

Summary
Aim
Pathological release of excess zinc ions has been implicated in ischemic brain cell death. However, the underlying mechanisms remain to be elucidated. In stroke, ischemia‐induced zinc release and hypoxia‐inducible factor‐1 (HIF‐1) accumulation concurrently occur in the ischemic tissue. The present study tests the hypothesis that the presence of high intracellular zinc concentration is a major cause of modifications to PARP‐1 and HIF‐1α during hypoxia, which significantly contributes to cell death during ischemia.
Methods
Primary cortical astrocytes and C8‐D1A cells were exposed to different concentrations of zinc chloride. Cell death rate and protein expression of HIF‐1 and Poly(ADP‐ribose) polymerase (PARP)‐1 were examined after 3‐h hypoxic treatment.
Results
Although 3‐h hypoxia or 100 μM of zinc alone did not induce noticeable cytotoxicity, their combination led to a dramatic increase in astrocytic cell death in a zinc‐concentration‐dependent manner. Exposure of astrocytes to hypoxia for 3 h remarkably increased the levels of intracellular zinc and HIF‐1α protein, which was further augmented by added exogenous zinc. Notably, HIF‐1α knockdown blocked zinc‐induced astrocyte death. Moreover, knockdown of PARP‐1, another important protein in the response of hypoxia, attenuated the overexpression of HIF‐1α and reduced the cell death rate.
Conclusions
Our studies show that zinc promotes hypoxic cell death through overexpression of the hypoxia response factor HIF‐1α via the cell fate determine factor PARP‐1 modification, which provides a novel mechanism for zinc‐mediated ischemic brain injury.
Keywords: Cell death, Hypoxia, Hypoxia‐inducible factor‐1, Poly(ADP‐ribose) polymerase‐1, Zinc
Introduction
Zinc is present at high concentrations in the brain and is crucial for cellular development and survival 1, 2. The majority of the brain's zinc is bound to proteins and 10–20% is concentrated within certain glutamatergic vesicles in a relatively free state 3, 4. Zinc is essential for normal cellular function and serves a signaling role in the central nerve system 5 by altering the behavior of several ion channels and receptors 6. Following ischemic stroke, zinc is released from a subset of glutamatergic terminals in the brain 7. A pathological release of excess zinc ions following ischemia contributes significantly to ischemic brain injury, but the mechanism of zinc neurotoxicity is not well understood 8, 9, 10, 11, 12.
Hypoxia, a cardinal feature of ischemic stroke, is involved in brain injury, whose dyshomeostasis has been recognized as an important mechanism for cell death in acute brain injury 4. While ischemic brain injury research mostly focuses on neuronal injury and death, studies have shown that astrocytes are also injured after ischemia and astrocytic death can even occur prior to neuronal death 13, 14, 15, 16. Exposing cells to low oxygen triggers hypoxic response pathways responsible for regulating the expression of hypoxia‐inducible transcription factor 1 (HIF‐1), which regulates many genes and plays an important role in the fate of cells under ischemic conditions 17, 18, 19, 20, 21. It is well known that the HIF‐1α subunit is sensitive to oxygen levels, and hypoxia leads to changes in its stability, subcellular localization, and transcription 22. At normal conditions, HIF‐1α protein level remains very low or undetectable due to the rapid degradation by the ubiquitin proteasome system. However, under hypoxia, HIF‐1α becomes stabilized, followed by translocation from the cytoplasm into the nucleus, where it heterodimerizes with HIF‐1β 23, 24, 25, 26. Overexpression of HIF‐1α has been shown to exacerbate hypoxia‐induced apoptotic cell death through stabilizing tumor suppressor protein p53 or interacting with calcium and calpain 27. Recent reports show that zinc is involved in HIF‐1α regulation, but its effect remains inconsistent in the literature. In human tumors, zinc has been shown to promote HIF‐1α degradation 28, but in normal prostate cells 29 and keratinocytes 30, it stabilizes HIF‐1α. In stroke, ischemia‐induced zinc release and HIF‐1α accumulation concurrently occur in the ischemic brain, but to date, little is known about their interaction and how this interaction may contribute to ischemic brain injury.
Poly(ADP‐ribose) polymerase‐1 (PARP‐1) is a protein that plays an important role in cell death. On the one hand, PARP‐1 directly participates in DNA repair through the formation of (poly(ADP)ribosylation) (PAR) 31 and effects other DNA‐repair and DNA‐damage checkpoint proteins responsible for regulating DNA repair. On the other hand, PARP‐1 activation can promote cell death when extensive DNA damage has occurred by releasing apoptosis‐inducing factor (AIF), as seen in inflammation and ischemia 32. PARP‐1 is also involved in caspase‐dependent apoptosis, and it can be inactivated and cleaved into two fragments (24 and 89 kDa) by caspase 33 during apoptosis. Being a zinc finger protein, PARP‐1 depends on the presence of zinc to exert its full function 34. Moreover, PARP‐1 is shown to regulate the transcriptional activity of HIF‐1α 35. Given this premise, it is reasonable to speculate that zinc, HIF‐1α, and PARP‐1 may interact with each other to contribute to ischemic brain injury.
In the present study, we tested the hypothesis that the presence of high intracellular zinc concentration is the major cause of the modifications to PARP‐1 and HIF‐1α during hypoxia, which contributes to cell death during ischemia. We found that zinc promotes hypoxic cell death and induces HIF‐1α overexpression in astrocytes by interacting with PARP‐1. Our results demonstrate that zinc may exacerbate astrocyte death by augmenting hypoxia‐induced HIF‐1α stabilization via PARP‐1. These findings provide novel insights to the potential mechanisms of zinc‐induced brain injury following cerebral ischemia.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), antibiotic–antimycotic solution, and FluoZin‐3 were purchased from Invitrogen, and zinc chloride was from Sigma. siRNA specific for mouse HIF‐1α and PARP‐1 were obtained from Santa Cruz (Dallas, TX, USA).
