

Abstract
Pancreatic cancer often presents in advanced stages and is unresponsive to conventional treatments. Thus, the need to develop novel treatment strategies for pancreatic cancer has never been greater. Here we report that combination of focal irradiation with hedgehog (Hh) signaling inhibition exerts better than additive effects on reducing metastases. In an orthotopic model, we found that focal irradiation alone effectively reduced primary tumor growth but did not significantly affect metastasis. We hypothesized that cancer stem cells (CSC) of pancreatic cancer are responsible for the residual tumors following irradiation, which may be regulated by Hh signaling. To test our hypothesis, we showed that tumor metastasis in our model was accompanied by increased expression of CSC cell surface markers as well as Hh target genes. We generated tumor spheres from orthotopic pancreatic and metastatic tumors, which have elevated levels of CSC markers relative to the parental cells and elevated expression of Hh target genes. Irradiation of tumor spheres further elevated CSC cell surface markers and increased Hh target gene expression. Combination of Hh signaling inhibition with radiation had more than additive effects on tumor sphere regeneration in vitro. This phenotype was observed in two independent cell lines. In our orthotopic animal model, focal radiation plus Hh inhibition had more than additive effects on reducing lymph node metastasis. We identified several potential molecules in mediating Hh signaling effects. Taken together, our data provide a rationale for combined use of Hh inhibition with irradiation for clinical treatment of pancreatic cancer patients.
INTRODUCTION
Pancreatic cancer continues to be the most difficult malignancy to treat, with the 5 year survival rate around 5% (1). Unlike most other malignancies, only 15–20% of pancreatic tumors are resectable, and there is an 80% chance of recurrence after surgery. In this patient population, survival averages 20 months with the use of standard gemcitabine chemotherapy (2). The use of radiotherapy alone on pancreatic cancer is disputed due to the high mortality rate and relatively small improvement with chemoradiotherapy (3). Since pancreatic cancer appears resistant to radiation, one strategy is to combine radiotherapy with another treatment option, such as a targeted drug.
Recent studies indicate that sonic hedgehog signaling can protect cancer cells against ionizing radiation therapy (4). The hedgehog (Hh) pathway, initially discovered in Drosophila, is a major regulator for cell differentiation, tissue polarity and cell proliferation (5). The seven transmembrane domain containing protein SMO serves as the key player for the Hh pathway, whose function is inhibited by another transmembrane protein Patched (PTC) in the absence of Hh ligands (6). Binding of Hh to its receptor PTC releases this inhibition, allowing SMO to signal downstream, eventually to Gli transcription factors, primarily Gli2 transcriptional factor. As transcription factors, Gli molecules can regulate target gene expression by direct association with a specific consensus sequence located in the promoter region of the target genes (7, 8). Activation of the Hh pathway has been reported in pancreatic cancer, but the exact role of this pathway in pancreatic tumorigenesis and progression remains largely unclear (9–15).
There is evidence to indicate that Hh signaling is involved in the maintenance of a variety of tissue stem cells and cancer stem cells (CSCs) (16–27). Growing evidence indicates that CSCs are inherently resistant to radiation and other therapeutic treatments (28–32). We hypothesized that inhibition of Hh signaling can reduce the regeneration potential of CSCs or cancer progenitor cells, which should facilitate focal radiotherapy of pancreatic cancer.
Using an orthotopic model of pancreatic cancer metastasis, we first tested the effect of irradiation on tumor growth and metastases. While irradiation was effective in reducing tumor growth, tumor metastasis was not significantly affected. We hypothesized that CSCs within the pancreatic tumor may be responsible for this resistance and tumor metastasis. To test our hypothesis, we examined activity of the Hh signaling pathway as well as CSC markers in these tumors because Hh signaling is known to regulate CSCs (33). We first tested our hypothesis in vitro using CSCs-enriched tumor spheres, and then in an orthotopic mouse model.
MATERIALS AND METHODS
Chemicals
Two smoothened signaling inhibitors were used in this study: CycT and BMS833923. CycT is a cyclopamine derivative provided by Logon Natural Products (Plano, Texas). CycT has been described in our previous study, including the structure and biological activities (34). BMS833923 was provided by Bristol-Meyers Squibb. BMS833923 is a potent synthetic small molecule (EC50=50 nM) with specific inhibition on smoothened signaling. BMS833923 was originally patented by Exelixis and is now licensed to Bristol-Meyers Squibb (35). Phase I clinical trial of BMS833923 has been completed, and further clinical trials are being planned.
Cell lines
AsPC1 & MIA PaCa2 were purchased from ATCC, authenticated by STR profiling, and cultured as instructed by the vendor. Pan02 was purchased from ATCC. MMC16 cell line was generated from a metastatic tumor of mouse pancreatic cancer model (36), and cultured in DMEM with 10% FBS.
