

Abstract
CD69 is a membrane molecule transiently expressed on activated lymphocytes, and its selective expression in inflammatory infiltrates suggests that it plays a role in the pathogenesis of inflammatory diseases. In this study, we used CD69-deficient (CD69 KO) mice to assess the role of CD69 in the pathogenesis of dextran sulphate sodium (DSS)-induced acute and chronic colitis. The severity of colitis was assessed by the survival rate, clinical signs, colon length, histological examination and the expression of cytokines and chemokines in the large intestines. Both acute and chronic colitis were attenuated in the CD69 KO mice, as reflected by the lower lethality, weight loss, clinical signs, and improved histological findings. CD69+ cells infiltrated extensively into the inflamed mucosa of the colon in WT mice after DSS treatment. Experiments with the transfer of WT CD4 T cells into CD69 KO mice restored the induction of colitis. The administration of an anti-CD69 antibody also inhibited the induction of the DSS-induced colitis. These results indicate that CD69 expressed on CD4 T cells plays an important role in the pathogenesis of DSS-induced acute and chronic colitis, and that CD69 could be a possible therapeutic target for colitis.
Introduction
Human inflammatory bowel diseases (IBDs), such as Crohn’s disease (CD) and ulcerative colitis (UC), are characterized by chronic inflammation of the intestinal tract. The pathogenesis of IBD is related to an inappropriate and exaggerated mucosal immune responses to constituents of the intestinal flora [1]–[5]. The inflamed IBD tissue is heavily populated by inflammatory cells, including lymphocytes, plasma cells, neutrophils and macrophages [6]. Dysregulated CD4 T cells involved in adaptive immunity have also been postulated to play an important role in the pathogenesis of IBD [7]–[10]. A dysregulated T cell response leads to alterations in the mucosal cytokine expression. The patients display an impaired cytokine profile, with high local production of inflammatory cytokines including IL-1β, IL-6, IFN-γ and TNF-α [11], [12].
Dextran sulphate sodium (DSS)-induced colitis in mice has been used as a model of colitis resembling human UC. Mice that are exposed to DSS in their drinking water develop inflammation of the colon and exhibit symptoms such as diarrhea, rectal bleeding, and weight loss. DSS-induced acute colitis has been reported to be a T cell-independent model [13]. However, in chronic colitis induced by multiple cycles or in the recovery phase of DSS, adaptive immunity plays an important role in the disease process [14]–[16].
Chemokines and their receptors are considered to be important factors in the pathogenesis of IBD. Several chemokines and their receptors, including CCL2, CCL3, CCL4, CCL5, CCL17, CCL22, CXCL8, CXCL10, CCR2 and CCR5 have been documented to be up-regulated in IBD tissue [17]–[24]. CCL2 is a potent chemoattractant and an activator of monocytes [25]. CCL3, CCL4 and CCL5 recruit memory and activated CD4 and CD8 T cells [26]. Intestinal epithelial cells can rapidly produce CCL2 and CCL5 upon exposure to inflammatory mediators [27], [28]. CCR2 and CCR5 are involved in both monocyte- and macrophage-mediated immune responses, and in the regulation of T cell migration and activation. Mice deficient in CCR2 or CCR5 are protected from DSS-induced colitis [29].
CD69 is a type II membrane protein expressed as a homodimer of heavily glycosylated subunits [30]. It is known as an early activation marker antigen of lymphocytes [31], [32], and its expression is upregulated on T cells in the inflamed mucosa [33]–[35]. CD69 is also involved in the regulation of T cell egress from the thymus [36], [37] and secondary lymphoid organs [38]. We and other groups have reported a role for CD69 in the regulation of arthritis [39], [40], asthma [41], [42], myocarditis [43] and tumor immunity [44], [45]. More recently, Radulovic et al. have reported a role for CD69 in the development of colitis using a CD45RBhigh CD4 T cell adaptive transfer model [46]. The transfer of CD69-deficient CD45RBhigh CD4 T cells into RAG-deficient hosts induced accelerated colitis. CD69-deficient CD4 T cells showed reduced potential to differentiate into FoxP3+ regulatory T cells in vivo and in vitro.
We herein investigated the role of CD69 using a DSS-induced acute and chronic colitis model. In CD69-deficient (CD69 KO) mice, both acute and chronic colitis were attenuated. Cell transfer of wild-type (WT), but not CD69-deficient CD4 T cells, restored the induction of acute colitis in CD69 KO mice, indicating a critical role of CD69 expression in CD4 T cells. The infiltration of FoxP3+ cells was similar in the colons of DSS-treated CD69 KO mice compared with that of DSS-treated WT mice. On the other hand, the IL-10 expression was significantly increased in the colons of DSS-treated CD69 KO mice. These results suggest that the function of CD69 in the inflammatory response is complicated, but that it may represent a potential therapeutic target for Crohn’s disease or other forms of IBD.
Materials and Methods
Ethics Statement
All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Yamaguchi University (Permit Number: 10–008).
Animal Studies
CD69-deficient (CD69 KO) mice [39] were backcrossed with BALB/c mice 15 times. The BALB/c were purchased from Charles River Laboratories (Tokyo, Japan). Mice were maintained under specific-pathogen-free conditions. All animal care was in accordance with the guidelines of Yamaguchi University.
Induction and General Assessment of Colitis
Acute colitis was induced in 7 to 8-week-old mice by adding 4% (w/v) DSS (MW 36,000–50,000; MP Biomedicals, Solon, Ohio, USA) to drinking water that was filter-purified (Millipore Corp., Billerica, Massachusetts, USA) for 7 days. From day 7 onwards, animals received normal drinking water. To induce chronic colitis, mice were administered 2% DSS on days 0–5, 10–15, and 20–25. The DSS consumption, body weight, stool consistency and fecal blood loss were recorded daily. A disease activity index (DAI) [47] was calculated as described in Table S1. On day 8, 20 or 30, mice were sacrificed. After measuring the colon length, one half of the colon was fixed in 10% (vol./vol.) formalin, paraffin embedded and stained for a histological examination. The other half was frozen in liquid nitrogen and used for cytokine measurements and RNA extraction. For the anti-CD69 antibody treatment, mice were intraperitoneally injected with an anti-CD69 mAb (H1.2F3, 500 µg/mouse) on day 0. The data presented are representative of at least three individual experiments.