Primary Culture of Rat Cortical Astrocytes
Primary cortical astrocytes were isolated from the cortices of postnatal day 1 Sprague‐Dawley rats as previously described 36. The animals were purchased from Charles River Laboratories, Wilmington, MA, USA, and the animal study has been approved by the University of New Mexico Institutional Animal Care and Use Committee (IACUC). Briefly, animals were decapitated and their brains immediately excised. Meninges and blood vessels were removed. Then, the forebrains were placed in DMEM. The tissue was minced and incubated in 0.05% trypsin for 30 min at 37°C. Trypsinization was completed by adding DMEM containing 10% (v/v) FBS. Cells were obtained by dissociating the tissue with pipette and passing through a 40 μM strainer. The cells were seeded in the flasks at a density of 1.5 × 105 cells/cm2. Cells were cultured in growth medium (85% Dulbecco's Modified Eagle medium containing 4.5 g/L glucose, and 15% fetal bovine serum) at 37°C with 95% air/5% CO2. Cells were used for experiments 18–20 days after seeding. More than 95% of cells were glial fibrillary acidic protein (GFAP)‐positive astrocytes, as described previously 37, 38.
Cell line Culture
C8‐D1A astrocyte cell line was obtained from American Type Culture Collection. The cells were cultured in DMEM containing 10% FBS and 1% (v/v) antibiotic–antimycotic solution at 37°C with 95% air/5% CO2 in an incubator.
Hypoxic Cellular Model
Before hypoxic treatment, the cell culture medium was replaced with oxygen‐free experimental media (DMEM containing different zinc chloride concentration at 0, 50, 100, and 150 μM), which had previously been bubbled with nitrogen for 15 min. Cells were then incubated in a polymer hypoxic glove chamber (Coy Laboratory Products Inc. Grass Lake, MI, USA) with 1% O2 at 37°C for 3 h.
Intracellular Zinc Detection by FluoZin‐3
Intracellular zinc was measured with FluoZin‐3, a selective fluorescent probe for zinc 39. Astrocytes were plated onto polylysine‐coated glass coverslips. After reaching 70% confluence, cells were washed with DMEM to remove extracellular zinc before incubating with DMEM containing 2.5 μM FluoZin‐3 for 45 min at room temperature. After washing in DMEM, the coverslips were mounted on a glass slide. Images were acquired using an inverted microscope with a GFP dichroic mirror to visualize FluoZin‐3 fluorescence.
Western Blot Analysis for HIF‐1α and PARP‐1
At the end of the indicated treatments, cells were quickly scraped down, collected, and lysed in RIPA buffer (Santa Cruz). Cell extracts were centrifuged at 18,000 g for 15 min at 4°C, and protein concentrations in supernatants were determined using protein assay reagents (Bio‐Rad, Hercules, CA, USA). After heating at 100°C for 5 min, samples of 20 μg protein were electrophoretically separated on 10% sodium dodecyl sulfate‐polyacrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA, USA). Membranes were blocked with Odyssey Blocking Buffer (Li‐cor, Lincoln, NE, USA) and then incubated at 4°C overnight with antibodies against HIF‐1α (diluted 1:1,000, Novus) or PARP‐1 (diluted 1:5,000, Cell Signaling, Danvers, MA, USA) followed by incubation with RDye 800CW goat antirabbit and IRDye 680 goat antimouse secondary antibodies (diluted 1:10,000, Li‐cor) for 1 h at room temperature. Immunoblots were photographed using the Odyssey® Infrared Imaging System (Li‐cor) with Molecular Imaging Software V4.0.
Cytotoxicity Assay
Cell death rate was measured using Cytotox 96 nonradioactive cytotoxicity assay kit (Promega), which quantitatively measures lactate dehydrogenase (LDH) release from dead cells. Cells (5 × 103 cells/well) were seeded into 96‐well microtiter plates. Following treatment, 50 μL of the reconstituted substrate mixture was added to each well of the plate. Thirty min later, 50 μL of the stop solution was added to each well, and the absorbance was measured at 490 nm in a Bio‐Rad 3350 microplate reader. Triton‐X 100‐treated cells were used as 100% cell death control. The cell death rate was calculated by using the formula: Cell death rate (%) = (Experimental absorbance value − culture medium absorbance value)/(Triton‐X 100‐treated absorbance value − culture medium absorbance value) × 100.
HIF‐1α and PARP‐1 Knockdown by Transfection of siRNA
Transfection of siRNA was performed according to the protocol provided by Santa Cruz. C8‐D1A cells were seeded in DMEM containing 10% FBS without antibiotics at a density of 2 × 106 cells/dish in 60‐mm dishes the day before the transfection. Transfection of siRNA was carried out using siRNA Transfection Reagent (Santa Cruz). Five microlitre siRNA Transfection Reagent and 5 μL siRNA were separately diluted in 100 μL siRNA Transfection Medium (Santa Cruz). 0.8 mL siRNA Transfection Medium was added after the two mixtures were combined and incubated at room temperature for 30 min. Cells were washed once with 2 mL of siRNA Transfection Medium before adding the siRNA transfection mixture. Six hours later, 1 mL of DMEM containing 20% FBS and 2% (v/v) antibiotic–antimycotic solution were added without removing the transfection mixture. Forty‐eight hours after transfection, cells were treated with zinc. Specific silencing was confirmed by Western blot analysis.
Data Analysis
Each experiment was repeated at least four times. Data were presented as means ± SE. Statistical analysis was carried out using Student's t‐tests. A value of P < 0.05 was considered statistically significant.