Orthotopic mouse model of pancreatic cancer metastasis
Cells (AsPC-1, MIA PaCa2, Panc02 and MMC16) with stable expression of GFP and luciferase were harvested in single cell suspension at a concentration of 4 X 106 cells/ ml. A total of 2 X 105 cells (in 50 μl of growth medium) were injected into the mouse pancreas using a 27-gauge needle according to a protocol developed in Fidler’s laboratory (37). For the human cell lines AsPC1 and MIA PaCa2, we used NOD/scid/IL2Rγnull mice (NSG). For mouse cell lines Panc02 and MMC16, we used inbred C57Bl/6 mice. Twelve mice were used for each group. Bioluminescent imaging was used to monitor tumor growth. GFP-based whole body imaging was used to visualize metastases after animal sacrifice. Tumor lesions in pancreas, liver, lung and lymph nodes were harvested and divided into several portions. Some were used for primary culture; some were snap-frozen in liquid nitrogen for mRNA extraction; some were fixed in 10% buffered formalin and embedded in paraffin for H&E staining and immunohistochemistry. Nu/Nu mice were purchased from Charles River, and NSG mice were provided by the In Vivo Therapeutics (IVT) Core in the IU Simon Cancer Center. Mice were treated with Hh signaling inhibitors [CycT at 25mg/Kg body weight via intra-peritoneal injection (34), or BMS833932 (labeled as BMS in the figures, provided by Bristol Myers Squib, 30mg/Kg body weight via oral gavage). All animal experiments were performed following protocols approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
X-rays-based radiation Studies
Cells or tumor spheres were irradiated at a 50 cm distance from the X-ray head with a Precision 320 kVp X-ray machine set at 250 kVp, 12.5mA, with 2 mm Al filtration with a dose rate of 1.6 Gy per minute as previously reported (38). For the in vivo pancreas irradiation studies the animals were set up at 50 cm from the X-ray head (confirmed by laser alignment) and irradiated with a 2 x 2 cm lighted collimated field at a dose rate of 1.3 Gy per minute. X-ray dosimetry of the Precision X-ray unit is tested every 6 months with both ionization chamber and TLD measurements by the Department of Radiation Oncology at IU School of Medicine.
Imaging analyses
Fluorescence-based whole body imaging was performed using an illumatool LT-9900 Bright Light System (LightTools, Optical Research) according to manufacturer’s instruction that has been previously described by others (39). Bioluminescent imaging was conducted using a Berthold Nightowl system (Berthold Technologies, USA). Animals were anesthetized with 2 to 4% isoflurane and administered 150 mg/kg D-luciferin (Caliper Life Sciences) intraperitoneally. Mice were immediately transferred to the heated stage (40±1 °C) and imaged at 2 min intervals for 44 minutes with image integration times ranging from 1 to 5 sec/image. Animals were imaged weekly for 5 weeks beginning on day 7 after cell implant. Areas of interests were drawn at each time-point using a semi-automated Maximum Entropy algorithm and the peak intensity determined. Data were expressed as the fold-increase in the peak intensity (photons/sec·mm2) from week 1 for each mouse, and then averaged for each treatment group.
Single cell isolation and sphere formation
Pancreatic tumor tissues were cut with 21-blade scalpel into < 1mm3 size and placed in 1 mg ml−1 collagenase IV (Worthington) for 2 hrs at 37 °C with constant agitation. Single cells were pelleted at 800 x g after passing the digested tissues through a 70 μm cell strainer filter (BD Falcon). The cell pellets were subsequently washed twice with sphere growing medium (Neural Basal medium with B-27, 10μg/ml of EGF, 10μg/ml of bFGF and 679u/ml of heparin), then plated in 6-well non-adherent culture plates with 2X106 cells/ml. Spheres generally appear 3 days after culture, and medium was changed every other day. Secondary spheres were generated by obtaining single cells from tumor spheres through gentle incubation with accutase (Innovative Cell Technology) with constant trituration for 5 min, and the cells re-seeded into sphere growing medium for an additional 3–6 days.
Flow cytometry analyses
Single cells from primary and metastatic tumors were incubated with fluorescence conjugated antibody against human ESA, CD44 and CD24 (all from eBioscience) for 30min at 4°C. After washing, cells were stained with 1μg/ml DAPI to detect dead cells.
The ALDEFLUOR kit (StemCell Technologies, Durham, NC, USA) was used to isolate cells with high ALDH enzymatic activity according to the manufacturer’s instruction. Briefly, new isolated single cells were suspended in ALDEFLUOR assay buffer containing ALDH substrate (BAAA, 1 μmol l−1 per 1 × 106 cells) and then incubated for 45 min at 37°C. For each sample, an aliquot of cells was stained under identical conditions with 15 mmol l−1 diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor, as a negative control. In all experiments, the ALDEFLUOR-stained cells treated with DEAB served as ALDH-negative controls.