Colon Histology and Immunohistochemistry
Mice were sacrificed by CO2 asphyxiation on day 8, and the colons were fixed with 10% (vol./vol.) formalin in PBS and embedded in paraffin. The samples were sectioned and stained with hematoxylin and eosin to examine the pathological changes under a light microscope. Colon specimens were embedded in Tissue-Tek OCT compound, frozen in liquid nitrogen, and cut by a cryostat into 6 µm thick sections. The endogenous peroxidase activity, as well as nonspecific protein binding, was sequentially blocked using 0.6% hydrogen peroxide and the Biotin-Blocking System reagent (DAKO, Glostrup, Denmark), respectively. The sections were incubated with a rabbit anti-CD3 antibody (DAKO), hamster anti-CD69 mAb (AbD, Oxford, Uk) or a rat anti-FoxP3 mAb (eBioscience, San Diego, California, USA) at 10 µg/ml overnight at 4°C and were then washed in TBST. Bound Ab was detected by sequential incubation with biotinylated rabbit anti-hamster IgG and streptavidin-HRP, followed by 3,3-diaminobenzidine (DAKO). Slides were then washed in water and counterstained with hematoxylin.
Adoptive Transfer of CD4 T Cells and Neutrophils
Splenic CD4+ T cells from wild-type (WT) BALB/c mice were purified using a CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch Glad-bach, Germany) and an Auto-MACS sorter (Miltenyi Biotec), yielding a purity of >98%. These cells were administered intravenously through the tail vain to CD69-KO mice (3×107 cells/mouse) on day –1. For neutrophil preparation, wild-type BALB/c mice were injected intraperitoneally with 2 ml of 4% thioglycolate (Merck, Darmstadt, Germany), and peritoneal neutrophils were recovered 4 h later by collecting the peritoneal lavage with 5 ml of saline [48]. Neutrophils in the peritoneal lavage were stained with biotin-conjugated Gr-1 and streptavidin microbeads, then purified using an Auto-MACS sorter, yielding a purity of >90%. Fifteen million neutrophils were injected intravenously into CD69 KO mice on days 0 and 2.
Cytokine and Chemokine Expression in the Colon Determined by Quantitative RT-PCR
The total colonic RNA was extracted using Trizol (Invitrogen, Carlsbad, California, USA). The RNA concentration was determined spectrophotometrically, and the sample quality was assessed after agarose electrophoresis. The cDNA synthesis and Quantitative RT-PCR were performed as described previously [49]. The primers and Taq Man probes used for the detection of IL-1β, IL-6, IL-10, CCR2, CCR3, CCL2, CCL4, CCL5 and HPRT were purchased from Applied Biosystems. The expression was normalized to the HPRT signal.
Isolation of Lamina Propria (LP) Cells
The colons were removed, washed in HBSS containing 5% FCS, cut into small pieces, and incubated with HBSS containing 5% FCS and 1 mM DTT for 40 min with gentle agitation to remove epithelial cells. Tissue specimens were then incubated while being shaken in HBSS containing 5% FCS with 1.5 mg/ml collagenase IV (SIGMA-Aldrich, St. Louis, Missouri, USA) at 37°C for 40 min. The supernatant was centrifuged and the pellet was washed with RPMI 1640. LP cells were isolated by Percoll (GE Healthcare, Uppsala, Sweden) density gradient centrifugation (800×g for 25 min) and collected at the interface. If necessary, CD4+ cells were isolated by magnetic negative selection with the CD4 MACS system (Miltenyi Bictec).
Immunofluorescent Staining and Flow Cytometric Analysis
In general, one million cells were incubated on ice for 30 min with the appropriate staining reagents, according to a standard method [50]. The intracellular staining of IL-10 was performed as described previously [51]. A PE-conjugated anti-IL-10 mAb (JES5-16E3, BD Biosciences, San Jose, California, USA), FITC-conjugated anti-Gr-1 mAb (RB6-8C5, BD Biosciences), APC-conjugated anti-CD4 mAb (RM4-5, BD Biosciences), anti-CD8a mAb (53-6.7, BD Biosciences), anti-CD11b mAb (M1/70, eBioscience) and anti-CD11c mAb (N418, BioLegend, San Diego, California, USA) were used for detection of the target proteins.
Proliferation Assay
Splenic CD4+ T cells (2×105) prepared by the AutoMACS sorter were stimulated in 200 µl cultures for 40 h with immobilized anti-TCRβ mAb (H57-597, BD Biosciences), PMA (50 ng/ml) and ionomycin (500 nM). [3H]Thymidine (37 kBq/well) was added to the stimulation culture for the last 16 h, and the incorporated radioactivity was measured on a beta plate [51].
Analysis of the Efficiency of Cell Division
CD4+ T cells isolated from lamina propria were labeled with CFSE (Invitrogen) as described previously [52]. The cells were cultured in complete medium in the presence of Con A (2 µg/ml) (WAKO, Osaka, Japan) for 48 h, and then were analyzed on a FACSCalibur instrument (BD Biosciences).
Statistical Analysis
The disease activity indices were statistically analyzed using the Mann-Whitney U test. Differences in parametric data were evaluated by Student’s t-test. Differences of p<0.05 were considered to be statistically significant. The variance of the groups was tested for equality by the F test prior to the t-test.
Results
Improvement of DSS-induced Acute Colitis in CD69 KO Mice
In order to evaluate the role of CD69 in the development of experimental colitis, acute colitis was induced in CD69 KO and control wild-type (WT) mice by adding 4% DSS to their drinking water. The survival rates were significantly higher in CD69 KO mice compared with those in control WT mice after DSS administration (Fig. 1A). CD69 KO mice showed significant protection against DSS-induced acute colitis, as indicated by the weight loss and clinical scores for weight loss, bleeding, and diarrhea (Fig. 1B and 1C). On day 8, the DSS-treated WT mice lost more than 20% of their body weight (maintaining 76.5±2.8% of their original weight, Fig. 1C). In contrast, the DSS-treated CD69 KO mice remained active, and their body weight was 99.0±2.0% of their original weight on day 8.
