 12 | 23 | 9 | 88.0% | 2.20 | 45 |
| D157S | 18 | 34 | 26 | 80.8% | 3.26 | 351 |
| D157W | 10 | 15 | 12 | 89.5% | 2.48 | 78 |
| D221SP | 18 | 36 | 19 | 81.5% | 2.94 | 190 |
| D221S | 15 | 37 | 19 | 84.0% | 2.94 | 190 |
| D221W | 10 | 13 | 11 | 90.5% | 2.40 | 66 |
| D224SP | 21 | 41 | 25 | 78.0% | 3.22 | 325 |
| D224S | 17 | 41 | 24 | 82.4% | 3.18 | 300 |
| D224W | 20 | 27 | 18 | 79.5% | 2.89 | 171 |
| D1512SP | 12 | 23 | 2 | 88.0% | 0.69 | 3 |
| D1512S | 27 | 63 | 38 | 72.0% | 3.71 | 741 |
| D1512W | 13 | 20 | 15 | 87.0% | 3.64 | 120 |
a Shannon-Weaver diversity index (Hʹ = - ΣPi log Pi N).
The representative 491 clones from all the ARDRA patterns were sequenced. Rarefaction curves were drawn for the spring groups, summer groups and winter groups (Figure S1), which indicated that the major members of the community had been sampled, and there is likely a long tail of rare taxa which are not included in the study. Total number of representative clones analyzed for each sample varied between 13 and 63, with an average of 32. Based on a taxa cutoff set at 97% similarity, the sequences were further grouped into 282 OTUs. The number of OTUs for each sample ranged from 2 to 38 with average of 20. The Shannon diversity index and the Chao1 estimator of species diversity for each sample were calculated (Table 2). The lowest diversity indices in the spring libraries indicated that its microbial community was composed of a few phylotypes, while in the summer libraries, the diversity indices showed a higher level of species richness. Tables 2 also showed that, for the D151 site and D221 site, the species diversity was higher than that of other sites. The lowest species diversity was observed in the D1512 site.
Bacterial community composition analysis of 16S rDNA clone libraries
The phylum composition of each clone library was shown in Figure 2 and Figure 3a. All of our sequenced clones fell into the nine major lineages of the bacterial domain: Alpha-, Gamma-, Delta-, and Betaproteobacteria; Cyanobacteria; Bacteriodetes; Verrucomicrobia; Actinobacteria; Unindentified bacteria. The percentage of 16S rDNA sequences from each group indicated that unindentified bacteria dominated D151, D157, D1512 sites, while Cyanobacteria dominated the D221, D224 sites. For the bacterial communities from each group, Alphaproteobacteria was the major group at D151, D221, D224 sites, while Gammaproteobacteria was the major group at D1512 site and Cyanobacteria was the major group at D157 site. Actinobacteria also occurred in all of the samples but was not abundant. Bacteroidetes, Betaproteobacteria, Verrucomicrobia and Deltaproteobacteria were detected in the clone libraries but not in all the sample sites. Others (Flavobacteria, Sphingobacteria, Planctomycetes and Firmicutes) were also detected as minor groups in a few of the samples
Figure 2.
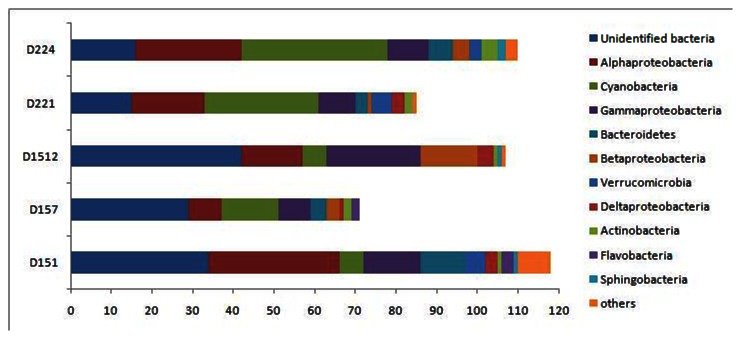
The spatial profiles of the bacterial community structure based on 16S rRNA gene clone libraries from the five sampling sites. D151, samples collected from D151 site; D157, samples collected from D157 site; D1512, samples collected from D1512 site; D221, samples collected from D221 site; D224, samples collected from D224 site.
Figure 3.

Pie charts of the relative abundance of the bacterial 16S rRNA gene clusters a, all the sequenced clones; b, water samples collected in spring; c, water samples collected in summer; d, water samples collected in winter.
Phylogenetic analysis of bacterial 16S rRNA genes across all sites and samples
A phylogenetic tree was constructed to show relationships between the dominant OTUs (52 OTUs, representing 334 sequences) and their closest neighbors (Figure 4). Based on the valid reference tree, eight different phyla were identified, with the majority of OTUs being classified as Proteobacteria, followed by Cyanobacteria , Bacteroidetes, Verrucomicrobia and Actinobacteria, respectively. The phylogenetic tree of dominant OTUs did not include any of these unidentified bacteria in Figure 3, which indicated the unidentified bacteria were rare taxa, and less dominant in the community.
Figure 4.