Results
Hypoxia Greatly Amplifies Zinc Cytotoxicity
Cerebral ischemia causes tissue hypoxia and synaptic activation 4. Moreover, extracellular zinc increases from nanomolar to micromolar levels after synaptic activation 6. Thus, we speculated that zinc and hypoxia may work together to exacerbate ischemic brain injury. To test this possibility, we compared zinc's cytotoxicity under normoxic and hypoxic conditions. 300 μM of zinc is often cited and used as the “physiological” concentration of free zinc for stimulating neural tissue 40. However, due to the elevated toxicity of 300 μM zinc to cells, we used the concentration range of 0–150 μM in our experiments. Primary astrocytes were treated with different concentrations of ZnCl2 (0, 50, 100, and 150 μM) for 3 h under normoxia or hypoxia. Then, the mortality of astrocytes was assessed by Cytotox 96 nonradioactive cytotoxicity assay kit. As shown in Figure 1A, under normoxic condition, treatment of astrocytes with zinc for 3 h did not cause significant cell death at the concentration of 0, 50, or 100 μM ZnCl2, and 12% cell death was observed at 150 μM. In contrast, under hypoxia, treatment with 50, 100, or 150 μM ZnCl2 led to dramatic zinc‐concentration‐dependent cell death with rates being 8%, 37%, and 68%, respectively. Similar results were observed using the C8‐D1A astrocytic cell line (Figure 1B). These findings clearly indicate that under hypoxic conditions, cytotoxicity of zinc is greatly amplified.
Figure 1.
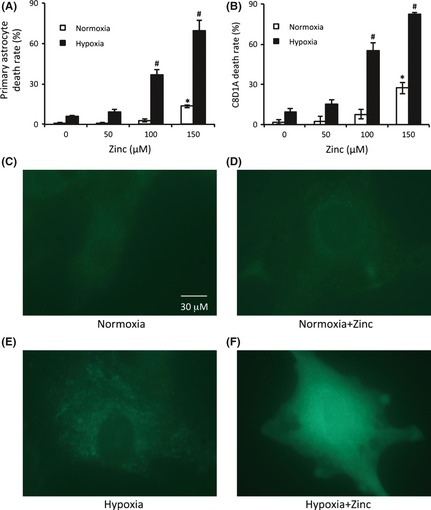
Extracellular zinc promoted hypoxic astrocytes death through the increase of intracellular zinc. Primary astrocytes (A) or C8‐D1A cells (B) were incubated with indicated concentrations of zinc chloride for 30 min before hypoxic treatment. After 3 h of hypoxic treatment, cell death rates were assayed by the Cytotox 96 nonradioactive cytotoxicity assay kit. The intracellular free zinc was visualized using FluoZin‐3 fluorescence probe after the hypoxic treatment. (C) C8‐D1A cells in normoxia. (D) C8‐D1A cells treated with 100 μM zinc chloride for 3 h in normoxia. (E) C8‐D1A cells exposed to hypoxia for 3 h. (F) C8‐D1A astrocytes treated with 100 μM zinc chloride plus hypoxia for 3 h. The experiments were repeated 4 times (n = 4). Data were presented as means ± SE. *P < 0.05 compared with normoxia without zinc; # P < 0.05 compared with hypoxia without zinc.
Hypoxia Increases Intracellular Free Zinc
Emerging evidence indicates that the rise in intracellular free zinc contributes to neuronal and astrocytic cell death following cerebral ischemia 4, 41, 42, 43, 44, 45, 46. We speculated that intracellular zinc concentrations would increase after the addition of exogenous zinc to the culture media under hypoxic conditions. To prove this, we used the Zn‐specific fluorophore FluoZin‐3 to visualize intracellular free zinc. We found that under normoxia, little free zinc in C8‐D1A astrocytes was detectable (Figure 1C), while adding 100 μM extracellular zinc (this concentration was chosen because it caused about 50% cell death under hypoxia; Figure 1B) barely increased intracellular free zinc (Figure 1D). However, exposure of astrocytes to hypoxia for 3 h without zinc administration significantly increased the intracellular free zinc (Figure 1E), which is consistent with literature reports 47. Most importantly, unlike the normoxic cells, the FluoZin‐3 fluorescence intensity was further augmented by the 100 μM zinc extracellular administered under hypoxia (Figure 1F). These results demonstrate that whereas under normoxia the addition of extracellular zinc does not necessarily increase intracellular free zinc, hypoxia can dramatically enhance the intracellular level of free zinc following zinc treatment.
Zinc chloride Induces Overexpression of HIF‐1α
To elucidate the mechanism behind zinc‐induced hypoxic cell death, we examined the expression of HIF‐1α, which plays a key role in the fate of cells under ischemic conditions. To determine the effect of zinc on HIF‐1α expression under hypoxia, primary and C8‐D1A astrocytes were treated with different concentrations of zinc chloride (0, 50, 100, and 150 μM). As shown in Figure 2, a remarkable zinc‐concentration‐dependent increase in HIF‐1α protein level was observed following 3 h of hypoxia in both primary (Figure 2A,B) and C8‐D1A (Figure 2C,D) astrocytes. As expected, no or little HIF‐1α was detected at normoxia as it could easily be hydrolyzed through ubiquitination. These results suggest that zinc synergistically acts with hypoxia to increase the expression of HIF‐1α.
Figure 2.
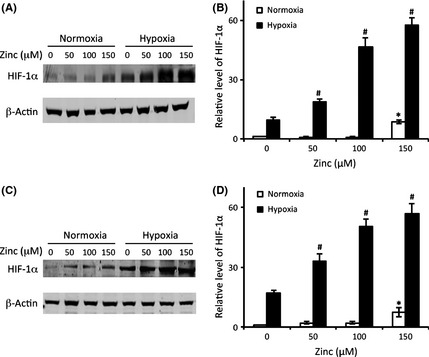
The effect of zinc on HIF‐1α protein levels. Astrocytes were treated with zinc chloride at indicated concentrations under hypoxia or normoxia for 3 h. Representative immunoblots showing HIF‐1α protein levels in primary astrocytes (A) and C8‐D1A cells (C). β‐actin served as a loading control. Relative HIF‐1α protein levels in primary astrocytes (B) and in C8‐D1A cells (D) were quantitated after normalization to β‐actin. The experiments were repeated four times (n = 4). Data were presented as means ± SE. *P < 0.05 compared with normoxia without zinc; # P < 0.05 compared with hypoxia without zinc.