Figure 1. Inhibition of dextran sulphate sodium (DSS)-induced acute colitis in CD69-deficient (CD69 KO) mice.
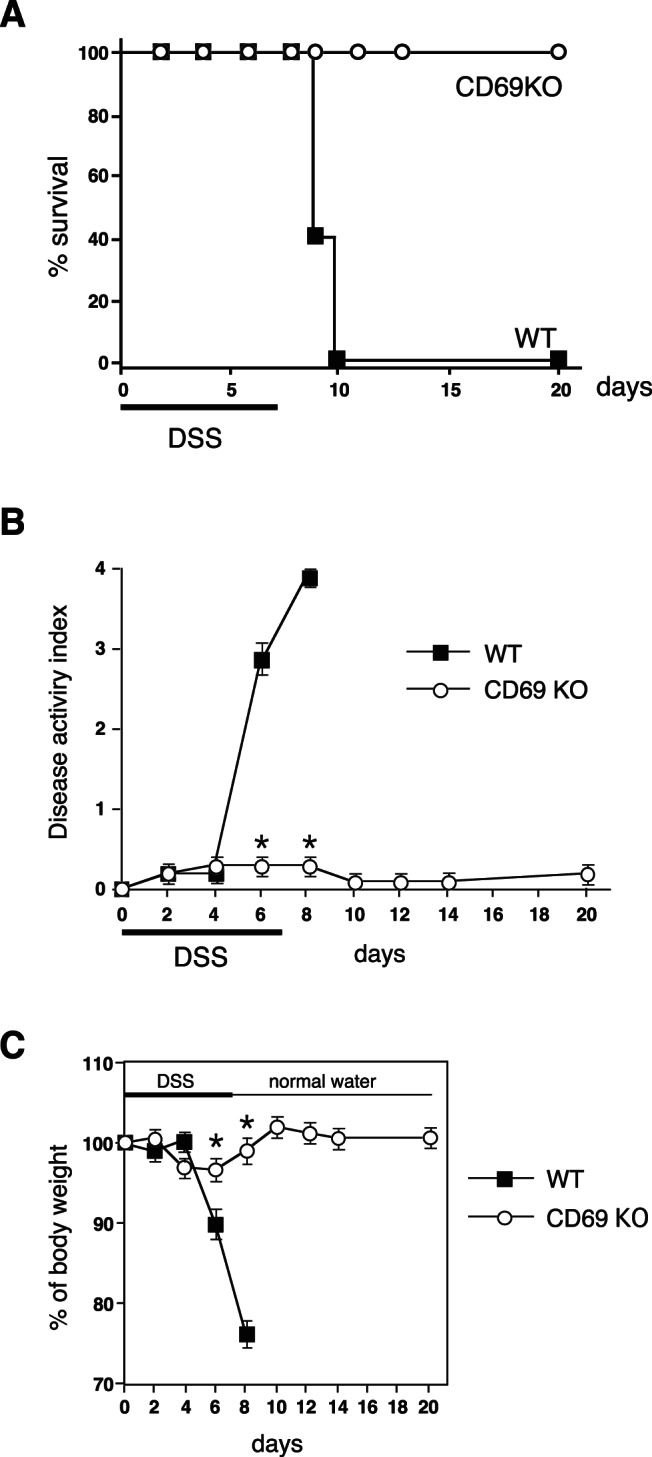
Acute colitis was induced by giving animals 4% DSS in drinking water for 7 days, followed by normal drinking water. (A) The survival rates of CD69 KO and wild-type (WT) mice after the initiation of during DSS-induced acute colitis. The survival was recorded daily (n = 10 per group). (B, C) Changes in the disease activity index (B) and body weight (%) (C) over the course of DSS treatment in CD69 KO and WT mice. The data are presented as the means (SD) (n = 10 per group). *p<0.01. Three independent experiments were performed with similar results.
The colon length progressively shortened and luminal bleeding was observed in WT mice on day 8 after DSS administration (Fig. 2A and 2B). The severity of changes in the gross appearance and in luminal bleeding was significantly lower and the colon length was maintained in the CD69 KO mice (Fig. 2A and 2B). During the histological examination, crypt damage, ulceration, and infiltration of inflammatory cells were observed in the colons of DSS-treated WT mice (Fig. 2C). On the other hand, the histological analysis of colons from DSS-treated CD69 KO mice showed greatly reduced numbers of infiltrating cells, a lower degree of mucosal injury, and less edema. These results indicate that the DSS-induced acute colitis was attenuated in CD69 KO mice.
Figure 2. DSS-induced colocecal damage was reduced in CD69 KO mice.

(A) The colon length. On day 8, the colon length of DSS-exposed wild-type (WT) and CD69 KO mice and control mice was measured. The data are presented as the means (SEM) (n = 12 per group). *p<0.01. (B) A representative photograph showing the gross appearance of the colon from each group is shown. (C) Histological sections of inflamed colons. Colons were taken on day 8 from WT and CD69 KO mice that received DSS in drinking water. Sections were prepared and stained with hematoxylin and eosin (original magnification 40×). (D) Immunohistochemical staining of CD69-positive cells in the colonic tissues of DSS-treated WT mice (original magnification 300× and 600×). (E, H) Immunohistochemical staining of CD3-positive cells (E) and FoxP3-positive cells (H) in the colonic tissues of DSS-treated WT and CD69 KO mice (original magnification 200×). (F, I) Summary of the accumulation of CD3-positive cells (F) and FoxP3-positive cells (I). Data are from 20 fields from 5 mice. *p<0.01. (G) The expression of CD69 on electronically gated lamina propria CD4 T cells from control (green line) and DSS-treated (red line) WT mice. Background staining is shown as hatched areas.