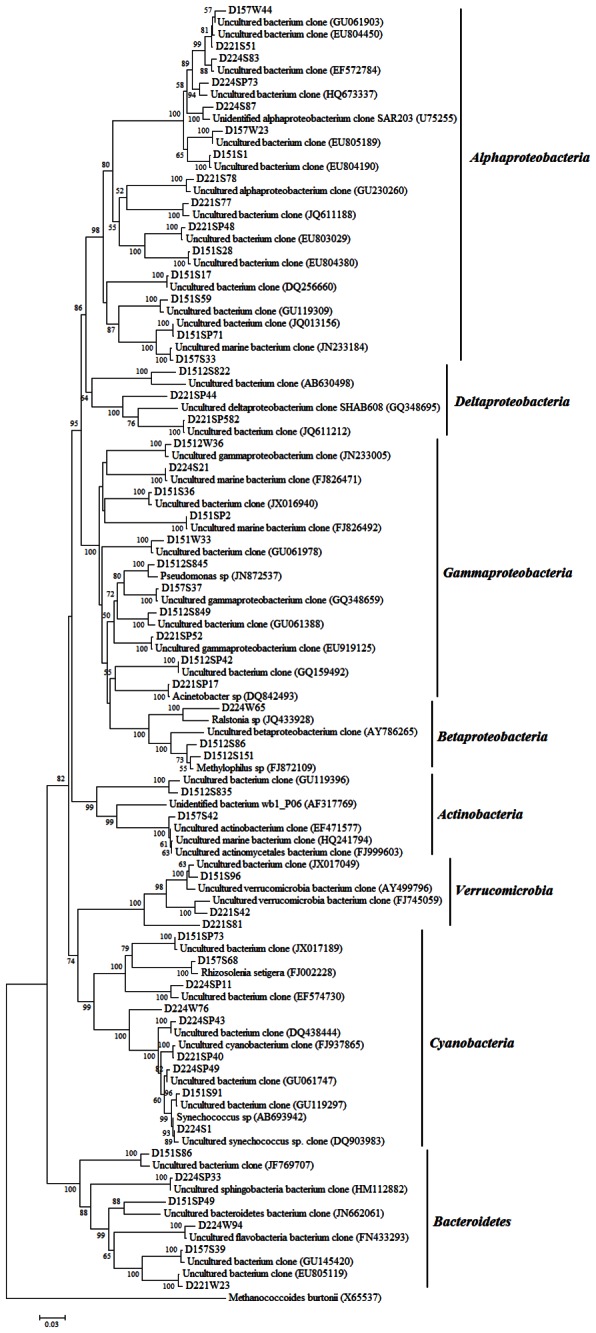
Phylogenetic tree based on analysis of the representative 16S rRNA gene sequences obtained from the 15 bacterial clone libraries. The tree was constructed using the neighbor-joining method in MEGA. Bootstrap analysis was conducted using 1000 replicates. Bootstrap values are shown for branches with > 50% bootstrap support.
A total of 32 OTUs representing 200 sequences were classified within the phylum Proteobacteria (Table 3). Four bacterial subphyla were identified: Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria and Deltaproteobacteria. The two predominant subphyla were Alphaproteobacteria and Gammaproteobacteria. Within the subphylum Alphaproteobacteria (15 OTUs, representing 102 sequences), OTUs were closely related to uncultured. The subphylum Gammaproteobacteria was comprised of 11 OTUs (76 sequences), most of them were related to the uncultured Gammaproteobacteria. The other two OTUs (22 sequences) were classified within the order Acinetobacteria (6 sequences), Pseudomonas (3 sequences), respectively. Six OTUs (22 sequences) were classified within the subphylum Betaproteobacteria and Deltaproteobacteria, respectively.
Table 3. Phylogenetic affiliation of library clones obtained from coastal waters of the South China Sea as deduced from BLAST searched.
| Representative Clones | No. of similar sequences | Phylogenetic ascription | Closest relative (accession number) | identity |
|---|---|---|---|---|
| D157W44 | 3 | Alphaproteobacteria | Uncultured bacterium clone S-5m-75(GU061903) | 99% |
| D221S51 | 11 | Alphaproteobacteria | Uncultured bacterium clone 6C232378(EU804450) | 99% |
| D224S83 | 38 | Alphaproteobacteria | Uncultured bacterium clone S23_883(EF572784) | 99% |
| D224SP73 | 2 | Alphaproteobacteria | Uncultured bacterium clone F9P41300_A04(HQ673337) | 98% |
| D224S87 | 3 | Alphaproteobacteria | Unidentified alpha proteobacterium clone SAR203(U75255) | 98% |
| D157W23 | 2 | Alphaproteobacteria | Uncultured bacterium clone(EU805189) | 99% |
| D151S1 | 12 | Alphaproteobacteria | Uncultured bacterium clone 6C232086(EU804190) | 99% |
| D221SP48 | 4 | Alphaproteobacteria | Uncultured bacterium clone 4C230433(EU803029) | 99% |
| D151S28 | 2 | Alphaproteobacteria | Uncultured bacterium clone 6C232292(EU804380) | 99% |
| D221S78 | 4 | Alphaproteobacteria | Uncultured alpha proteobacterium clone ARTE1_103(GU230260) | 99% |
| D221S77 | 2 | Alphaproteobacteria | Uncultured bacterium clone KSTye-PF1-B-003(JQ611188) | 99% |
| D151S17 | 3 | Alphaproteobacteria | Uncultured bacterium clone Fitz2_28(DQ256660) | 99% |
| D151S59 | 2 | Alphaproteobacteria | Uncultured bacterium clone Reef_N07(GU119309) | 98% |
| D151SP71 | 3 | Alphaproteobacteria | Uncultured bacterium clone DMS16SrDNA22(JQ013156) | 99% |
| D157S33 | 11 | Alphaproteobacteria | Uncultured marine bacterium clone IMS3D32(JN233184) | 99% |
| D1512S822 | 5 | Deltaproteobacteria | Uncultured bacterium clone MPB1-116(AB630498) | 94% |
| D221SP582 | 2 | Deltaproteobacteria | Uncultured bacterium clone KSTye-VF1-B-020(JQ611212) | 99% |
| D221SP44 | 3 | Deltaproteobacteria | Uncultured delta proteobacterium clone SHAB608(GQ348695) | 92% |
| D1512S151 | 3 | Betaproteobacteria | Uncultured beta proteobacterium clone 161GNFL6(AY786265) | 92% |
| D1512S86 | 7 | Betaproteobacteria | Methylophilus sp. Mim(FJ872109) | 99% |