HIF‐1α Plays a Significant Role in Zinc‐Induced Astrocyte Death
As zinc treatment remarkably increased the protein level of HIF‐1α (Figure 2), we next wanted to determine whether the increased HIF‐1α expression by zinc was responsible for zinc‐induced cell death. As shown in Figure 3, after knockdown of HIF‐1α by siRNA‐HIF‐1α (Figure 3A), the zinc‐induced hypoxic astrocytic death was partially reversed with cell death rate decreasing from 43% to 27% (Figure 3B). This result suggests that overexpression of HIF‐1α plays, at least in part, a significant role in zinc‐induced cell death.
Figure 3.
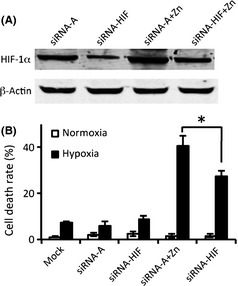
Zinc‐induced overexpression of HIF‐1α in astrocytes was involved with zinc‐induced cytotoxicity. siRNA‐HIF was used to downregulate the expression of HIF‐1α in C8‐D1A astrocytes before treatment with 100 μM zinc chloride and exposure to hypoxia. Control siRNA‐A, a nontargeting 20–25 nt siRNA, was used as a negative control. (A) The expression of HIF‐1α was determined by Western blot. (B) The effect of HIF‐1α on zinc‐induced cell death was measured by Cytotox 96 nonradioactive cytotoxicity assay kit. The experiments were repeated four times (n = 4). Data were presented as means ± SE. *P < 0.05. Mock: cells untreated with siRNA; siRNA‐A: cells were transfected with control siRNA before hypoxia/normoxia treatment; siRNA‐HIF: cells were transfected with HIF‐1α siRNA before hypoxia/normoxia treatment; siRNA‐A + zinc: cells were first transfected with control siRNA and then treated with hypoxia/normoxia in the presence of zinc; siRNA‐HIF + zinc: cells were first transfected with HIF siRNA and then treated with hypoxia/normoxia in the presence of zinc.
Zinc Chloride Modifies the Expression and Activity of PARP‐1
Besides HIF‐1, PARP‐1 is another important protein contributing to hypoxic cell death 48. In addition, zinc has been reported to activate PARP‐1 49, and therefore, it was reasonable to speculate that PARP‐1 may be involved in zinc‐induced hypoxic cell death.
To investigate whether PARP‐1 could be regulated by zinc during hypoxia, primary astrocytes were treated with different concentrations of zinc chloride under 3 h of hypoxia or normoxia conditions. We determined the activity of PARP‐1 by measuring the level of PAR, and PARP‐1 cleavage by measuring the cleaved PARP. As shown in Figure 4A,B, under normoxia conditions, little cleaved PARP protein was observable, whereas after 3 h of hypoxia, the cleaved PARP protein level increased dramatically with increasing zinc concentration. Interestingly, we found that when the cells were exposed to zinc alone, that is, without hypoxia, the activity of PARP‐1 increased with increasing zinc concentration. However, under hypoxia, when the cells were exposed to the same zinc concentrations, PARP‐1 activity decreased with increasing zinc concentration (Figure 4A,C). These findings can be explained by the dual role of PARP‐1 function in DNA repair and cell death regulation. Under normoxia conditions, or hypoxia with low zinc concentrations, the DNA damage was relatively low, which can be repaired through PARP‐1 activation. This is indicated by increased PARP‐1 activity and little cleaved PARP‐1 protein. However, at a higher concentration of zinc under hypoxic conditions, the cell injury may be fatal due to the severe damage that was beyond the repair ability of PARP‐1. Thus, PARP‐1 activity was reduced (Figure 4C), PARP‐1 cleavage was increased (Figure 4B), and cell death ensued (Figure 1A). Together, these results suggest that PARP‐1 plays an important role in zinc‐induced cell death. We also obtained very similar results using C8‐D1A astrocytes (Figure 4D, E, and F). As the results from primary astrocytes and cell line C8‐D1A are identical (Figures 1, 2, and 4), we chose to use C8‐D1A cells to perform our subsequent experiments.
Figure 4.
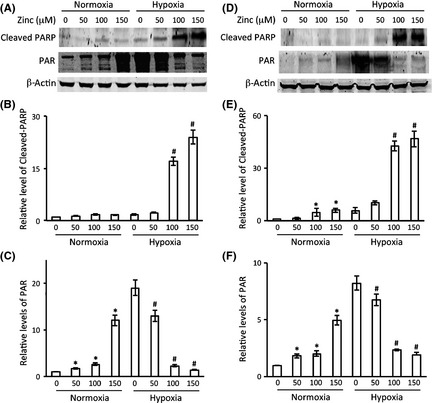
The effects of zinc and hypoxia on PARP‐1 expression and activity. Astrocytes were treated with zinc chloride at indicated concentrations under hypoxia or normoxia for 3 h. Representative immunoblots showing cleaved PARP and PAR protein levels in primary astrocytes (A) and C8‐D1A cells (D). β‐actin served as a loading control. Relative cleaved PARP (B, E) and PAR protein levels (C, F) in primary astrocytes and in C8‐D1A cells were quantitated after normalization to β‐actin, respectively. The experiments were repeated 4 times (n = 4). Data were presented as means ± SE. *P < 0.05 compared with normoxia without zinc; # P < 0.05 compared with hypoxia without zinc.
