

Abstract
There are few effective therapies for high-risk sarcomas. Initial chemosensitivity is often followed by relapse. In vitro, mTOR inhibition potentiates the efficacy of chemotherapy on resistant sarcoma cells. Although sarcoma trials using mTOR inhibitors have been disappointing, these drugs were used as maintenance. We conducted a Phase I/II clinical trial to test the ability of temsirolimus to potentiate the cytotoxic effect of liposomal doxorubicin and present here the dose-finding portion of this study. Adult and pediatrics patients with recurrent or refractory sarcomas were treated with increasing doses of liposomal doxorubicin and temsirolimus using a continual reassessment method for escalation, targeting a dose-limiting toxicity rate of 20%. Blood samples were drawn before and after the first dose of temsirolimus in Cycles 1 and 2 for pharmacokinetic analysis. The maximally tolerated dose combination was liposomal doxorubicin 30 mg/m2 monthly with temsirolimus 20 mg/m2 weekly. Hematologic toxicity was common but manageable. Dose-limiting toxicities were primarily renal. Concurrent administration of liposomal doxorubicin resulted in increased exposure to sirolimus, the active metabolite of temsirolimus. Thus, the combination of liposomal doxorubicin and temsirolimus is safe for heavily pretreated sarcoma patients. Coadministration with liposomal doxorubicin did not alter temsirolimus pharmacokinetics, but increased exposure to its active metabolite.
Keywords: mTOR, cancer stem cell, chemoresistance, sarcoma, clinical trial
Introduction
There are few effective treatments for high risk bone and soft tissue sarcomas, and most patients diagnosed with these tumors die of their disease. Although many sarcomas respond initially to chemo- and radiation therapy, recurrence with refractory disease is frequent, and is the most common cause of death in these patients. The cancer stem cell hypothesis provides a possible explanation for the discrepancy between response to therapy and overall survival. This model predicts the existence of a small subpopulation of tumor cells that is inherently resistant to treatment and is capable of regrowing the original tumor and metastasizing, thus causing relapse and patient death 1, 2. Therapies that target cancer stem cells would be expected to dramatically improve the survival of patients with high risk sarcomas.
Work in our laboratory has identified a population of Ewing sarcoma cells enriched for a cancer stem cell phenotype. We found that cells expressing high levels of aldehyde dehydrogenase (ALDHhigh) exhibit stem cell properties: they are enriched for the ability to form colonies in soft agar, to grow as spheres when cultured under nonadherent conditions, and for the expression of so-called “stem cell genes” such as oct4 and nanog. Importantly, as few as 160 ALDHhigh cells injected into immune-deficient mice are sufficient to generate a tumor, as compared to 800,000 unsorted cells 3. Compared to unsorted cells, ALDHhigh Ewing sarcoma cells are relatively resistant to doxorubicin and etoposide in vitro. Our preliminary work (presented below) demonstrated that inhibition of mammalian target of rapamycin (mTOR) by sirolimus not only causes synergistic cytotoxicity with doxorubicin when the bulk tumor population is treated, but also increases the sensitivity of the ALDHhigh cells to chemotherapy in vitro, suggesting that the chemoresistance of sarcoma stem cells can be overcome, at least in vitro.
Based on these findings, we designed a clinical trial of the combination of liposomal doxorubicin with temsirolimus, an mTOR inhibitor that is converted to sirolimus in vivo, for patients with recurrent and refractory bone and soft tissue sarcomas. Encapsulating doxorubicin in pegylated liposomes allows improved localization of doxorubicin to tumors, leading to activity in chemotherapy-refractory disease 4. Pegylated liposomal doxorubicin is generally better tolerated than “naked” doxorubicin, allowing treatment of patients who have already received 450 mg/m2 of doxorubicin (the typical dose used in the initial therapy of patients with Ewing sarcoma and osteosarcoma). Although previous studies of mTOR inhibition in patients with sarcoma have been generally disappointing 5, 6, this may be because these agents were used as cytotoxics or as maintenance (administered to patients after completion of planned cytotoxic chemotherapy in order to prevent recurrence). Our approach is different – using mTOR inhibition to augment sensitivity to a more standard cytotoxic agent. Because this drug combination has not been administered together previously, we conducted a Phase 1/2 study to determine the maximally tolerated dose (MTD) combination, collect pharmacokinetic data on temsirolimus given in conjunction with liposomal doxorubicin, and determine the efficacy of this combination. We are testing the hypothesis that this combination will be provide a safe, effective treatment for sarcoma patients with relapsed or refractory disease. We report here the results of the Phase 1 portion of the study, including temsirolimus pharmacokinetics.