| D224W65 | 2 | Betaproteobacteria | Ralstonia sp. LT3(JQ433928) | 99% |
| D224S21 | 2 | Gammaproteobacteria | Uncultured marine bacterium clone A6-5-63(FJ826471) | 99% |
| D221SP17 | 6 | Gammaproteobacteria | Acinetobacter sp. EN96(DQ842493) | 99% |
| D151S36 | 5 | Gammaproteobacteria | Uncultured bacterium clone HglFeb5F7(JX016940) | 99% |
| D1512W36 | 3 | Gammaproteobacteria | Uncultured gamma proteobacterium clone OS3SD61(JN233005) | 99% |
| D151SP2 | 5 | Gammaproteobacteria | Uncultured marine bacterium clone A6-5-84(FJ826492) | 99% |
| D1512SP42 | 40 | Gammaproteobacteria | Uncultured bacterium clone 16slp92-01d01.q1k(GQ159492) | 99% |
| D151W33 | 2 | Gammaproteobacteria | Uncultured bacterium clone S-DCM-17(GU061978) | 98% |
| D1512S845 | 3 | Gammaproteobacteria | Pseudomonas sp. SAP34_1(JN872537) | 99% |
| D157S37 | 2 | Gammaproteobacteria | Uncultured gamma proteobacterium clone SHAB561(GQ348659) | 99% |
| D1512S849 | 6 | Gammaproteobacteria | Uncultured bacterium clone CE1-5m-107(GU061388) | 99% |
| D221SP52 | 2 | Gammaproteobacteria | Uncultured gamma proteobacterium clone SW45(EU919125) | 99% |
| D151S96 | 2 | Verrucomicrobia | Uncultured bacterium clone HglFeb5G9m(JX017049) | 98% |
| D221S81 | 2 | Verrucomicrobia | Uncultured Verrucomicrobia bacterium clone Dover396(AY499796) | 97% |
| D221S42 | 8 | Verrucomicrobia | Uncultured Verrucomicrobiae bacterium clone SHWN (FJ745059) | 97% |
| D224W76 | 2 | Cyanobacteria | Uncultured Synechococcus sp. clone PR12 (DQ903983) | 98% |
| D151SP73 | 3 | Cyanobacteria | Uncultured bacterium clone HglFeb6C1m(JX017189) | 99% |
| D157S68 | 2 | Cyanobacteria | Rhizosolenia setigera isolate C22(FJ002228) | 99% |
| D224SP11 | 2 | Cyanobacteria | Uncultured bacterium clone S25_1074(EF574730) | 97% |
| D221SP40 | 7 | Cyanobacteria | Uncultured cyanobacterium clone MWLSA52(FJ937865) | 99% |
| D224SP43 | 15 | Cyanobacteria | Uncultured bacterium clone ECS-P7-C9(DQ438444) | 99% |
| D224SP49 | 4 | Cyanobacteria | Uncultured bacterium clone CEP-5m-60(GU061747) | 99% |
| D151S91 | 4 | Cyanobacteria | Uncultured bacterium clone Reef_G16(GU119297) | 99% |
| D224S1 | 52 | Cyanobacteria | Synechococcus sp.(AB693942) | 99% |
| D157S42 | 2 | Actinobacteria | Uncultured marine bacterium clone Sp02sw-15(HQ241794) | 99% |
| D1512S835 | 13 | Actinobacteria | Uncultured bacterium clone Reef_M14(GU119396) | 99% |
| D151S86 | 5 | Bacteroidetes | Uncultured bacterium clone REP6-45(JF769707) | 98% |
| D224SP33 | 2 | Bacteroidetes | Uncultured Sphingobacteria bacterium clone SHOF496 | 99% |
| D151SP49 | 2 | Bacteroidetes | Uncultured Bacteroidetes bacterium clone LF8CBb87(JN662061) | 91% |
| D224W94 | 3 | Bacteroidetes | Uncultured Flavobacteria bacterium(FN433293) | 98% |
| D157S39 | 2 | Bacteroidetes | Uncultured bacterium clone BS035(GU145420) | 99% |
| D221W23 | 2 | Bacteroidetes | Uncultured bacterium clone 6C233107(EU805119) | 99% |
Nine OTUs representing 91 sequences were classified within the phylum Cyanobacteria . Two subphyla were identified: Synechococcus (5 OTUs, 82 sequences) and Rhizosolenia (3 OTUs, 7 sequences). In comparison with Rhizosolenia, Synechococcus has a high relative abundance. The less prominent 9 OTUs (28 sequences) were classified within the phylum Verrucomicrobiae and Bacteroidetes. The remaining two OTUs (15 sequences) were classified within the phylum Actinobacteria.