Expression of CD69 Molecules on Infiltrating Cells in the Colon
Next, we examined the expression of CD69 by the infiltrating cells in the colon of WT mice treated with DSS. Immunohistochemical staining was performed on the colons from DSS-treated WT mice. As shown in Fig. 2D, significant expression of CD69 was detected on the infiltrating cells in the colons of DSS-treated WT mice. However no significant expression of CD69 was observed in the colons of untreated WT mice (Fig. 2D).
Infiltration of CD69-deficient T Cells into the Colon
To evaluate the ability of CD69-deficient T cells to infiltrate into the inflamed colon tissue, we performed immunohistochemical staining of colonic cross sections from DSS-treated WT and CD69 KO mice. As can be seen in Fig. 2E, substantial numbers of CD3+ cells had infiltrated into the colons of DSS-treated WT mice on day 8. The infiltration of CD3+ cells was significantly reduced in the colons of CD69 KO mice (Fig. 2E and 2F). These results indicate that the infiltration of CD69-deficient CD3+ cells in the inflamed colon was reduced in comparison to that of WT CD3+ cells.
We next assessed the cell surface expression of CD69 on the infiltrating CD4 T cells by flowcytometry. As shown in Fig. 2G, the cell surface expression of CD69 on the lamina propria CD4 T cells was upregulated in DSS-treated WT mice.
We also performed immunohistochemical staining for FoxP3 in the colonic cross sections from DSS-treated WT and CD69 KO mice. As shown in Fig. 2H, FoxP3+ cells had infiltrated into the colons of DSS-treated WT mice on day 8. The infiltration of FoxP3+ cells was similar in the colons of DSS-treated CD69 KO mice compared with that of DSS-treated WT mice (Fig. 2H and 2I). These results indicate that the impaired infiltration of CD69-deficient CD3+ cells in the inflamed colon was not due to the increased infiltration of FoxP3+ regulatory T cells in the inflamed colon.
Cytokine, Chemokine and Receptor Expression in the Colon
DSS treatment strongly induces several inflammatory cytokines and chemokines, such as CCL2, CCL3 and CCL5. In order to examine the pattern of cytokine and chemokine expression in the DSS-treated animals, we also performed reverse transcription (RT) PCR using RNA derived from whole colonic tissue. There were no significant increases in the mRNA expression of any of these molecules in either the untreated WT or CD69 KO mice (data not shown). However, DSS treatment caused strong induction of IL-1β, IL-6, CCL2, CCL4 and CCL5 mRNA in DSS-treated WT mice in comparison with untreated mice (data not shown). The expression of IL-1β, IL-6, CCL2, and CCL4 was decreased in the colons of DSS-treated CD69 KO mice in comparison to the DSS-treated WT mice (Fig. 3A). The expression of CCR3 was partially inhibited in the colons of DSS-treated CD69 KO mice. In contrast, the expression levels of CCR2 and CCL5 were similar in the colons of DSS-treated CD69 KO mice and DSS-treated WT mice. The IL-10 expression was significantly increased in the colons of DSS-treated CD69 KO mice in comparison to DSS-treated WT mice. These results suggest that CD69 KO mice are protected from severe colitis because of the reduced induction of proinflammatory cytokines and chemokines, such as IL-1β, IL-6, CCL2 and CCL4. In addition, the higher levels of IL-10 in the colon may be involved in the protection of CD69 KO mice from severe colitis.
Figure 3. The expression of cytokines, chemokines and their receptors in the colons of DSS-treated mice.

(A) The expression of cytokines, chemokines and their receptor mRNA in the colons of DSS-treated mice. Mice were sacrificed on day 8 and the colon tissues were harvested. Whole colonic RNA was isolated, reverse-transcribed into cDNA, and the expression levels of IL-1β, IL-6, IL-10, CCR2, CCR3, CCL2, CCL4 and CCL5 were determined by real-time quantitative PCR. Data are expressed as the ratios of the target mRNA levels to the HPRT mRNA level (n = 5 per group). *p<0.05. Data are presented as the means (SD). The data are representative of three independent experiments. (B) The intracellular staining profiles of IL-10 in the lamina propria CD4 T cells (CD4+), CD8 T cells (CD8+), macrophages (CD11b+, Gr-1int) and dendritic cells (CD11c+) are shown as the percentages of cells in each area. The ratios of IL-10+ cells/total cells of each cell subset are shown over the area bar. The ratios of IL-10+ cells in each subset/total cells of all subsets are shown under the area bar. The results are representative of three independent experiments.
In order to identify the major sources of IL-10 in the colon, we performed intracellular IL-10 staining of the lamina propria cells in the colon from DSS-treated mice. The percentages of total IL-10+ cells in CD69 KO mice were higher than those in WT mice even in the untreated mice (Fig. 3B). The number of IL-10+ cells increased in the DSS-treated mice in comparison to untreated WT and CD69 KO mice. We also performed intracellular staining of CD4 T cells, CD8 T cells, macrophages and dendritic cells. The percentages of IL-10+ cells in all subsets in CD69 KO mice were higher than those in WT mice. However, the number of IL-10+ cells among these four cell subsets was less than 20% of the total IL-10+ cells in CD69 KO mice. Thus, the major source of IL-10 in the colon of CD69 KO mice seems to be a cell subset other than CD4 T cells, CD8 T cells, macrophages and dendritic cells.
Requirement of CD69 on CD4 T Cells for the Induction of DSS-induced Colitis
To investigate the cellular basis underlying the requirement of CD69 in the pathogenesis of DSS-induced acute colitis, we performed cell transfer experiments in which splenic CD4 T cells from WT and CD69 KO mice were adoptively transferred into CD69 KO mice. As shown in Fig. 4A, the survival rates were significantly restored by the transfer of WT CD4 T cells to the CD69 KO mice. Although the weight loss did not show any significant difference following the transfer of WT CD4 T cells to the CD69 KO mice (Fig. 4C), the clinical scores for weight loss, bleeding, and diarrhea were all worsened by the transfer of WT CD4 T cells to the CD69 KO mice (Fig. 4B).
Figure 4. DSS-induced colitis was restored by adoptive transfer of wild-type (WT) CD4 T cells into CD69 KO mice.