Methods
In Vitro Studies
TC71 and MHH-ES Ewing’s sarcoma cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. Cell lines were obtained from Dr. J Toretsky (Georgetown University) in 2010 and expression of EWS-FLI1 confirmed by RT-PCR upon receipt and again in 2012. As previously described 3, cells were treated with Aldefluor according to the manufacturer’s instructions and separated by flow cytometry into populations with high aldehyde dehydrogenase expression (ALDHhigh), low expression (ALDHlow), and flow through cells (cells passed through the flow cytometer but not sorted). Individual populations of cells were incubated concurrently for 48 hours with 250 nM doxorubicin and increasing concentrations of sirolimus. Cell survival was determined using an MTT assay according to the manufacturer’s instructions. For synergy experiments, 104 unsorted cells were plated in 96 well plates and incubated with doxorubicin, sirolimus, or both for 24 hours. Viability was assessed using the CCK-8 kit (Dojindo Molecular Technologies, Rockville, MD). Ewing sarcoma cells were chosen for this in vitro work because the identity of stem cells from other sarcoma types is less firmly established.
Patient Eligibility Requirements
Patients were eligible if they were at least 1 year of age and had a histologically confirmed diagnosis of a bone or soft tissue sarcoma that was recurrent or refractory to conventional therapy and for which standard curative options did not exist. Patients were required to have measurable disease that was amenable to percutaneous image-guided biopsy. In addition, patients were required to have an ECOG performance status ≤ 2 (Karnofsky or Lansky ≥ 60% for children), a life expectancy greater than 3 months, and adequate organ function (absolute neutrophil count ≥ 1500/μl, platelet count ≥ 100,000/μl, total bilirubin ≤ 1.5× institutional upper limit of normal, AST and ALT ≤ 2.5× institutional upper limit of normal, and creatinine ≤ 1.5× institutional upper limit of normal for age or a creatinine clearance ≥ 60 ml/min/1.73 m2). Because most patients were pretreated with anthracyclines and the study includes further anthracycline treatment, patients were evaluated with an electrocardiogram and echocardiogram prior to study entry and after every second dose of liposomal doxorubicin. Because temsirolimus can affect lipid metabolism, patients were also required to have a fasting cholesterol ≤ 350 mg/dl, fasting serum triglycerides ≤ 400 mg/dl, and amylase and lipase within normal limits (unless elevations were related to tumor involving the pancreas). Additionally, because of the effect of temsirolimus on glucose metabolism, patients were required to have a hemoglobin A1c ≤ 10%. Patients were excluded if they had a history of pulmonary hypertension or pneumonitis, prior therapy with an mTOR inhibitor, uncontrolled brain metastases, a history of hypersensitivity to macrolide antibiotics, or grade 3 or 4 proteinuria. The study was approved by the Johns Hopkins University Institutional Review Board and patients signed written informed consent according to institutional standards. The trial was registered with ClinicalTrials.gov (trial registration ID: NCT00949325).