Community structures of bacteria with seasons and between locations
The distribution of bacteria clones with seasons was represented in Figure 3a. Unidentified bacteria dominated the spring libraries, followed by Alphaproteobacteria, Cyanobacteria , Gammaproteobacteria, Bacteroidetes, Actinobacteria, Delataproteobacteria and Betaproteobacteria (Figure 3b). The unidentified bacteria were also the largest phylum in the winter libraries, followed by Cyanobacteria , Gammaproteobacteria, Alphaproteobacteria, Betaproteobacteria, Actinobacteria, Flavobacteria, Sphingobacteria and Verrucomicrobia (Figure 3d). While in the summer libraries, Alphaproteobacteria was the largest phylum, followed by Cyanobacteria , Gammaproteobacteria, Betaproteobacteria, Bacteroidetes, Verrucomicrobia, Delataproteobacteria, Actinobacteria, Flavobacteria and Sphingobacteria (Figure 3c) .
The partitioning of bacterial diversity among the three groups based on seasons was analyzed using LIBSHUFF analysis (10,000 randomizations). The result revealed that there were significant differences (P<0.008) in phylogenetic composition between all the three groups (Table 4), indicated an interesting partitioning of bacterial diversity responding to season variations. To identify the spatial variations, libraries were divided into five groups based on the five sample sites. The LIBSHUFF analysis (10,000 randomizations) was performed to determine statistically differences between the five groups (Table 5). The results showed that most of the groups were significantly different (P<0.0025), with the exception of the community structures of D157 and D224, D221 and D224.
Table 4. LIBSHUFF analysis of the community structures of spring samples, summer samples and winter samples at an OTU definition level of 97%.
| Spring | Summer | Winter | ||||
|---|---|---|---|---|---|---|
| dCXYScore | P value* | dCXYScore | P value* | dCXYScore | P value* | |
| Spring | 0.001 | 0.036 | 0.012 | <0.001 | ||
| Summer | 0.023 | <0.001 | 0.025 | <0.001 | ||
| Winter | 0.001 | 0.009 | 0.001 | 0.027 | ||
* The significance values should be below the critical threshold (0.05/6 = 0.008)
Table 5. LIBSHUFF analysis of the community structures of samples collected at D151, D157, D1512, D221, D224 at an OTU definition level of 97%.
| D151 | D157 | D1512 | D221 | D224 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dCXYScore | P value* | dCXYScore | P value* | dCXYScore | P value* | dCXYScore | P value* | dCXYScore | P value* | |
| D151 | 0.009 | <0.001 | 0.023 | <0.001 | 0.008 | <0.001 | 0.014 | <0.001 | ||
| D157 | 0.007 | <0.001 | 0.002 | 0.0039 | 0.002 | 0.0035 | 0.002 | 0.0030 | ||
| D1512 | 0.065 | <0.001 | 0.014 | <0.001 | 0.043 | <0.001 | 0.022 | <0.001 | ||
| D221 | 0.001 | 0.0032 | 0.006 | <0.001 | 0.005 | <0.001 | 0.002 | 0.0056 | ||
| D224 | 0.004 | <0.001 | 0.003 | 0.0028 | 0.005 | <0.001 | 0.003 | 0.0043 | ||
* The significance values should be below the critical threshold (0.05/20 = 0.0025)
The community assemblages were clustered using the UPGMA algorithm in the program MOTHUR at the OTU definition level of 97% (Figure 5). In the UPGMA tree, bacterial communities collected in spring and summer displayed less variation across the spatial profile, on the other hand, those collected in winter were more heterogeneous. The D221 and D224 samples collected at spring, summer and winter clustered with the D1512 sample collected at spring and summer (Figure 5). Unlike the spring and summer samples, the winter samples collected from D151, D157, D1512 were clearly separated from the spring and summer samples except for the D157SP sample (Figure 5). The D157SP and D157W samples were nearly identical, yet differed greatly from most of the other communities. The D151S sample was nearly as different from all of the other communities as D151W and D1512W samples.
Figure 5.

UPGMA cluster of the samples collected from the five locations in spring, summer and winter at OTU definition of 0.03.
Relationships of environmental factors and bacterial community structures
RDA was used to determine how environmental parameters influenced the bacterial community structures, after initial analysis by detrended correspondence analysis (Figure 6). The first two RDA axes explained 64.0% of the cumulative variance of the bacteria-environment relationship. The water salinity (P=0.001) and TOC (P=0.089) contributed most to this distinction and contributed significantly to the bacteria-environment relationship. The concentration of salinity contributed to the distribution of the water samples, especially for D1512SP, D1512S, D157SP, while the separation of D221S, D221SP, D224S, D224W was a result of the temperature (Figure 6).
Figure 6.
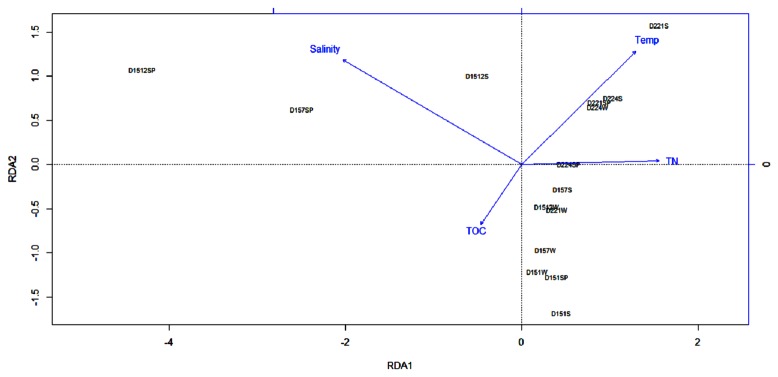
RDA ordination plots for the environmental parameters and the bacterial communities represented by 16S rRNA gene sequences.