The side population was detected by staining cells with Hoechst 33342 dye (Molecular Probes-Invitrogen) using the methods described by Goodell et al. (40). Briefly, cells were resuspended at 1 × 106/mL in DMEM with 2% serum and 10 mmol/L HEPES buffer. Hoechst 33342 dye (final concentration 5 μg/mL) was added, and the mixture incubated at 37°C for 90 min with shaking. The cells were then washed with ice-cold HBSS with 2% FCS and 10 mmol/L HEPES, centrifuged down at 4°C, and resuspended in ice-cold HBSS containing 2% FCS and 10 mmol/L HEPES. An aliquot of cells was used as a negative control by adding ABC transporter inhibitor fumitremorgin C (FTC, 10μM) (to block the uptake of the dye) 30min before addition of Hoechst 33342.
Flow cytometry was performed on a LSR407 device (Beckton Dickinson) and data analyzed using flowjo software.
RNA extraction, RT-PCR and real-time PCR
Total RNAs of cells were extracted using Tri-RNA reagent from Sigma according to the manufacturer, and real-time quantitative PCR analyses were performed according to a previously published procedure (41–43). Triplicate CT values were analyzed in Microsoft Excel using the comparative CT(ΔΔCT) method as described by the manufacturer (Applied Biosystems, Foster City, CA). The amount of target (2−ΔΔCT) was obtained by normalization to an endogenous reference (GAPDH) and relative to a calibrator. All primers and probes were purchased from Applied Biosystems Inc. and the sequences are available upon request.
Statistical Methods
Comparison of the difference between two groups was carried out using the chi-square test, Dunnett’s test or Student’s t test. P value <0.05 was regarded as statistically significant. A Bliss independence model was employed to evaluate combination effects. The Bliss expectation was calculated with the equation (A+B)-(A x B), in which A and B are the fractional growth inhibitions induced by agents A and B at a given dose, respectively. The difference between the Bliss expectation and the observed growth inhibition induced by the combination of agent A and B at the same dose is the Bliss excess (44).
RESULTS
Effects of irradiation on tumor growth and metastases in orthotopic mouse model
In order to monitor tumor growth and metastasis in mouse models, we generated cells with expression of GFP-luciferase fusion protein, which allowed us to monitor tumor growth by bioluminescence imaging and to detect metastases by fluorescence-based whole body imaging. As shown in Fig. 1, the level of luciferase activity increased steadily in the pancreas area following pancreatic injection of GFP-luciferase expressing AsPC1 cells in NSG mice, indicating rapid tumor growth in this model. Tumor metastasis is not very easily detected using luciferase activity due to sporadic metastases in organs like lymph nodes, peritoneum, diaphragm, mesentery, liver and lung. Tumor metastases can be detected by GFP expression after dissection (Fig. 2). Using this mouse model, we examined the effect of radiation on tumor growth and metastases of pancreatic cancer.
Fig. 1. Effects of radiation on tumor growth in an orthotopic mouse model of pancreatic cancer.
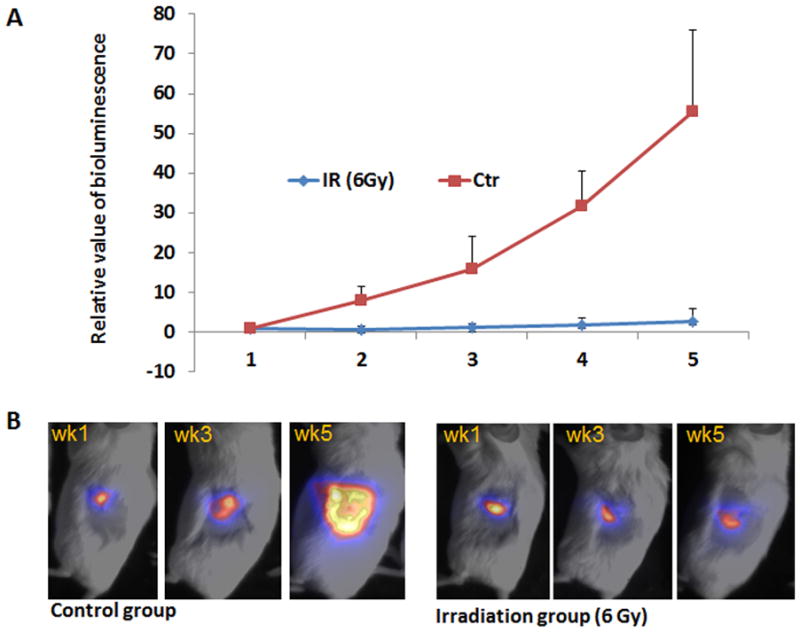
AsPC1 cells were infected with lentiviruses expressing luciferase-green fluorescent protein (GFP). Three days after pancreatic injection of these cells, mice were focally irradiated at 6 Gy around the pancreas site (area: 2cm X 2cm) or left untreated. Tumor growth was monitored by bioluminescence imaging (Fig. 1A/1B). For bioluminescent imaging analysis, we treated the value from the first imaging (week 1) as 1. Each group had 12 or more mice. We monitored the mice for 5 weeks. A shows the quantitative analysis of relative bioluminescent imaging values over 5 weeks, and B shows typical imagines for each group.