Study Design and Treatment Plan
Temsirolimus and liposomal doxorubicin were both administered intravenously in the outpatient clinic. Temsirolimus was given weekly, and liposomal doxorubicin was administered every 28 days. On days when both drugs were administered, liposomal doxorubicin was given first. All dosing was based on body surface area to allow the concurrent enrollment of children and adults. The starting doses (designated Dose Level 3) were 30 mg/m2 of liposomal doxorubicin and 15 mg/m2 of temsirolimus. A liposomal doxorubicin dose of 30 mg/m2 is lower than typically administered as a single agent to women with ovarian cancer, who receive 50 mg/m2, but is the dose typically administered to patients with multiple myeloma receiving concurrent bortezomib. To allow both children and adults to be treated on the same study with comparable dosing, the starting dose of temsirolimus was chosen by converting the standard adult dose of 25 mg to a body surface area-based dose by dividing by 1.7 m2. Patients were pretreated with diphenhydramine to avoid infusional hypersensitivity reactions.
A modified continual reassessment method (CRM) was used to evaluate the safety and tolerability of the combination of liposomal doxorubicin and temsirolimus and to determine the MTD. Toxicities were graded according to the NCI-CTCAE v3.0. Subjects were enrolled in cohorts of three, and after each member of the cohort had completed one cycle, the CRM model was run to determine the dose level for the next cohort. When six patients had been treated at the current dose level and the CRM no longer dictated a change in dose, the current dose was designated the “recommended phase 2 dose.” The MTD was defined as the dose level that produced a rate of DLTs, documented in the first 2 cycles, of 20%. We used CRM v2 7, 8) to implement a novel graded toxicity model, with a fatal toxicity being scored as a full DLT, each reversible grade 4 toxicity being scored as 0.5 event, and each reversible grade 3 toxicity being scored 0.25 event, and a total event score limited to 1 per patient. Reversible hematologic toxicities were not scored as DLTs. The CRM allows more patients to be treated near the MTD for the regimen than traditional dose finding designs, and this graded toxicity model can account for a range of severity of dose limiting toxicities.
Pharmacokinetics
Blood samples were drawn from patients at hours 0, 1, 2, 6, and 24 after the first dose of temsirolimus and then at hours 0, 1, 2, 6, 24, 96, and 120 after the first dose of temsirolimus during Cycle 2. Blood samples were 2 ml, collected in EDTA tubes, and stored at −70°C until ready for analysis. Whole blood concentrations of temsirolimus and sirolimus were measured using a validated LC/MS/MS method. Data were analyzed using a single compartment model for temsirolimus and a noncompartmental model for sirolimus to determine peak concentration (Cmax), area under the concentration-vs-time curve (AUC), clearance, and average steady state concentration (WinNonlin v. 5.2, Pharsight Corp., Cary, NC).
Treatment Efficacy
Both event-free and progression-free survival were determined from the date of the first dose of study drug. Event-free survival (EFS) was defined as the time to either documentation of disease progression or withdrawal from the study due to unacceptable toxicity. Progression-free survival (PFS) was defined as the time to documentation of disease progression as defined by RECIST 1.1 criteria. Subjects who withdrew from the study were censored for PFS on the date of withdrawal.
Statistical Analysis
In vitro drug synergy data were analyzed using Calcusyn (Biosoft, Inc.). All other data were analyzed using Prism 5.0 software (GraphPad, Inc.). Survival differences in the in vitro assays were analyzed by ANOVA, differences in pharmacokinetic parameters were analyzed by one-sided Student’s t test, and EFS and PFS were analyzed using the method of Kaplan and Meier.
Results
Inhibition of mTOR Synergizes with Doxorubicin and Augments Stem Cell Cytotoxicity
MHH-ES Ewing sarcoma cells were treated with varying concentrations of doxorubicin and sirolimus, and cell viability analyzed after 48 hours. At each doxorubicin concentration, there was a dose-dependent increase in cytotoxicity in the presence of sirolimus (Figure 1A). Similar results were seen with TC71 cells (data not shown). Evaluation for synergy showed a combinatorial index (CI) <1 at every concentration of doxorubicin tested (Figure 1B), demonstrating that the addition of sirolimus causes a synergistic, not just additive, increase in cell death. Next, TC71 cells were treated with Aldefluor and sorted by flow cytometry into the ALDHhigh stem cell population, and the ALDHlow population (depleted of stem cells); unsorted cells (flow through) were also collected. Cells were incubated in growth medium alone or with 250 nM doxorubicin and increasing concentrations of sirolimus (Figure 1C). As we have previously shown, the ALDHhigh stem cell population was resistant to doxorubicin, whereas less than 50% of the ALDHlow and unsorted cells were alive after 48 hours. Inclusion of the mTOR inhibitor sirolimus resulted in a dose-dependent decrease in cell viability, both in the ALDHhigh stem cell population and in the ALDHlow and unsorted populations. Thus, mTOR inhibition results in synergistic toxicity directed toward the bulk tumor population, and renders the chemotherapy-insensitive stem cell population sensitive to doxorubicin.