Discussion
Our results demonstrate that the biodiversity of marine bacteria communities at the SCS exhibit distinctly spatial and temporal patterns. These patterns in distribution and abundance of marine bacteria were influenced by a variety of environmental variables. Our data and analyses show that marine bacterial diversity is higher in summer and that bacterial community composition exhibits a spatial gradient of increasing diversity from offshore to coastal sea (Table 2, Table 5). The UPGMA analysis for our samples showed that community-level changes in 16S-based diversity were better explained by spatial patterns than by temporal patterns (Figure 5, Table 4, Table 5). These patterns found in community composition can add new knowledge to bacterioplankton abundance and distribution in coastal marine ecosystems of the SCS.
In this study, although we have explored only a limited number of sets of OTUs at 97% sequence similarity, a number of strong patterns are apparent. The association with the environmental variables of temperature, salinity, nutrients and organic matter, suggest a large number of possible ecological mechanisms responsible for these patterns (Figure 6). Our sample location (D151, D157, D1512, D221, D224) off Hainan Island is within a system of currents that exhibit strong seasonality, with generally southwestward flow (China Coastal current) in spring, northeastward flow (western Pacific warm current) in summer, and weak southwestward flow in early winter [29]. Because the region northeast of our site is the Pearl River and the Jianjiang River estuaries, one might expect the China Coastal current from northeast to bring more eutrophic conditions and associated organisms of these estuaries in spring, whereas western Pacific warm current from southwest would be associated with more oligotrophic conditions and associated organisms. As higher rates of resource supply can potentially support larger numbers and more specialized kinds of organisms, the diversity of organism increases with increasing productivity [30,31]. It is likely that these seasonal hydrographic conditions and their influence on bacteria, contributed significantly to our observed patterns. Such variations in hydrography are reflected in several parameters we investigated, such as temperature, salinity and nutrients.
Our data showed bacterial diversity in the coastal waters (D151, D221) was higher than that in the offshore waters (D157, D1512, D224). As terrestrially impacted seawater has a higher concentration of particles than offshore seawater, which receives a low input of organic carbon and the associated particles, the environmental variables (e.g., nutrient, salinity) display lateral gradient patterns related with the distance to shore [32]. The geochemical variables suggested the primary production at the coastal regions (D151, D221) was higher than that at offshore regions (D157, D1512, D224) (Table 1, Table 2). The primary production of the coastal waters is known to commonly exceed the consumption of herbivores. Therefore, a large fraction of primary organic matter becomes available to consumers as detritus [33]. Most of this detritus is degraded by heterotrophic bacteria before entering higher trophic levels [34,35], and this results in the more diverse bacterial communities in the coastal waters than the offshore waters.
Both the coastal sites D151 and D221 exhibited similar spatial patterns with higher bacterial diversity than that of the offshore sites. However, the diversity of bacterial communities in the coastal site D151 and D221 showed different temporal patterns (Table 2), which might suggest the mixing of two seasonal currents was blocked by Hainan Island. As the blocking of the currents by Hainan Island, the temporal pattern of the coastal site D151 might be mainly contributed by China Coastal current, while that of the coastal site D221 might be mainly contributed by west Pacific warm current. In addition, the results of UPGMA analysis showed bacterial communities collected during spring and summer displayed less variation across the spatial profile (Figure 5), while those collected during summer were more heterogeneous (Figure 5, Table 4). Due to the block of currents by Hainan Island, samples collected in coastal site D151 clustered separately, while samples collected in coastal site D221 clustered together. Thus, our results indicate that spatial patterns show more heterogeneous than temporal patterns in the coastal waters of the SCS.
Past studies have found some similar results in comparison to those we report here. For example, Gilbert et al. [36] reported repeatable seasonal patterns occurred in surface water microbial community and the driver of this pattern was day length. Morris et al. [37] found certain bacterial groups tended to be more common during certain seasons. Fuhrman et al. [17] showed repeatable temporal pattern in distribution and abundance of microbial taxa was highly predictable. Their temporal pattern was most strongly correlated to parameters related to the strong seasonality of their sites. These reports were similar with our results suggesting temporal patterns in the SCS might be due to the seasonal currents. Additionally, Gao et al. [38] reported spatial diversity of microbial community in Hawaiian coastal waters and showed coastal waters had the greatest diversity, which was consistent with our study indicating higher diversity in the coastal waters of the SCS. However, none of these studies reported temporal pattern together with spatial pattern, so we do not know which pattern is predominant in sites reported.