PARP‐1 is Involved in Zinc‐Induced Astrocyte Death and Regulates the Overexpression of HIF‐1α During Hypoxia
To directly determine the role of PARP‐1 in zinc‐induced hypoxic cell death, we measured the cell death rate during hypoxia in zinc‐treated astrocytes after knockdown of PARP‐1 by siRNA (Figure 5A). We found that silencing PARP‐1 partially blocked zinc‐induced cell death, reducing the death rate from 45% to 25% (Figure 5B). These results demonstrate that, in addition to HIF‐1α, PARP‐1 also plays an important role in zinc‐induced cell death.
Figure 5.
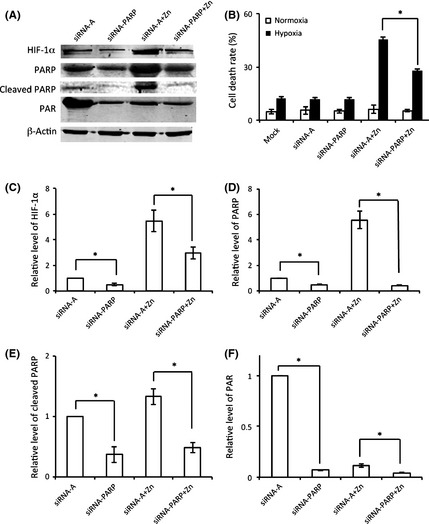
Poly(ADP‐ribose) polymerase‐1 contributed to HIF‐1α regulation and zinc‐induced cytotoxicity. C8‐D1A astrocytes were incubated with siRNA‐PARP to inactive PARP‐1 before the zinc and hypoxia treatment. (A) The expression and activity of PARP‐1 and the expression of HIF‐1α were determined by Western blot. (B) The effect of PARP on zinc‐induced cell death was measured by Cytotox 96 nonradioactive cytotoxicity assay kit. Levels of HIF‐1α (C), PARP (D), cleaved PARP (E), and PAR (F) were quantified and normalized by β‐actin. The experiments were repeated 4 times (n = 4). Data were presented as means ± SE. *P < 0.05. Mock: cells untreated with siRNA; siRNA‐A: cells were transfected with control siRNA before hypoxia/normoxia treatment; siRNA‐PARP: cells were transfected with PARP siRNA before hypoxia/normoxia treatment; siRNA‐A + zinc: cells were first transfected with control siRNA and then treated with hypoxia/normoxia in the presence of zinc; siRNA‐PARP + zinc: cells were first transfected with PARP siRNA and then treated with hypoxia/normoxia in the presence of zinc.
As both PARP‐1 and HIF‐1α were found to be regulated by zinc and both contributed to zinc‐induced cell death, next we wanted to determine whether PARP‐1 is upstream of HIF‐1α, or HIF‐1α is upstream of PARP‐1, on the zinc‐induced cell death pathway. C8‐D1A astrocytes were treated with siRNA‐PARP to inhibit the expression of PARP‐1 before hypoxia. The inhibition of siRNA‐PARP was confirmed by Western blot for PARP expression (Figure 5A,D) and activity (Figure 5A,F). After inhibition of PARP‐1, the expression of HIF‐1α was significantly decreased (Figure 5A,C). Decrease of HIF‐1α in response to PARP‐1 knockdown indicates that PARP‐1 regulated HIF‐1α and PARP‐1 maybe on the upstream of HIF‐1α.
To further confirm that PARP‐1 was on the upstream side of this cascade, siRNA was also used to inhibit HIF‐1α to examine any effects of HIF‐1α on PARP‐1. As shown in Figure 6, no significant changes were detected in PARP‐1 protein expression, cleaved PARP protein level, or PARP activity (Figure 6A,C–E) after HIF‐1α knockdown by siRNA‐HIF (Figure 6A,B). These results indicate that HIF‐1α is a downstream gene from PARP‐1. These findings clearly suggest that PARP‐1 modifies the expression of HIF‐1α and is upstream of HIF‐1α in the zinc‐induced cell death pathway.
Figure 6.
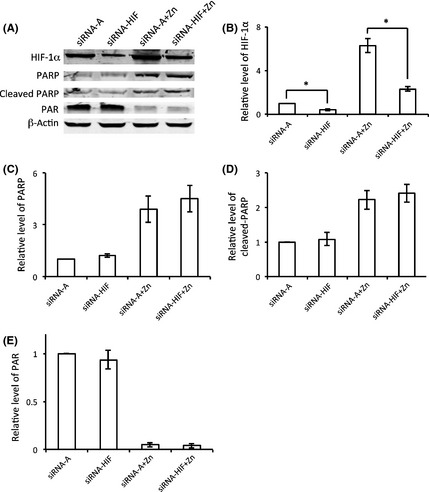
The effect of HIF‐1 on PARP‐1 regulation. (A) The expressions of HIF‐1α and PARP‐1 were determined by Western blot. HIF‐1α (B), PARP (C), cleaved PARP (D), and PAR (E) were normalized by β‐actin. The experiments were repeated 4 times (n = 4). Data were presented as means ± SE. *P < 0.05. siRNA‐A: cells were transfected with control siRNA before hypoxia/normoxia treatment; siRNA‐HIF: cells were transfected with HIF‐1α siRNA before hypoxia/normoxia treatment; siRNA‐A + zinc: cells were first transfected with control siRNA and then treated with hypoxia/normoxia in the presence of zinc; siRNA‐HIF + zinc: cells were first transfected with HIF siRNA and then treated with hypoxia/normoxia in the presence of zinc.