Figure 1.

A) MHH-ES cells were treated with the indicated concentration of doxorubicin with (dotted line) or without (solid line) 200 nM sirolimus (Rapamycin). Cell survival was quantified using an MTT assay. The difference between the curves was statistically significantly different by 2-way ANOVA (p <0.0001). We also performed a t-test comparing cell survival in 100 nM doxorubicin with or without sirolimus, and the difference was statistically significant (p=0.003). Experiments were performed in triplicate and repeated twice, and error bars represent standard error of the mean. B) The combination index (CI) was calculated for each combination of doxorubicin and sirolimus and plotted against the combinatorial effect. Numbers represent the concentration of doxorubicin in each experiment. CI < 1 indicates synergy 12. C) TC71 cells were sorted as previously described into a population enriched in stem cells (ALDHhigh), a population depleted of stem cells (ALDHlow), and an unsorted population (Flow Through). Cells were incubated for 48 hours with 250 nM doxorubicin and the indicated concentration of sirolimus (Rapamycin), and cell viability was determined using the MTT assay. The ALDHhigh cells were resistant to doxorubicin, and this resistance was overcome by sirolimus in a dose-dependent manner. Experiments were performed in triplicate and repeated twice, and error bars represent standard error of the mean.
Patient Characteristics
Fifteen patients were enrolled in the Phase I portion of this study (Table 1). All were treated long enough to be evaluable. The median age was 39, with a range from 9 to 70. Five patients had rhabdomyosarcoma, three had leiomyosarcoma, two had spindle cell sarcoma, and the remainder had other sarcomas (detailed in Table 1). Two of the patients had primary refractory disease, and the rest had relapsed. Four of the relapses were refractory to an attempt at salvage chemotherapy. Most of the patients were heavily pretreated. Five patients had only had a single prior treatment regimen, but two had 2 prior regimens, five had 3 prior regimens, and two had 4 or more prior regimens. One patient had a previous autologous peripheral blood stem cell transplant. Eleven of the patients had had previous surgery, eleven had previously been treated with radiation therapy, two patients had been treated with cryoablation, and two with chemoembolization.
Table 1.
Patient Characteristics
| Number | Age | Gender | Diagnosis | Disease Status | Prior Treatments |
|---|---|---|---|---|---|
| 1 | 61 | M | Pleiomorphic Spindle Cell Sarcoma | Second Relapse | Surgery x2 Ifos/Adria XRT |
| 2 | 18 | M | Alveolar Rhabdomyosarcoma | Refractory Relapse | Surgery VAC VIT Auto PBSCT Cryoablation XRT IGF-1R Antibody Trabectedin Brivanib |
| 3 | 39 | F | Desmoplastic Small Round Cell Tumor | Primary Refractory | VAdriaC/IE |
| 4 | 19 | M | Mesenchymal Chondrosarcoma | Refractory Relapse | Surgery XRT VAdriaC/IE VIT Doxil/Temodar Gem/Tax Carbo/Tax Doxil/Avastin Cryoablation Chemoembolization |
| 5 | 43 | F | Malignant Fibrous Histiocytoma | First Relapse | Surgery Gem/Tax Ifos/Adria XRT |
| 6 | 39 | F | Leiomyosarcoma | First Relapse | Surgery Adriamycin XRT Gem/Tax |
| 7 | 18 | M | Embryonal Rhabdomyosarcoma | First Relapse | Surgery VAC |
| 8 | 9 | F | Embryonal Rhabdomyosarcoma | Second Relapse | Surgery VAC ICE XRT Vincristine/Irinotecan |