Typical marine clades, such as Alphaproteobacteria, Gammaproteobacteria and Cyanobacteria were more represented in marine coastal and open sea samples [36,39], and Alphaproteobacteria was more abundant in marine water than in freshwater [40]. In our study, Alphaproteobacteria SAR203 was the most abundant clade in marine water samples (Figure 4). The clones were very distantly related to the uncultured organisms that are frequent in mangrove sediments, marine waters, marine sediments [18,41,42]. Interestingly, an OTU D221S78 was only detected in summer samples, which indicated this OTU might be summer-associated species (Figure 4). Gammaproteobacteria was one of the dominant phyla in the samples. Being metabolically versatile, this phylum is ecologically very successful [43]. Two clades Pseudomonas and Acinetobacteria were retrieved from our marine libraries and clustered with sequences retrieved from water of the Sargasso Sea [12], the East China Sea [32], the SCS and North Pacific Ocean [44], marine sediment [18], marine plankton [45] and hot springs [46]. A dominant OTU D1512SP42 mainly existed in spring samples, which suggested this OTU might be spring-associated species (Figure 4, Table 3). These findings indicated that Alphaproteobacteria and Gammaproteobacteria were widely distributed and formed large cluster in the sea areas surveyed. Our study was different from previous studies that found a high relative abundance of phototrophic Cyanobacteria [47,48]. The results revealed Cyanobacteria was the most abundant groups in the D221 site and the D224 site (Figure 2), especially in the samples collected at summer (Figure 3c). Synechococcus and Rhizosolenia were common in the summer waters (Figure 4). The OTU D224SP49 was only detected at the D224 site and could be recognized as a site special species. From the results, it could be suggested that the high abundance and proportion of Cyanobacteria were an important feature of the planktonic bacteria in the two sites, and indicated the ongoing deterioration water quality in the area was due to the rapid development of the tourism and sea farming [49].
Betaproteobacteria have been commonly detected in freshwater lakes worldwide, where they are the most abundant group [50]. Recovery of 16S rRNA gene clones affiliated to Betaproteobacteria is common in libraries constructed from coastal samples [51], but few to no Betaproteobacteria have been reported by open ocean surveys [39,52–54]. These findings lead to the idea that bacterioplankton represented by these lineages have a probable freshwater origin and are adapted to coastal marine environments and could be representative bacterioplanckton phylotypes that transit between freshwater and marine habitats [55]. In this study, our data also support this proposal, since Ralstonia and Methylophilus were detected in our marine libraries, owing to the contribution of freshwater. This result indicates that freshwater bacteria have affected the bacterial community structure of this marine system.
The ecological significance of Bacteroidetes has been brought to light because of their proficiency in degrading various biopolymers such as cellulose, chitin and pectin [56]. The Bacteroidetes clade was well represented in both saline and freshwater environments [53]. This might be a consequence of the presence of closely related marine phylotypes of common freshwater taxa [48]. In this study, several Bacteroidetes related OTUs clustered with sequences from marine habits of different geographic areas, indicating that Bacteroidetes are distributed worldwide. A OTU D151S86 clustered with uncultured bacteria was only obtained in summer samples, which suggested this OTU to be summer-associated species.
The number of OTUs identified from D1512SP and D157SP is quite small (Table 2). We thought this was not merely an error in the cloning and sequencing. The D1512SP and D157SP samples were separated from others, and had a positive correlation with the concentration of salinity (Figure 6). However, in spring the China Coastal currents together with the fresh water from the Pearl river and Jianjiang river flowed south-westwards, which contributed to the lower concentration of salinity at the D1512 and D157 site. Due to the lower concentration of salinity, it is not surprising to observe less OTUs in the D1512 and D157 sites.
This study has confirmed that temporal and spatial patterns occur in water bacterial community of the SCS and that the environmental factors governing diversity and structure of bacteria could be identified from the analysis. Due to complicated biogeochemical processes and hydrodynamic conditions, the spatiotemporal diversity of bacterial communities would be very complicated. To better understand the bacterial community structure response to spatiotemporal variations, more intensive microbial measurements using new technologies including metatranscriptomic, metaproteomic approaches should be conducted in future.
Supporting Information
The phylotypes were determined with a 97% similarity cutoff value.
Acknowledgments
Many thanks to the three anonymous reviews and the Academic Editor whose pertinent comments have greatly improved the quality of this paper.
Funding Statement
This research was supported by grants from China Marine Commonweal Research Project (201005022-2) and Guangdong Provincial Science and Technique Major Project (2011A080403006). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Copley J (2002) All at sea. Nature 415: 572-574. doi:10.1038/415572a. PubMed: 11832910. [DOI] [PubMed] [Google Scholar]
- 2. Karl DM (2002) Hidden in a sea of microbes. Nature 415: 590-591. doi:10.1038/415590b. PubMed: 11832923. [DOI] [PubMed] [Google Scholar]