(A) The survival rates of WT, CD69 KO mice without cell transfer, and CD69-KO mice that received WT CD4 T cells (WT-CD4T), CD69 KO CD4 T cells (KO-CD4T), or WT neutrophils (WT-Neutro) during DSS treatment. Survival was recorded daily (n = 5 per group). (B, C) Changes in the disease activity index (B) and body weight (%) (C) over the course of DSS treatment. The data are presented as the means (SD) (n = 5 per group). *p<0.05. (D) Histological sections of inflamed colons. Colons were taken on day 8 from mice receiving DSS in their drinking water. Sections were fixed and stained with hematoxylin and eosin (original magnification 40×). Two independent experiments were performed with similar results.
On the other hand, the transfer of WT neutrophils or CD69 KO CD4 T cells to the CD69KO mice failed to change the survival rates or clinical scores. The histological analysis revealed that the transfer of WT CD4 T cells resulted in a substantial increase in crypt damage, ulceration, and infiltration of inflammatory cells in the colon, whereas transfer of WT neutrophils or CD69 KO CD4 T cells failed to induce the inflammatory cell infiltration (Fig. 4D). These results indicate that CD69 molecules on CD4 T cells play an important role in the induction of DSS-induced acute colitis.
With regard to the CD4 T cell function, the anti-TCRβ mAb-induced proliferative responses of splenic CD4 T cells were similar between WT and CD69 KO mice (Fig. S1A). We next examined the Con A-induced cell division of lamina propria CD4 T cells. After 48 h of culture, the cells had divided two to three times in the case of both WT and CD69 KO mouse T cells. The rate of cell division of the lamina propria CD4 T cells was indistinguishable between WT and CD69 KO mice (Fig. S1B and S1C). Similar results were obtained using splenic CD4 T cells (data not shown). These results that there are no obvious differences in the proliferations of CD4 T cells from the CD69 KO mice.
The in vivo Treatment with an Anti-CD69 mAb Inhibited the Induction of DSS-induced Acute Colitis
In order to explore the potential therapeutic effect of the administration of an anti-CD69 monoclonal antibody (mAb) during DSS-induced acute colitis, WT BALB/c mice were treated with 500 µg of anti-CD69 mAb or control antibody (Ab) on day 0. The survival rates were significantly increased in the anti-CD69 mAb-treated WT mice compared with control Ab-treated WT mice (Fig. 5A). Anti-CD69 mAb treated-WT mice showed significant protection against DSS-induced acute colitis, as indicated by their decreased weight loss and better clinical scores for weight loss, bleeding, and diarrhea (Fig. 5B and 5C). Furthermore, a histological analysis of the colons from anti-CD69 mAb-treated WT mice showed greatly reduced numbers of infiltrating cells, a lower degree of mucosal injury, and less edema (Fig. 5D). These results suggest that the development of DSS-induced acute colitis can be inhibited by treatment with an anti-CD69 mAb.
Figure 5. Effect of in vivo treatment with an anti-CD69 monoclonal antibody (mAb) on DSS-induced colitis.

(A) Wild-type (WT) BALB/c mice were treated with an anti-CD69 mAb or control hamster IgG on day 0. The survival of each group during DSS-induced colitis was recorded daily (n = 5 per group). (B, C) Changes in the disease activity index (B) and body weight (%) (C) over the course of DSS treatment in WT mice treated with the anti-CD69 mAb or control hamster IgG. The data are presented as the means (SD) (n = 5 per group). *p<0.01. **p<0.05. (D) Histological sections. Colons were taken on day 8 from control and DSS-exposed WT mice treated with the anti-CD69 mAb or control hamster IgG. Sections were fixed and stained with hematoxylin and eosin (original magnification 40×). Three independent experiments were performed with similar results.
Improvement of DSS-induced Chronic Colitis in CD69 KO Mice
To investigate the role of CD69 in the pathogenesis of DSS-induced chronic colitis, CD69 KO mice were administered 2% DSS on days 0–5, 10–15, and 20–25. As shown in Fig. 6A and 6B, DSS-induced chronic colitis was dramatically attenuated in CD69 KO mice, as indicated by the reduced weight loss and disease activity index of weight loss, bleeding, and diarrhea. The disease activity index was especially improved in the first recovery phase (days 6) of the exaggerated colitis induced by DSS. These results indicate that the DSS-induced chronic colitis is attenuated in CD69 KO mice.
Figure 6. DSS-induced chronic colitis in CD69 KO mice.

CD69 KO mice and wild-type (WT) mice were administered 2% DSS on days 0–5, 10–15, and 20–25. The body weight (A) and disease activity index (B) were monitored every day, and the values for body weight are expressed as the percentage of body weight on day 0. The data are presented as the means with SD (n = 5 per group). *p<0.01. Three independent experiments were performed with similar results.
Discussion
In this study, we used CD69 KO mice to assess the role of CD69 in a DSS-induced colitis model. We have herein demonstrated that both acute and chronic colitis were attenuated in CD69 KO mice (Fig. 1 and 6), and that the CD69 expressed on CD4 T cells plays an important role in the development of colitis (Fig. 4). The attenuated colitis in CD69 KO mice appeared to be due largely to the reduced infiltration of T cells into the inflamed colon (Fig. 2E). In addition, a therapeutic effect of administering an anti-CD69 mAb was revealed (Fig. 5), indicating that CD69 could represent a new target for mAb treatment in IBD patients.
CD69 KO mice showed reduced colitis in both acute and chronic DSS-induced colitis models (Fig. 1, 2A, 2B, 2C and 6), and CD69 was highly expressed on the infiltrating CD4 T cells in DSS-treated WT mice (Fig. 2G). The infiltration of CD3+ cells was significantly reduced in the colons of CD69 KO mice (Fig. 2E). The DSS-induced colitis was restored by adoptive transfer of WT CD4 T cells, but not CD69-deficient CD4 T cells, to the CD69 KO mice (Fig. 4). These results indicate that CD69 molecules on CD4 T cells play an important role in the induction of DSS-induced acute colitis. The proliferative responses of CD4 T cells from the spleen and colonic lamina propria were indistinguishable between WT and CD69 KO mice (Fig. S1). Thus, the regulation of the proliferative activity of CD4 T cells may not be the mechanism by which the CD69 on CD4 T cells plays a role in inducing colitis. Although DSS-induced acute colitis has been reported to be a T cell-independent model [13], adaptive immunity plays an important role in the disease progression of chronic colitis and/or in the recovery phase following the administration of DSS [14]–[16]. Thus, CD69+ CD4 T cells may be involved in the disease progression of chronic colitis or the recovery from acute colitis. On the other hand, the survival rates and the clinical scores were significantly restored by the transfer of WT CD4 T cells to CD69 KO mice, but the weight loss did not show any significant differences following this restoration. These results suggest that CD69-expressing cells other than CD4 T cells may also contribute to the pathogenesis of DSS-induced colitis.