| 9 | 20 | F | Alveolar Rhabdomyosarcoma | First Relapse | XRT Ifos/Etoposide/Vincristine/Irinotecan/Doxorubicin/Cytoxan/Actinomycin D |
| 10 | 63 | F | Leiomyosarcoma | Second Relapse | Surgery Ifos/Adria Chemoembolization Gem/Tax Brivanib |
| 11 | 21 | M | Spindle Cell Rhabdomyosarcoma | Refractory Relapse | VAC/VI VAdriaC/IE XRT Temozolomide |
| 12 | 70 | M | Spindle Cell Sarcoma | First Relapse | XRT Surgery Gem/Tax |
| 13 | 59 | F | Leiomyosarcoma | Refractory Relapse | Surgery XRT Ifos/Adria/Avastin Gem/Tax AP23573 |
| 14 | 57 | F | Myxoid Chondrosarcoma | Second Relapse | Surgery XRT |
| 15 | 10 | M | Osteoblastic Osteosarcoma | Primary Refractory | CDDP/Adria/MTX/IE Carbo/Doxo Gem/Tax |
Toxicities
The MTD, or recommended phase 2 dose combination, as calculated by the CRM model, was dose level 4: liposomal doxorubicin 30 mg/m2 monthly with temsirolimus 20 mg/m2 weekly. Hematologic toxicity was common, and dose-limiting toxicities were primarily renal (electrolyte abnormalities; Table 2). Four patients experienced either grade 3 or 4 hematologic toxicity, and five patients experienced electrolyte abnormalities (hypokalemia, hypophosphatemia, and hypocalcemia). Only one patient experienced grade 3 stomatitis, and no one had grade 4 mouth sores. Other dose-limiting toxicities included sub-clinical pancreatitis (1 grade 3), hyperlipidemia (1 grade 3), and anorexia (1 grade 4). All toxicities were reversible after discontinuation of treatment.
Table 2.
Severe Toxicities
| Toxicity | Grade1 | Number2 | Dose Level3 |
|---|---|---|---|
| Neutropenia | 3 | 2 | 3, 4 |
| Anemia | 3 | 1 | 5 |
| Thrombocytopenia | 3, 4 | 2 | 4, 4 |
| Hypokalemia | 3 | 2 | 3, 5 |
| Hypophosphatemia | 3 | 3 | 4, 5, 5 |
| Hypocalcemia | 3 | 1 | 4 |
| Elevated transaminases | 3 | 2 | 4, 4 |
| Hyperlipidemia | 3 | 1 | 5 |
| Stomatitis | 3 | 1 | 4 |
| Anorexia | 4 | 1 | 5 |
| Pancreatitis | 3 | 1 | 4 |
Maximum CTCAE v3.0 grade of each toxicity
The cumulative number of patients with grade 3 or higher toxicity in thefirst two cycles
Dose levels at which severe toxicitieswere observed.
Pharmacokinetics
The disposition of temsirolimus and its active metabolite, sirolimus, was studied in all patients. Pharmacokinetic parameters of temsirolimus (Cmax, AUC, and Cl) at steady state were similar to those obtained in subjects given the drug as a single agent (Table 3). As the dose of temsirolimus increased from level 3 (15 mg/m2) to level 5 (27 mg/m2), its clearance increased, so its AUC did not increase significantly. However, the exposure to sirolimus did increase with dose level. At all dose levels, patients achieved therapeutic whole blood sirolimus concentrations rapidly, within 1 hr after the temsirolimus infusion. However, at dose level 3, trough whole blood sirolimus levels 1 week after the last dose were low, whereas at dose levels 4 and 5, trough whole blood sirolimus levels 1 week after the last dose remained therapeutic. At dose level 5, peak sirolimus levels were high (mean 104, range 79 – 124 ng/ml), corresponding to dose-limiting toxicity (Table 4). Interestingly, Cmax and AUC of sirolimus were substantially higher in subjects treated at the recommended Phase 2 dose (dose level 4) than have been reported for subjects treated with temsirolimus alone (Table 3).