Discussion
Hypoxia plays a fundamental role in the pathophysiology of common causes of mortality, including stroke 50. Zinc release from a subset of glutamatergic terminals heightens under ischemia 7, leading to extracellular zinc increase from nanomolar to micromolar levels 51, 52. During synaptic transmission, zinc can be released into the surrounding milieu making it available for entry into cells through gated zinc channels on neighboring cells 5. Growing evidence suggests that elevated intracellular free zinc levels contribute significantly to ischemic brain injury 53, 54, 55, 56. Understanding the effect of the interaction of zinc with hypoxia on cell death, and the underlying mechanism, is critically important for ischemic stroke research. The present study demonstrates a synergistic cytotoxicity by zinc and hypoxia using cultured astrocytic cells. Notably, the normal total plasma zinc is 13 ± 3 μM 57, and 150 μM zinc has been chosen as the highest zinc concentration to treat the cells in our experiments. Thus, the concentration of the additional zinc and endogenous zinc is much less than 300 μM, which is often cited and used as the “physiological” concentration of free zinc for stimulating neural tissue 40. Although 100 μM zinc or 3 h of hypoxia alone does not cause significant cell death, the combination of these two treatments dramatically increases cell death in both primary (Figure 1A) and C8‐D1A (Figure 1B) astrocytes, which is attributed to increased intracellular free zinc levels under hypoxic conditions (Figure 1F). These increases of intracellular free zinc may come through several pathways: (1) increased zinc influx via Zip transporters 58; (2) reduced zinc efflux via ZnT transporters 58, 59; (3) increased zinc release from zinc‐binding proteins in particular MT 60, 61; (4) other channels, such as calcium‐permeable AMPA and/or kainite channels (Ca‐A/K) and voltage‐sensitive calcium channels 62. However, the mechanism of intracellular free zinc increase still requires further investigation. In conclusion, our results presented here provide direct evidence that hypoxia promotes the increase of intracellular zinc when cells are exposed to high concentrations of extracellular zinc, leading to a cascade of molecule processes that contribute to ischemic brain damage.
Hypoxia‐inducible factor‐1α is a transcription factor that binds to a specific DNA consensus sequence 5′‐RCGTG‐3′ within the hypoxia‐responsive elements and promotes the transcription of target genes encoding several proteins important for cell survival such as EPO, VEGF, HO‐1, among others 63, 64, 65, 66, 67, 68. Some studies have indicated that HIF‐1 exerts both antiapoptotic and proapoptotic effects, depending on the cell type 69. The effect of zinc on HIF‐1α has been studied and discussed in recent years but controversy remains. Some researchers believe that zinc affects the stability of HIF‐1α 29, 30, but others insist zinc has no effect on HIF‐1α stabilization but on nuclear migration of HIF‐1α 70. Notably, most of these reported experiments were performed under normoxia, so the regulation of zinc on HIF‐1α during hypoxia has been poorly understood. However, hypoxia is a primary consequence of cerebral ischemia. It is critically important to understand the interaction of zinc and HIF‐1α under hypoxic conditions and its role in zinc‐induced hypoxic cell death. Thus, we examined the expression of HIF‐1α in astrocytes exposed to both zinc and hypoxia. We found that while zinc had little effect on HIF‐1α in normoxia, the expression of HIF‐1α increased dramatically in a zinc‐concentration‐dependent manner in hypoxia (Figure 2), following exactly the same pattern of zinc‐induced cell death in hypoxia (Figure 1A,B). Moreover, zinc's cytotoxicity was significantly attenuated by knocking down HIF‐1α with siRNA (Figure 3). Together, these findings support the conclusion that under hypoxia conditions, zinc greatly increases HIF‐1α protein levels, which contributes to cell death. To our knowledge, this is the first report linking excessive accumulation of HIF‐1α protein to zinc‐induced hypoxic cell death, providing a novel mechanism of brain injury by zinc following cerebral ischemia.
It has been reported that PARP‐1 inhibition reduces the transcriptional activity of HIF‐1α 35. Unfortunately, whether or not the interaction between PARP‐1 and HIF‐1α is involved in zinc‐induced hypoxic cell death remains unclear. We show here that the expression of PARP‐1 regulates HIF‐1α protein levels, and HIF‐1α is downstream of PARP‐1, as demonstrated by the significant decrease of HIF‐1α expression by knocking down PARP‐1 (Figure 5). In contrast, knockdown of HIF‐1α had no effect on either expression or activity of PARP‐1 (Figure 6). Our data suggest that zinc increases hypoxia‐induced HIF‐1α stabilization through PARP‐1 modification. Our study presents direct evidence that PARP‐1 has the ability to modulate HIF‐1α in astrocytes.
In conclusion, our results demonstrate that although exposure of astrocytes to hypoxia or to zinc alone did not induce significant cell death, the combination of these two treatments leads to a dramatic increase in astrocytic death. The hypoxic response protein HIF‐1α and the cell fate determination factor PARP‐1 are both involved in zinc‐induced hypoxic cell death. Zinc may exacerbate astrocyte death by augmenting hypoxia‐induced HIF‐1α stabilization via PARP‐1. These findings provide a novel mechanism accounting for zinc‐mediated ischemic brain injury.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported in part by grants from NIH (P20RR15636, P30GM103400, and R01AG031725). We thank Jerry Hou for editing the manuscript.