- 3. Pomeroy LR (1974) The ocean’s food web, a changing paradigm. BioScience 24: 499–504. [Google Scholar]
- 4. Azam F, Long RA (2001) Sea snow microcosms. Nature 414: 497-498. PubMed: 11734832. [DOI] [PubMed] [Google Scholar]
- 5. Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L et al. (2011) Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13: 135-144. doi:10.1111/j.1462-2920.2010.02315.x. PubMed: 21199253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azam F, Fenchel T, Field JG, Gray JS, Meyer-Reil LA et al. (1983) The ecological role of water-column microbes in the sea. Mar Ecol Prog S 10: 257-263. doi:10.3354/meps010257. [Google Scholar]
- 7. Pomeroy LR, Williams PJ, Azam F (2007) The Microbial loop. J Oceanogr Vol. 20 no. 2. [Google Scholar]
- 8. Briée C, Moreira D, López-García P (2007) Archaeal and bacterial community composition of sediment and plankton from a suboxic freshwater pond. Res Microbiol 158: 213–227. doi:10.1016/j.resmic.2006.12.012. PubMed: 17346937. [DOI] [PubMed] [Google Scholar]
- 9. Kanokratana P, Chanapan S, Pootanakit K, Eurwilaichitr L (2004) Diversity and abundance of Bacteria and Archaea in the Bor Khlueng Hot Spring in Thailand. J Basic Microbiol 44: 430–444. doi:10.1002/jobm.200410388. PubMed: 15558824. [DOI] [PubMed] [Google Scholar]
- 10. Pace NR (1997) A molecular view of microbial diversity and the biosphere. Science 276: 734–740. doi:10.1126/science.276.5313.734. PubMed: 9115194. [DOI] [PubMed] [Google Scholar]
- 11. Schwarz JI, Eckert W, Conrad R (2007) Community structure of Archaea and Bacteria in a profundal lake sediment Lake Kinneret (Israel). Syst Appl Microbiol 30: 239–254. doi:10.1016/j.syapm.2006.05.004. PubMed: 16857336. [DOI] [PubMed] [Google Scholar]
- 12. Treusch AH, Vergin KL, Finlay LA, Donatz MG, Burton RM et al. (2009) Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J 3: 1148-1163. doi:10.1038/ismej.2009.60. PubMed: 19494846. [DOI] [PubMed] [Google Scholar]
- 13. Danovaro R, Pusceddu A (2007) Biodiversity and ecosystem functioning in coastal lagoons: does microbial diversity play any role? Estuar Coast Shelf Sci 75: 4-12. doi:10.1016/j.ecss.2007.02.030. [Google Scholar]
- 14. Andersson AF, Riemann L, Bertilsson S (2010) Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J 4: 171-181. doi:10.1038/ismej.2009.108. PubMed: 19829318. [DOI] [PubMed] [Google Scholar]
- 15. Crump BC, Armbrust EV, Baross JA (1999) Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 65: 3192-3204. PubMed: 10388721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilbert JA, Field D, Swift P, Thomas S, Cummings D et al. (2010) The Taxonomic and Functional Diversity of Microbes at a Temperate Coastal Site: A ‘Multi-Omic’ Study of Seasonal and Diel Temporal Variation. PLOS ONE 5(11): e15545. doi:10.1371/journal.pone.0015545. PubMed: 21124740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fuhrman JA, Hewson I, Schwalbach MS, Steele JA, Brown MV et al. (2006) Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci U S A 103: 13104-13109. doi:10.1073/pnas.0602399103. PubMed: 16938845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Du J, Xiao K, Huang Y, Li H, Tan H et al. (2011) Seasonal and spatial diversity of microbial communities in marine sediments of the South China Sea. Antonie Van Leeuwenhoek 100: 317-331. doi:10.1007/s10482-011-9587-9. PubMed: 21604204. [DOI] [PubMed] [Google Scholar]
- 19. Feingersch R, Suzuki MT, Shmoish M, Sharon I, Sabehi G et al. (2010) Microbial community genomics in eastern Mediterranean Sea surface waters. ISME J 4: 78-87. doi:10.1038/ismej.2009.92. PubMed: 19693100. [DOI] [PubMed] [Google Scholar]
- 20. Grasshoff K, Kremling K, Erhardt M (1999) Methods of seawater analysis, 3rd ed. Germany: Wiley-VGH Verlag. [Google Scholar]
- 21. Lai X, Cao L, Tan H, Fang S, Huang Y et al. (2007) Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea. ISME J 1: 756-762. doi:10.1038/ismej.2007.51. PubMed: 18059498. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261-5267. doi:10.1128/AEM.00062-07. PubMed: 17586664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537-7541. doi:10.1128/AEM.01541-09. PubMed: 19801464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876-4882. doi:10.1093/nar/25.24.4876. PubMed: 9396791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5: 150-163. doi:10.1093/bib/5.2.150. PubMed: 15260895. [DOI] [PubMed] [Google Scholar]
- 26. Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40: 237-264. doi:10.2307/2333344. [Google Scholar]
- 27.Oksanen Jari (2013) Multivariate Analysis of Ecological Communities in R: vegan tutorial.
- 28. Magalhães C, Bano N, Wiebe WJ, Bordalo AA, Hollibaugh JT (2008) Dynamics of nitrous oxide reductase genes (nosZ) in intertidal rocky biofilms and sediments of the Douro River estuary (Portugal), and their relation to N-biogeochemistry. Microb Ecol 55: 259-269. doi:10.1007/s00248-007-9273-7. PubMed: 17604988. [DOI] [PubMed] [Google Scholar]
- 29. Xie SP, Xie Q, Wang DX, Liu WT (2003) Summer upwelling in the South China Sea and its role in regional climate variations. J Geophys Res 108: 3261. doi:10.1029/2003JC001867. [Google Scholar]
- 30. Connell JH, Orias E (1964) The ecological regulation of species diversity. Am Nat 98: 399–414. doi:10.1086/282335. [Google Scholar]
- 31. Wright DH, Currie DJ, Maurer BA (1993) Energy supply and patterns of species richness on local and regional scales. Species Diversity in Ecological Communities: Historical and Geographical Perspectives, Ricklefs RE, Schluter D. Chicago: University of Chicago Press; pp 66–74. [Google Scholar]