Table 3.
Steady state pharmacokinetics of temsirolimus administered in combination with liposomal doxorubicin compared to historical single agent results
| Temsirolimus parent drug | Sirolimus active metabolite | |||
|---|---|---|---|---|
| Combination (n=7) | Single Agent (n=11) | Combination (n=7) | Single Agent (n=11) | |
| Cmax (ng/ml) | 346.2 ± 250 | 443 ± 109.2 | 69.6 ± 23.2 | 34.5 ± 19.3 (p=0.003) |
| AUC (ng*hr/ml) | 1210 ± 340 | 1349 ± 232 | 7499.8 ± 4591.1 | 3793 ± 1466 (p=0.02) |
| Cl (L/hr/m2) | 17.8 ± 7.1 | 19.0 ± 3.0 | ||
Whole blood temsirolimus and sirolimus levels were measured by LC/MS/MS after the first temsirolimus dose of Cycle 2 for patients treated at the recommended phase 2 dose, Dose Level 4. Control data are from cancer patients treated with temsirolimus alone as reported in the Investigator’s Brochure. The numbers in parentheses indicate the number of subjects evaluated. AUC and clearance of Temsirolimus were calculated using a single compartment model. AUC and Cmax of sirolimus were calculated using a non-compartmental model. Data are mean ± standard deviation.
Table 4.
Steady State Pharmacokinetics at Each Dose Level
| Dose Level | 3 | 4 | 5 |
| N | 3 | 7 | 3 |
| Temsirolimus dose (mg/m2) | 15 | 20 | 27 |
| Temsirolimus | |||
| Cmax (ng/ml) | 359 ± 160 | 346 ± 250 | 488 ± 321 |
| AUC (ng*hr/ml) | 1000 ± 361 | 1210 ± 340 | 1273 ± 315 |
| Clearance (L/hr/m2) | 16.7 ± 6 | 17.8 ± 7.1 | 22.3 ± 4.7 |
| Sirolimus | |||
| Cmax | 37 ± 11 | 70 ± 23 | 104 ± 23 |
| Cmin | 3.3 ± 1 | 14 ± 8 | 13 ± 4 |
| AUC | 3063 ± 1714 | 7500 ± 4591 | 5258 ± 1105 |
| Css (ng/ml) | 14 ± 1 | 37 ± 15 | 41 ± 8 |
Whole blood levels of Temsirolimus and Sirolimus were measured by LC/MS/MS after the first temsirolimus dose of Cycle 2. AUC and clearance of Temsirolimus were calculated using a single compartment model. AUC and steady state Cmax of sirolimus were calculated using a non-compartmental model. Css, average steady state concentration. Data are mean ± standard deviation.
Efficacy
Although the primary objective of this study was to determine the recommended dosing combination for evaluation in a Phase II study, event-free and progression-free survival were also determined (Figure 2). Median EFS for the entire study population was 76 days, and median PFS was 118 days. For the subgroup of subjects treated at the MTD, median EFS was 49 days and median PFS was 118 days. The range of survival times was the same for all 4 analyses: 28–353 days.
Figure 2.
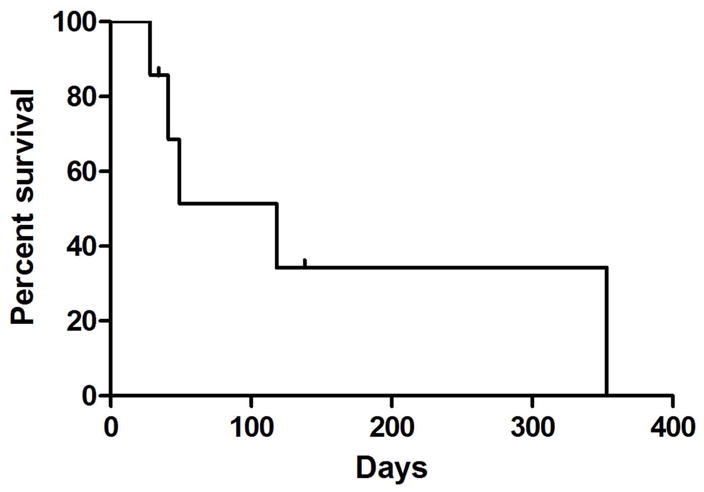
Progression-free Survival of patients treated at the MTD. A Kaplan-Meier curve indicating the time from the beginning of treatment until the first objective evidence of disease progression, censored at discontinuation of drug.