References
- 1. MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr 2000;130(5S Suppl.):1500S–1508S. [DOI] [PubMed] [Google Scholar]
- 2. Prasad AS. Zinc in growth and development and spectrum of human zinc deficiency. J Am Coll Nutr 1988;7:377–384. [DOI] [PubMed] [Google Scholar]
- 3. Frederickson CJ, Sun SW, Silva D, Thompson RB. Importance of zinc in the central nervous system: the zinc‐containing neuron. J Nutr 2000;130(5S Suppl.): 1471S–1483S. [DOI] [PubMed] [Google Scholar]
- 4. Lee SJ, Koh JY. Roles of zinc and metallothionein‐3 in oxidative stress‐induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol Brain 2010;3:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bitanihirwe BK, Cunningham MG. Zinc: the brain's dark horse. Synapse 2009;63:1029–1049. [DOI] [PubMed] [Google Scholar]
- 6. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 2009;10:780–791. [DOI] [PubMed] [Google Scholar]
- 7. Galasso SL, Dyck RH. The role of zinc in cerebral ischemia. Mol Med 2007;13:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nguyen T, Hamby A, Massa SM. Clioquinol down‐regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc Natl Acad Sci U S A 2005;102:11840–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci 2000;21:395–401. [DOI] [PubMed] [Google Scholar]
- 10. Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci 2005;6:449–462. [DOI] [PubMed] [Google Scholar]
- 11. Cherny RA, Atwood CS, Xilinas ME, et al. Treatment with a copper‐zinc chelator markedly and rapidly inhibits beta‐amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 2001;30:665–676. [DOI] [PubMed] [Google Scholar]
- 12. Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci 2008;28:5827–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu D, Smith CL, Barone FC, et al. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res 1999;68:29–41. [DOI] [PubMed] [Google Scholar]
- 14. Petito CK, Olarte JP, Roberts B, Nowak TS, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol 1998;57:231–238. [DOI] [PubMed] [Google Scholar]
- 15. Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci 2007;27:4253–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Danilov CA, Fiskum G. Hyperoxia promotes astrocyte cell death after oxygen and glucose deprivation. Glia 2008;56:801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakanaka M, Wen TC, Matsuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A 1998;95:4635–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A 2000;97:10242–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marti HJ, Bernaudin M, Bellail A, et al. Hypoxia‐induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 2000;156:965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bergeron M, Yu AY, Solway KE, Semenza GL, Sharp FR. Induction of hypoxia‐inducible factor‐1 (HIF‐1) and its target genes following focal ischaemia in rat brain. Eur J Neurosci 1999;11:4159–4170. [DOI] [PubMed] [Google Scholar]
- 21. Chavez JC, LaManna JC. Activation of hypoxia‐inducible factor‐1 in the rat cerebral cortex after transient global ischemia: potential role of insulin‐like growth factor‐1. J Neurosci 2002;22:8922–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 2004;5:343–354. [DOI] [PubMed] [Google Scholar]
- 23. Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia‐inducible factor 1alpha is mediated by an O2‐dependent degradation domain via the ubiquitin‐proteasome pathway. Proc Natl Acad Sci U S A 1998;95:7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kallio PJ, Wilson WJ, O'Brien S, Mkino Y, Poelinger L. Regulation of the hypoxia‐inducible transcription factor 1alpha by the ubiquitin‐proteasome pathway. J Biol Chem 1999;274:6519–6525. [DOI] [PubMed] [Google Scholar]
- 25. Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia‐inducible factor (HIF‐1)alpha: its protein stability and biological functions. Exp Mol Med 2004;36:1–12. [DOI] [PubMed] [Google Scholar]
- 26. Tekin D, Dursun AD, Xi L. Hypoxia inducible factor 1 (HIF‐1) and cardioprotection. Acta Pharmacol Sin 2010;31:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan X, Heijnen CJ, Kooij MA, Groenendaal F, Bel F. The role and regulation of hypoxia‐inducible factor‐1alpha expression in brain development and neonatal hypoxic‐ischemic brain injury. Brain Res Rev 2009;62:99–108. [DOI] [PubMed] [Google Scholar]
- 28. Nardinocchi L, Pantisano V, Puca R, et al. Zinc downregulates HIF‐1alpha and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010;5:e15048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yun YJ, Li SH, Cho YS, Park JW, Chun YS. Survivin mediates prostate cell protection by HIF‐1alpha against zinc toxicity. Prostate 2010;70:1179–1188. [DOI] [PubMed] [Google Scholar]
- 30. Hwang JJ, Kim HN, Kim J. et al. Zinc(II) ion mediates tamoxifen‐induced autophagy and cell death in MCF‐7 breast cancer cell line. Biometals 2010;23:997–1013. [DOI] [PubMed] [Google Scholar]
- 31. Gilliams‐Francis KL, Quaye AA, Naegele JR. PARP cleavage, DNA fragmentation, and pyknosis during excitotoxin‐induced neuronal death. Exp Neurol 2003;184:359–372. [DOI] [PubMed] [Google Scholar]
- 32. Alano CC, Swanson RA. Players in the PARP‐1 cell‐death pathway: JNK1 joins the cast. Trends Biochem Sci 2006;31:309–311. [DOI] [PubMed] [Google Scholar]
- 33. Koh DW, Dawson TM, Dawson VL. Poly(ADP‐ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci 2005;62:760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zahradka P, Ebisuzaki K. Poly(ADP‐ribose) polymerase is a zinc metalloenzyme. Eur J Biochem 1984;142:503–509. [DOI] [PubMed] [Google Scholar]
- 35. Martin‐Oliva D, Aguilar‐Quesada R, O'Valle F, et al. Inhibition of poly(ADP‐ribose) polymerase modulates tumor‐related gene expression, including hypoxia‐inducible factor‐1 activation, during skin carcinogenesis. Cancer Res 2006;66:5744–5756. [DOI] [PubMed] [Google Scholar]
- 36. Tabernero A, Bolanos JP, Medina JM. Lipogenesis from lactate in rat neurons and astrocytes in primary culture. Biochem J 1993;294:635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 1995;15:378–384. [DOI] [PubMed] [Google Scholar]