- 32. Feng BW, Li XR, Wang JH, Hu ZY, Meng H et al. (2009) Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol Ecol 70: 80-92. PubMed: 19780829. [DOI] [PubMed] [Google Scholar]
- 33. Newell RC (1982) The energetics of detritus utilisation in coastal lagoons and nearshore waters. In: Laserre P, Postma H. .Proceedings of International Symposium on Coastal Lagoos; Oceanologica Acta (Special issue):347-355 [Google Scholar]
- 34. Manni E, Fiordelmondo C, Gambi C, Pusceddu A, Danovaro R (2003) Benthic microbial loop functioning in coastal lagoons: a comparative approach. Oceanol Acta 26: 27-33. doi:10.1016/S0399-1784(02)01227-6. [Google Scholar]
- 35. Mou XZ, Sun SL, Edwards RA, Hodson RE, Moran MA (2008) Bacterial carbon processing by generalist species in the coastal ocean. Nature 451: 708-U704. doi:10.1038/nature06513. PubMed: 18223640. [DOI] [PubMed] [Google Scholar]
- 36. Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J et al. (2012) Defining seasonal marine microbial community dynamics. ISME J 6: 298-308. doi:10.1038/ismej.2011.107. PubMed: 21850055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris RM, Vergin KL, Cho JC, Rappe MS, Carlson CA et al. (2005). Limnol Oceanogr 50: 1687–1696. doi:10.4319/lo.2005.50.5.1687. [Google Scholar]
- 38. Gao Z, Johnson ZI, Wang G (2010) Molecular characterization of the spatial diversity and novel lineages of mycoplankton in Hawaiian coastal waters. ISME J 4: 111-120. doi:10.1038/ismej.2009.87. PubMed: 19641535. [DOI] [PubMed] [Google Scholar]
- 39. Brown MV, Philip GK, Bunge JA, Smith MC, Bissett A et al. (2009) Microbial community structure in the North Pacific ocean. ISME J 3: 1374–1386. doi:10.1038/ismej.2009.86. PubMed: 19626056. [DOI] [PubMed] [Google Scholar]
- 40. Glöckner FO, Fuchs BM, Amann R (1999) Bacterioplankton compositions of lakes and oceans: a first comparison based on fluorescence in situ hybridization. Appl Environ Microbiol 65: 3721-3726. PubMed: 10427073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Field KG, Gordon D, Wright T, Rappé M, Urback E et al. (1997) Diversity and depth-specific distribution of SAR11 cluster rRNA genes from marine planktonic bacteria. Appl Environ Microbiol 63: 63-70. PubMed: 8979340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allers E, Wright JJ, Konwar KM, Howes CG, Beneze E et al. (2013) Diversity and population structure of Marine Group A bacteria in the Northeast subarctic Pacific Ocean. ISME J 7: 256-268. doi:10.1038/ismej.2012.108. PubMed: 23151638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Madigan MT, Marrs BL (1997) Extremophiles. Sci Am 276: 82-87. doi:10.1038/scientificamerican0497-82. PubMed: 11536798. [DOI] [PubMed] [Google Scholar]
- 44. Du H, Jiao N, Hu Y, Zeng Y (2006) Diversity and distribution of pigmented heterotrophic bacteria in marine environments. FEMS Microbiol Ecol 57: 92-105. doi:10.1111/j.1574-6941.2006.00090.x. PubMed: 16819953. [DOI] [PubMed] [Google Scholar]
- 45. Fuhrman JA, McCallum K, Davis AA (1995) Phylogenetic diversity of subsurface marine microbial communities from the Atlantic and Pacific oceans. Appl Environ Microbiol 59: 1294–1302. PubMed: 85341227685997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ward DM, Ferris MJ, Nold SC, Bateson MM (1998) A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol Mol Biol Rev 62: 1353-1370. PubMed: 9841675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giovannoni SJ, Rappé MS (2000) Evolution, diversity and molecular ecology of marine prokaryotes. In: Kirchman DL. Microbial Ecology of the Oceans. Wiley & Sons; . pp. 47–84 [Google Scholar]
- 48. Pommier T, Canbäck B, Riemann L, Boström KH, Simu K et al. (2007) Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16: 867–880. PubMed: 17284217. [DOI] [PubMed] [Google Scholar]
- 49. Müller-Navarra DC, Brett MT, Liston AM, Goldman CR (2000) A highly unsaturated fatty acid predicts carbon transfer between primary producers and consumers. Nature 403: 74-77. doi:10.1038/47469. PubMed: 10638754. [DOI] [PubMed] [Google Scholar]
- 50. Mueller-Spitz SR, Goetz GW, McLellan SL (2009) Temporal and spatial variability in nearshore bacterioplankton communities of Lake Michigan. FEMS Microbiol Ecol 67: 511-522. doi:10.1111/j.1574-6941.2008.00639.x. PubMed: 19220863. [DOI] [PubMed] [Google Scholar]
- 51. Yeo SK, Huggett MJ, Eiler A, Rappé MS (2013) Coastal bacterioplankton community dynamics in response to a natural disturbance. PLOS ONE 8: e56207. doi:10.1371/journal.pone.0056207. PubMed: 23409156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S et al. (2007) The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLOS Biol 5: e77. doi:10.1371/journal.pbio.0050077. PubMed: 17355176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Silveira CB, Vieira RP, Cardoso AM, Paranhos R, Albano RM et al. (2011) Influence of salinity on bacterioplankton communities from the Brazilian rain forest to the coastal Atlantic Ocean. PLOS ONE 6: e17789. doi:10.1371/journal.pone.0017789. PubMed: 21408023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR et al. (2007) Microbial population structures in the deep marine biosphere. Sciene 318: 97-100. doi:10.1126/science.1146689. PubMed; : 17916733 [DOI] [PubMed] [Google Scholar]
- 55. Rappé MS, Giovannoni SJ (2003) The uncultured microbial majority. Annu Rev Microbiol 57: 369–394. doi:10.1146/annurev.micro.57.030502.090759. PubMed: 14527284. [DOI] [PubMed] [Google Scholar]
- 56. Kirchman DL (2002) The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91-100. doi:10.1111/j.1574-6941.2002.tb00910.x. PubMed: 19709188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The phylotypes were determined with a 97% similarity cutoff value.