We observed that the migration of CD3+ cells was reduced in the colons of CD69 KO mice (Fig. 2E). CCL2 and CCL4 recruit memory and activated CD4 and CD8 T cells [26]. The expression of both CCL2 and CCL4 was dramatically inhibited in the colons of DSS-treated CD69 KO mice (Fig. 3A). Therefore, it is possible that inhibiting the induction of CCL2 and CCL4 may reduce the migration of T cells in the colon and protect CD69 KO mice from severe colitis. Although the ligand for CD69 has not been identified, another possible scenario is that a putative ligand may be induced and expressed on the inflamed colon tissues. Activated CD4 T cells expressing CD69 then may migrate into the colon tissue, and remain at the inflammatory site efficiently via the CD69/CD69 ligand interaction. CD69-deficient CD4 T cells may not be retained at the inflammatory site very efficiently because there would be no interaction between CD69 and its ligand(s).
Another potential mechanism could be that the CD69 expressed on CD4 T cells may regulate the induction and/or maintenance of inflammation in the colon tissue (Fig. 4). Interestingly, difference in the migration of FoxP3+ regulatory T cells in the colon was not detected in the CD69 KO mice (Fig. 2H). On the other hand, IL-10 expression was significantly increased in the colons of DSS-treated CD69 KO mice (Fig. 3). Higher expression levels of IL-10 in the colon may be involved in the protection of CD69 KO mice from severe colitis. Intracellular IL-10 staining of the lamina propria cells in the colon revealed higher percentages of IL-10+ cells in even untreated CD69 KO mice (Fig. 3B). IL-10 is a cytokine that is predominantly secreted by CD4 memory and effector T cells, regulatory T cells and antigen-presenting cells, such as monocytes/macrophages. The number of IL-10+ cells in the total population of CD4 T cells, including CD4+ FoxP3+ regulatory T cells, and CD11b+Gr-1int macrophages was less than 20% of the total IL-10+ cells in CD69 KO mice. These results suggest that the major source of IL-10 in the colon of CD69 KO mice seems to be a cell subset other than these cells. Therefore, further investigations will be required to identify the major cell source of IL-10 and the molecular mechanism(s) underlying the high IL-10 expression in CD69 KO mice.
This study demonstrated the therapeutic effect of the administration of an anti-CD69 mAb (Fig. 5). The ligand for CD69 has not been identified, but it is possible that the anti-CD69 mAb may block the interaction between CD69 and putative CD69 ligands, resulting in reduced CD4 T cell migration and attenuated colitis. Another mechanism that may explain the inhibitory effect of anti-CD69 antibody treatment could be the downregulation of CD69 molecules on CD4 T cells. It is possible that CD69 may transduce some type of inhibitory signal to reduce CD4 T cell activity. A third potential mechanism could be antibody-dependent cellular cytotoxicity (ADCC) resulting from the anti-CD69 mAb, but this is unlikely, because no evidence supporting ADCC was obtained in our previous study [41].
Recently CD69 has been suggested to increase the incidence and severity of colitis in a T cell transfer colitis model [46]. In this model, colitis was induced by transplanting RAG KO mice with CD45RBhigh CD4 T cells. Radulovic et al. demonstrated that the CD4 T cells from CD69 KO mice produced higher amounts of the proinflammatory cytokines IFN-γ, TNF-α, and IL-21, whereas the production of TGF-β1 was decreased. The transfer of CD69-deficient CD45RBhigh CD4 T cells into RAG KO hosts induced accelerated colitis. CD69-deficient CD4 T cells showed a lower potential to become FoxP3+ regulatory T cells. In contrast, in our present study using a model of DSS-induced colitis, the FoxP3+ regulatory T cells migrated normally in the inflamed tissue (Fig. 2H), and impaired colitis was observed in both the acute and chronic models of colitis (Fig. 1, 4, and 6). It appears that the contribution of Treg cells to the pathogenesis of colitis in these two models may be different. This could be the reason why apparent contradictory results in CD69-deficient mice were obtained in these two models. CD69 may play an important role in the induction of inflammation as a result of its expression on effector T cells, and also in the inhibition of inflammation by Treg cells.
In summary, our results indicate that the CD69 expressed on CD4 T cells plays a critical role in the development of DSS-induced acute and chronic colitis via the efficient migration of activated CD4 T cells into the colon. Moreover, our mAb administration experiments revealed that CD69 could be a potential therapeutic target for inflammatory bowel disease.
Supporting Information
Functional characterization of CD69 KO mouse T cells. (A) Splenic CD4 T cells from WT and CD69 KO mice were stimulated with immobilized anti-TCRβ mAb or PMA (50 ng/ml) plus ionomycin (500 nM). The mean [3H]thymidine incorporation of each group is shown with SDs. (B, C) CD4 T cells isolated from the lamina propria were labeled with CFSE and stimulated with Con A (2 µg/ml). After culturing them for 48 h, the number of cell divisions (0 to 4) was assessed by flow cytometry (B), and the percentages of the cells in the gates representing the different numbers of cell divisions are shown (C). Three independent experiments were performed and similar results were obtained each time.
(TIFF)
Scoring of disease activity index. The disease activity index is the combined scores of weight loss, stool consistency and bleeding divided by three. aNormal stools = well-formed pellets, loose stools = pasty and semi-formed stools which do not stick to the anus, diarrhea = liquid stools that stick to the anus.