- 38. Seib TM, Patel SA, Bridges RJ. Regulation of the system x(C)‐ cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011;59:1387–1401. [DOI] [PubMed] [Google Scholar]
- 39. Gee KR, Zhou ZL, Qian WJ, Kennedy R. Detection and imaging of zinc secretion from pancreatic beta‐cells using a new fluorescent zinc indicator. J Am Chem Soc 2002;124:776–778. [DOI] [PubMed] [Google Scholar]
- 40. Frederickson CJ, Giblin LJ, Krezel A, et al. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol 2006;198:285–293. [DOI] [PubMed] [Google Scholar]
- 41. Chong ZZ, Kang JQ, Maiese K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid Redox Signal 2004;6:277–287. [DOI] [PubMed] [Google Scholar]
- 42. Koh JY, Choi DW. Zinc toxicity on cultured cortical neurons: involvement of N‐methyl‐D‐aspartate receptors. Neuroscience 1994;60:1049–1057. [DOI] [PubMed] [Google Scholar]
- 43. Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996;272:1013–1016. [DOI] [PubMed] [Google Scholar]
- 44. Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab 2003;23:263–274. [DOI] [PubMed] [Google Scholar]
- 45. Dressler J, Hanisch U, Kuhlisch U, Kuhlisch E, Geiger KD. Neuronal and glial apoptosis in human traumatic brain injury. Int J Legal Med 2007;121:365–375. [DOI] [PubMed] [Google Scholar]
- 46. Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 2007;10:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Malaiyandi LM, Dineley KE, Reynolds IJ. Divergent consequences arise from metallothionein overexpression in astrocytes: zinc buffering and oxidant‐induced zinc release. Glia 2004;45:346–353. [DOI] [PubMed] [Google Scholar]
- 48. Chiu SC, Huang SY, Tsai YC, et al. Poly (ADP‐ribose) polymerase plays an important role in intermittent hypoxia‐induced cell death in rat cerebellar granule cells. J Biomed Sci 2012;19:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suh SW, Aoyama K, Alano CC, Anderson CM, Hamby AM, Swanson RA. Zinc inhibits astrocyte glutamate uptake by activation of poly(ADP‐ribose) polymerase‐1. Mol Med 2007;13:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Semenza GL, Agani F, Feldser D, et al. Hypoxia, HIF‐1, and the pathophysiology of common human diseases. Adv Exp Med Biol 2000;475:123–130. [DOI] [PubMed] [Google Scholar]
- 51. Tomita M, Semenza GL, Michiels C, et al. Retraction. Activation of hypoxia‐inducible factor 1 in human T‐cell leukaemia virus type 1‐infected cell lines and primary adult T‐cell leukaemia cells. Biochem J 2011;434:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984;308:734–736. [DOI] [PubMed] [Google Scholar]
- 53. Chimienti F, Seve M, Richard S, Mathieu J, Favier A. Role of cellular zinc in programmed cell death: temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem Pharmacol 2001;62:51–62. [DOI] [PubMed] [Google Scholar]
- 54. Cho E, Hwang JJ, Han SH, Chung SJ, Koh JY, Lee JY. Endogenous zinc mediates apoptotic programmed cell death in the developing brain. Neurotox Res 2010;17:156–166. [DOI] [PubMed] [Google Scholar]
- 55. Filipiak M, Bilska E, Tylko G, Pyza E. Effects of zinc on programmed cell death of Musca domestica and Drosophila melanogaster blood cells. J Insect Physiol 2010;56:383–390. [DOI] [PubMed] [Google Scholar]
- 56. Hamatake M, Iguchi K, Hirano K, Ishida R. Zinc induces mixed types of cell death, necrosis, and apoptosis, in molt‐4 cells. J Biochem 2000;128:933–939. [DOI] [PubMed] [Google Scholar]
- 57. Akcil E, Tug T, Doseyen Z. Antioxidant enzyme activities and trace element concentrations in ischemia‐reperfusion. Biol Trace Elem Res 2000;76:13–17. [DOI] [PubMed] [Google Scholar]
- 58. Lichten LA, Cousins RJ. Mammalian zinc transporters: nutritional and physiologic regulation. Annu Rev Nutr 2009;29:153–176. [DOI] [PubMed] [Google Scholar]
- 59. Palmiter RD, Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Archiv: European J Physiol 2004;447:744–751. [DOI] [PubMed] [Google Scholar]
- 60. Pedersen MO, Larsen A, Stoltenberg M, Penkowa M. Cell death in the injured brain: roles of metallothioneins. Prog Histochem Cytochem 2009;44:1–27. [DOI] [PubMed] [Google Scholar]
- 61. West AK, Hidalgo J, Eddins D, Levin ED, Aschner M. Metallothionein in the central nervous system: roles in protection, regeneration and cognition. Neurotoxicology 2008;29:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Galasso SL, Dyck RH. The role of zinc in cerebral ischemia. Mol Med 2007;13:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 2003;3:721–732. [DOI] [PubMed] [Google Scholar]
- 64. Trisciuoglio D, Gabellini C, Desideri M, et al. Involvement of BH4 domain of bcl‐2 in the regulation of HIF‐1‐mediated VEGF expression in hypoxic tumor cells. Cell Death Differ 2011;18:1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dawn B, Bolli R. HO‐1 induction by HIF‐1: a new mechanism for delayed cardioprotection? Am J Physiol Heart Circ Physiol 2005;289:H522–H524. [DOI] [PubMed] [Google Scholar]
- 66. Komatsu DE, Hadjiargyrou M. Activation of the transcription factor HIF‐1 and its target genes, VEGF, HO‐1, iNOS, during fracture repair. Bone 2004;34:680–688. [DOI] [PubMed] [Google Scholar]
- 67. Yeligar SM, Machida K, Kalra VK. Ethanol‐induced HO‐1 and NQO1 are differentially regulated by HIF‐1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem 2010;285:35359–35373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Agrawal A, Guttapalli A, Narayan S, Albert TJ, Shapiro IM, Risbud MV. Normoxic stabilization of HIF‐1alpha drives glycolytic metabolism and regulates aggrecan gene expression in nucleus pulposus cells of the rat intervertebral disk. Am J Physiol Cell Physiol 2007;293:C621–31. [DOI] [PubMed] [Google Scholar]
- 69. Piret JP, Mottet D, Raes M, Michiels C. Is HIF‐1alpha a pro‐ or an anti‐apoptotic protein? Biochem Pharmacol 2002;64:889–92. [DOI] [PubMed] [Google Scholar]
- 70. Park SE, Park JW, Cho YS, Ryu JH, Paick JS, Chun YS. HIF‐1alpha promotes survival of prostate cells at a high zinc environment. Prostate 2007;67:1514–23. [DOI] [PubMed] [Google Scholar]