Discussion
In recent years, several clinical trials have explored the use of mTOR inhibitors for the treatment of high risk bone and soft tissue sarcomas. Results of these studies have been disappointing. One feature these trials all shared is the use of mTOR inhibitors as cytotoxic agents. Our laboratory work suggests a different use for these drugs – increasing the sensitivity of sarcoma cells to standard chemotherapeutics. Our in vitro data demonstrate synergistic cytotoxicity of the combination of sirolimus and doxorubicin, and that sirolimus reverses the innate chemoresistance of Ewing sarcoma stem cells. Based on these findings, we conducted a clinical trial aimed at demonstrating this same synergy in vivo.
In the current study, we found that the combination of liposomal doxorubicin and temsirolimus could be safely administered to patients with bone and soft tissue sarcomas that were refractory to conventional therapy or recurrent. Our dose-finding algorithm was designed to identify a dose combination that would have a DLT rate of 20% in the first 2 cycles of treatment. We found that the combination of liposomal doxorubicin given at 30 mg/m2/day once every 28 days in conjunction with temsirolimus given at 20 mg/m2/day weekly resulted in the target DLT rate in this cohort of heavily pretreated patients. As anticipated, toxicities were primarily hematologic and dose-limiting toxicities were primarily renal. Interestingly, although stomatitis is often a significant problem in patients treated with either of these agents, only 1 subject had significant stomatitis (grade 3). A Phase 2 study of this combination therapy is ongoing as an expansion of the MTD cohort.
Our study design incorporated a graded toxicity model as a novel enhancement of the continuous reassessment method for identifying the MTD. First introduced by O’Quigley et al., and subsequently refined by Goodman et al., 9, 10 the CRM has been demonstrated by statistical simulations to be less biased and more efficient for estimating MTD than ad hoc dose ranging methods such as the standard 3+3 study design. We used a graded toxicity model to account for a range of severity of dose limiting toxicities, thus more accurately reflecting clinical utility than traditional models that treat DLTs from grade 3 to death as binary outcomes. The CRM generally allows more patients to be treated near the MTD for the regimen than traditional dose-finding designs and the graded toxicity model allowed severity of each toxicity to inform the MTD. In fact, of the 15 subjects enrolled on this study, 9 were treated at the MTD, and the recommended phase 2 dose was determined without fatal toxicity.
Although not an aim of this portion of the study, event-free and progression-free survival data were collected. The median progression-free survival for the 9 patients treated at the MTD was 118 days. This survival time compares favorably with PFS reported for sarcoma patients treated with either agent alone. Okuno et al. reported a median PFS of 2.0 months in a phase 2 study of temsirolimus in 41 patients with soft tissue sarcoma 6, and Judson et al. reported a median PFS of 65 days for patients with advanced soft tissue sarcoma treated on the liposomal doxorubicin arm of a randomized phase 2 study of conventional versus liposomal doxorubicin 11. Although comparisons across studies, especially very small Phase 1 trials such as ours, are problematic, our results justify pursuing the Phase 2 portion of the trial.
Because liposomal doxorubicin and temsirolimus have not previously been administered together, we evaluated the pharmacokinetics of temsirolimus and its active metabolite, sirolimus, in each study subject. The distribution of temsirolimus in our study subjects was similar to that which has been reported for patients treated with temsirolimus alone, but co-administration with liposomal doxorubicin resulted in increases in Cmax and AUC of sirolimus. Thus co-administration of liposomal doxorubicin with temsirolimus did not alter the conversion of prodrug to active sirolimus, but did significantly increase exposure to the active metabolite. Moreover, our ability to increase the dose of temsirolimus and administer the drug with liposomal doxorubicin resulted in maintenance of a therapeutic sirolimus level throughout the treatment period, despite only weekly dosing, which may have contributed to the improved PFS seen in our subjects compared with similar patients treated with temsirolimus alone.