(TIFF)
Acknowledgments
The authors would like to thank Kaoru Sugaya, Toshihiro Ito and Mitsuaki Kimoto for their excellent technical assistance.
Funding Statement
This work was supported by JSPS KAKENHI (Japan) (http://www.jsps.go.jp/english/index.html) Grant Number 22591093 and MEXT KAKENHI (Japan) (http://www.mext.go.jp/english/) Grant Number 23111520. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, et al. (1998) CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J Exp Med 187: 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fiocchi C (1998) Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology 115: 182–205. [DOI] [PubMed] [Google Scholar]
- 3. Maaser C, Kagnoff MF (2002) Role of the intestinal epithelium in orchestrating innate and adaptive mucosal immunity. Z Gastroenterol 40: 525–529. [DOI] [PubMed] [Google Scholar]
- 4. McKay DM (1999) Intestinal inflammation and the gut microflora. Can J Gastroenterol 13: 509–516. [DOI] [PubMed] [Google Scholar]
- 5. Sartor RB (1997) The influence of normal microbial flora on the development of chronic mucosal inflammation. Res Immunol 148: 567–576. [DOI] [PubMed] [Google Scholar]
- 6. Brandtzaeg P, Haraldsen G, Rugtveit J (1997) Immunopathology of human inflammatory bowel disease. Springer Semin Immunopathol 18: 555–589. [DOI] [PubMed] [Google Scholar]
- 7. Bouma G, Strober W (2003) The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 3: 521–533. [DOI] [PubMed] [Google Scholar]
- 8. Cobrin GM, Abreu MT (2005) Defects in mucosal immunity leading to Crohn’s disease. Immunol Rev 206: 277–295. [DOI] [PubMed] [Google Scholar]
- 9. Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, et al. (1996) Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 157: 1261–1270. [PubMed] [Google Scholar]
- 10. Targan SR, Karp LC (2005) Defects in mucosal immunity leading to ulcerative colitis. Immunol Rev 206: 296–305. [DOI] [PubMed] [Google Scholar]
- 11. Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, et al. (1993) Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol 94: 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Podolsky DK (2002) Inflammatory bowel disease. N Engl J Med 347: 417–429. [DOI] [PubMed] [Google Scholar]
- 13. Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, et al. (1994) Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107: 1643–1652. [DOI] [PubMed] [Google Scholar]
- 14. Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, et al. (1998) Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 114: 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuchiya T, Fukuda S, Hamada H, Nakamura A, Kohama Y, et al. (2003) Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol 171: 5507–5513. [DOI] [PubMed] [Google Scholar]
- 16. Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, et al. (2002) The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest 109: 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Annunziato F, Cosmi L, Galli G, Beltrame C, Romagnani P, et al. (1999) Assessment of chemokine receptor expression by human Th1 and Th2 cells in vitro and in vivo. J Leukoc Biol 65: 691–699. [DOI] [PubMed] [Google Scholar]
- 18. Banks C, Bateman A, Payne R, Johnson P, Sheron N (2003) Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J Pathol 199: 28–35. [DOI] [PubMed] [Google Scholar]
- 19. Connor SJ, Paraskevopoulos N, Newman R, Cuan N, Hampartzoumian T, et al. (2004) CCR2 expressing CD4+ T lymphocytes are preferentially recruited to the ileum in Crohn’s disease. Gut 53: 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grimm MC, Elsbury SK, Pavli P, Doe WF (1996) Interleukin 8: cells of origin in inflammatory bowel disease. Gut 38: 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jugde F, Alizadeh M, Boissier C, Chantry D, Siproudhis L, et al. (2001) Quantitation of chemokines (MDC, TARC) expression in mucosa from Crohn’s disease and ulcerative colitis. Eur Cytokine Netw 12: 468–477. [PubMed] [Google Scholar]
- 22. Mazzucchelli L, Hauser C, Zgraggen K, Wagner HE, Hess MW, et al. (1996) Differential in situ expression of the genes encoding the chemokines MCP-1 and RANTES in human inflammatory bowel disease. J Pathol 178: 201–206. [DOI] [PubMed] [Google Scholar]
- 23. McCormack G, Moriarty D, O’Donoghue DP, McCormick PA, Sheahan K, et al. (2001) Tissue cytokine and chemokine expression in inflammatory bowel disease. Inflamm Res 50: 491–495. [DOI] [PubMed] [Google Scholar]
- 24. Uguccioni M, Gionchetti P, Robbiani DF, Rizzello F, Peruzzo S, et al. (1999) Increased expression of IP-10, IL-8, MCP-1, and MCP-3 in ulcerative colitis. Am J Pathol 155: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ (1989) Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med 169: 1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schrum S, Probst P, Fleischer B, Zipfel PF (1996) Synthesis of the CC-chemokines MIP-1alpha, MIP-1beta, and RANTES is associated with a type 1 immune response. J Immunol 157: 3598–3604. [PubMed] [Google Scholar]
- 27. Kolios G, Wright KL, Jordan NJ, Leithead JB, Robertson DA, et al. (1999) C-X-C and C-C chemokine expression and secretion by the human colonic epithelial cell line, HT-29: differential effect of T lymphocyte-derived cytokines. Eur J Immunol 29: 530–536. [DOI] [PubMed] [Google Scholar]
- 28. Yang SK, Eckmann L, Panja A, Kagnoff MF (1997) Differential and regulated expression of C-X-C, C-C, and C-chemokines by human colon epithelial cells. Gastroenterology 113: 1214–1223. [DOI] [PubMed] [Google Scholar]
- 29. Andres PG, Beck PL, Mizoguchi E, Mizoguchi A, Bhan AK, et al. (2000) Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated Th2-type immune response in the intestine. J Immunol 164: 6303–6312. [DOI] [PubMed] [Google Scholar]
- 30. Ziegler SF, Ramsdell F, Hjerrild KA, Armitage RJ, Grabstein KH, et al. (1993) Molecular characterization of the early activation antigen CD69: a type II membrane glycoprotein related to a family of natural killer cell activation antigens. Eur J Immunol 23: 1643–1648. [DOI] [PubMed] [Google Scholar]