In summary, we determined that the combination of 30 mg/m2 of liposomal doxorubicin delivered monthly with weekly temsirolimus at a dose of 20 mg/m2 can be safely administered to heavily pretreated pediatric and adult patients with recurrent or refractory bone and soft tissue sarcomas. Toxicity of this combination was manageable and reversible. Although pharmacokinetics of the temsirolimus parent drug were not substantially altered by concurrent administration of liposomal doxorubicin, exposure to sirolimus, its active metabolite, was increased compared with previous reports. The Phase 2 expansion portion of this study is ongoing, along with pharmacodynamic studies to determine whether temsirolimus overcomes the inherent chemoresistance of sarcoma stem cells in vivo, as we have shown in vitro.
Acknowledgments
This study was approved and funded by the National Comprehensive Cancer Network (NCCN) from general research support from Pfizer, Inc. We are also grateful for the support of The Heather Brooke Foundation.
Footnotes
Disclosure
The authors have declared no conflicts of interest.
References
- 1.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer Stem Cells--Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 2.Rasheed ZA, Kowalski J, Smith BD, Matsui W. Concise review: Emerging concepts in clinical targeting of cancer stem cells. Stem Cells. 2011;29:883–7. doi: 10.1002/stem.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Awad O, Yustein JT, Shah P, Gul N, Katuri V, O’Neill A, Kong Y, Brown ML, Toretsky JA, Loeb DM. High ALDH activity identifies chemotherapy-resistant Ewing’s sarcoma stem cells that retain sensitivity to EWS-FLI1 inhibition. PLoS One. 2010;5:e13943. doi: 10.1371/journal.pone.0013943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muggia FM. Clinical efficacy and prospects for use of pegylated liposomal doxorubicin in the treatment of ovarian and breast cancers. Drugs. 1997;54 (Suppl 4):22–9. doi: 10.2165/00003495-199700544-00006. [DOI] [PubMed] [Google Scholar]
- 5.Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ, Blay JY, Mita MM, Sankhala KK, Berk L, Rivera VM, Clackson T, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 30:78–84. doi: 10.1200/JCO.2011.35.6329. [DOI] [PubMed] [Google Scholar]
- 6.Okuno S, Bailey H, Mahoney MR, Adkins D, Maples W, Fitch T, Ettinger D, Erlichman C, Sarkaria JN. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas: a study of the Mayo phase 2 consortium (P2C) Cancer. 2011;117:3468–75. doi: 10.1002/cncr.25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garrett-Mayer E. The continual reassessment method for dose-finding studies: a tutorial. Clin Trials. 2006;3:57–71. doi: 10.1191/1740774506cn134oa. [DOI] [PubMed] [Google Scholar]
- 8.Piantadosi S, Fisher JD, Grossman S. Practical implementation of a modified continual reassessment method for dose-finding trials. Cancer Chemother Pharmacol. 1998;41:429–36. doi: 10.1007/s002800050763. [DOI] [PubMed] [Google Scholar]
- 9.Goodman SN, Zahurak ML, Piantadosi S. Some practical improvements in the continual reassessment method for phase I studies. Stat Med. 1995;14:1149–61. doi: 10.1002/sim.4780141102. [DOI] [PubMed] [Google Scholar]
- 10.O’Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. [PubMed] [Google Scholar]
- 11.Judson I, Radford JA, Harris M, Blay JY, van Hoesel Q, le Cesne A, van Oosterom AT, Clemons MJ, Kamby C, Hermans C, Whittaker J, Donato di Paola E, et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2001;37:870–7. doi: 10.1016/s0959-8049(01)00050-8. [DOI] [PubMed] [Google Scholar]
- 12.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]