- 31. Testi R, D’Ambrosio D, De Maria R, Santoni A (1994) The CD69 receptor: a multipurpose cell-surface trigger for hematopoietic cells. Immunol Today 15: 479–483. [DOI] [PubMed] [Google Scholar]
- 32. Sancho D, Gomez M, Sanchez-Madrid F (2005) CD69 is an immunoregulatory molecule induced following activation. Trends Immunol 26: 136–140. [DOI] [PubMed] [Google Scholar]
- 33. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA (2001) IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 98: 13249–13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guehler SR, Bluestone JA, Barrett TA (1999) Activation and peripheral expansion of murine T-cell receptor gamma delta intraepithelial lymphocytes. Gastroenterology 116: 327–334. [DOI] [PubMed] [Google Scholar]
- 35. Aranda R, Sydora BC, McAllister PL, Binder SW, Yang HY, et al. (1997) Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol 158: 3464–3473. [PubMed] [Google Scholar]
- 36. Nakayama T, Kasprowicz DJ, Yamashita M, Schubert LA, Gillard G, et al. (2002) The generation of mature, single-positive thymocytes in vivo is dysregulated by CD69 blockade or overexpression. J Immunol 168: 87–94. [DOI] [PubMed] [Google Scholar]
- 37. Feng C, Woodside KJ, Vance BA, El-Khoury D, Canelles M, et al. (2002) A potential role for CD69 in thymocyte emigration. Int Immunol 14: 535–544. [DOI] [PubMed] [Google Scholar]
- 38. Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, et al. (2006) CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440: 540–544. [DOI] [PubMed] [Google Scholar]
- 39. Murata K, Inami M, Hasegawa A, Kubo S, Kimura M, et al. (2003) CD69-null mice protected from arthritis induced with anti-type II collagen antibodies. Int Immunol 15: 987–992. [DOI] [PubMed] [Google Scholar]
- 40. Sancho D, Gomez M, Viedma F, Esplugues E, Gordon-Alonso M, et al. (2003) CD69 downregulates autoimmune reactivity through active transforming growth factor-beta production in collagen-induced arthritis. J Clin Invest 112: 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miki-Hosokawa T, Hasegawa A, Iwamura C, Shinoda K, Tofukuji S, et al. (2009) CD69 controls the pathogenesis of allergic airway inflammation. J Immunol 183: 8203–8215. [DOI] [PubMed] [Google Scholar]
- 42.Martin P, Gomez M, Lamana A, Matesanz Marin A, Cortes JR, et al.. (2010) The leukocyte activation antigen CD69 limits allergic asthma and skin contact hypersensitivity. J Allergy Clin Immunol 126: 355–365, 365 e351–353. [DOI] [PubMed]
- 43. Cruz-Adalia A, Jimenez-Borreguero LJ, Ramirez-Huesca M, Chico-Calero I, Barreiro O, et al. (2010) CD69 limits the severity of cardiomyopathy after autoimmune myocarditis. Circulation 122: 1396–1404. [DOI] [PubMed] [Google Scholar]
- 44. Esplugues E, Sancho D, Vega-Ramos J, Martinez C, Syrbe U, et al. (2003) Enhanced antitumor immunity in mice deficient in CD69. J Exp Med 197: 1093–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Esplugues E, Vega-Ramos J, Cartoixa D, Vazquez BN, Salaet I, et al. (2005) Induction of tumor NK-cell immunity by anti-CD69 antibody therapy. Blood 105: 4399–4406. [DOI] [PubMed] [Google Scholar]
- 46. Radulovic K, Manta C, Rossini V, Holzmann K, Kestler HA, et al. (2012) CD69 Regulates Type I IFN-Induced Tolerogenic Signals to Mucosal CD4 T Cells That Attenuate Their Colitogenic Potential. J Immunol 188: 2001–2013. [DOI] [PubMed] [Google Scholar]
- 47. Cooper HS, Murthy SN, Shah RS, Sedergran DJ (1993) Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 69: 238–249. [PubMed] [Google Scholar]
- 48. Ajuebor MN, Das AM, Virag L, Flower RJ, Szabo C, et al. (1999) Role of resident peritoneal macrophages and mast cells in chemokine production and neutrophil migration in acute inflammation: evidence for an inhibitory loop involving endogenous IL-10. J Immunol 162: 1685–1691. [PubMed] [Google Scholar]
- 49. Nigo YI, Yamashita M, Hirahara K, Shinnakasu R, Inami M, et al. (2006) Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc Natl Acad Sci U S A 103: 2286–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakayama T, June CH, Munitz TI, Sheard M, McCarthy SA, et al. (1990) Inhibition of T cell receptor expression and function in immature CD4+CD8+ cells by CD4. Science 249: 1558–1561. [DOI] [PubMed] [Google Scholar]
- 51. Yamashita M, Kimura M, Kubo M, Shimizu C, Tada T, et al. (1999) T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc Natl Acad Sci U S A 96: 1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hasegawa A, Miki T, Hosokawa H, Hossain MB, Shimizu C, et al. (2006) Impaired GATA3-dependent chromatin remodeling and Th2 cell differentiation leading to attenuated allergic airway inflammation in aging mice. J Immunol 176: 2546–2554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Functional characterization of CD69 KO mouse T cells. (A) Splenic CD4 T cells from WT and CD69 KO mice were stimulated with immobilized anti-TCRβ mAb or PMA (50 ng/ml) plus ionomycin (500 nM). The mean [3H]thymidine incorporation of each group is shown with SDs. (B, C) CD4 T cells isolated from the lamina propria were labeled with CFSE and stimulated with Con A (2 µg/ml). After culturing them for 48 h, the number of cell divisions (0 to 4) was assessed by flow cytometry (B), and the percentages of the cells in the gates representing the different numbers of cell divisions are shown (C). Three independent experiments were performed and similar results were obtained each time.
(TIFF)
Scoring of disease activity index. The disease activity index is the combined scores of weight loss, stool consistency and bleeding divided by three. aNormal stools = well-formed pellets, loose stools = pasty and semi-formed stools which do not stick to the anus, diarrhea = liquid stools that stick to the anus.
(TIFF)