Fig. 2. Effects of irradiation on pancreatic cancer metastasis.
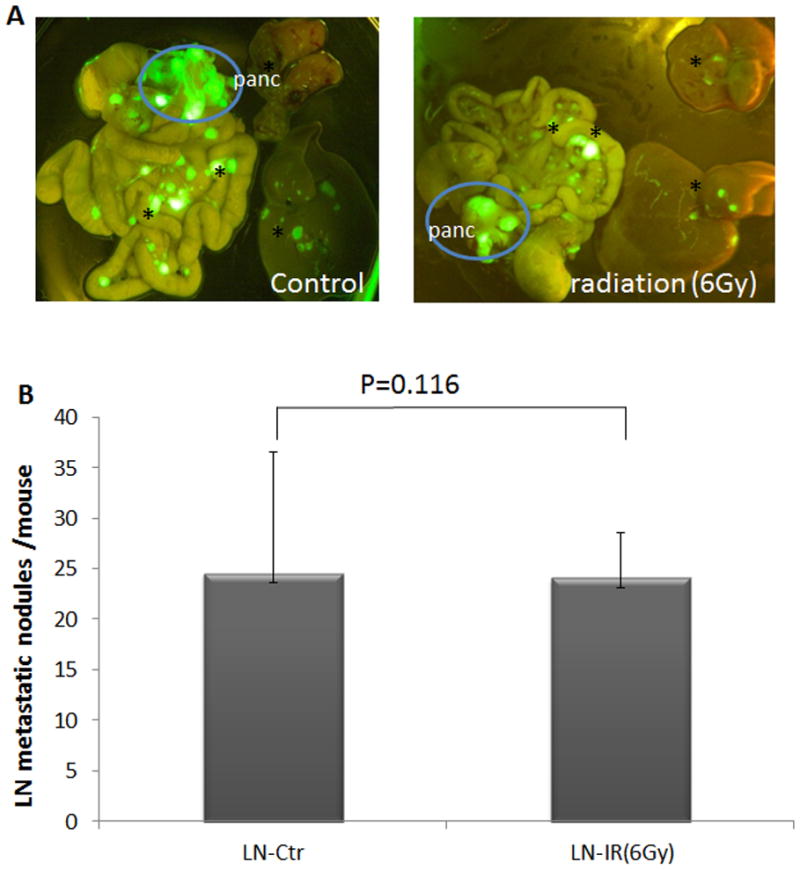
The mouse model was same as described in Fig. 1. At the end of the study (day 35 after pancreatic injection), mice were sacrificed, and the site of tumor metastases were detected by the appearance of GFP using GFP-based imaging system (A). Panc indicates pancreatic tumor, and * indicates metastatic nodules. B shows the quantitative analysis of metastatic nodules in lymph nodes (indicated as LN). AsPC1 cells without luciferase-GFP gave similar patterns of tumor metastasis and similar pancreatic tumor size, indicating that luciferase-GFP expression did not alter the property of the cells.
Mice with pancreatic tumors were subjected to 6Gy focal radiation once, 3 days after pancreatic injection of cancer cells, and were examined for tumor size by bioluminescence on a weekly basis, and for tumor metastasis by GFP-based imaging at the end of the study. The value of the bioluminescence (indicating primary tumor growth) decreased in magnitude one week after irradiation, and was not significantly increased five weeks after irradiation (Fig. 1A, 1B). In contrast, the bioluminescence value in the control group increased over 50 fold during the same period of time, indicating that radiation was effective in reducing primary tumor growth (Fig. 1A, 1B). The irradiated mice had no signs of body weight change or reduction of survival in the first three weeks, suggesting that focal radiation at 6Gy had limited damages in mice. Starting from week 4, mice with or without radiation started to show signs of weakness, loss of weight and eventually succumbed to pancreatic cancer. We noticed that the number and the size of metastatic nodules were not significantly decreased in the irradiated mice (Fig. 2). This result indicates that despite effective reduction in primary tumor size, focal radiation did not affect tumor metastases of pancreatic cancer.
Metastases are associated with increased expression of CSC markers and Hh target genes
It is widely accepted that pancreatic cancer is hierarchically organized with a small population of CSCs that possess a capacity to self-renewal (45, 46). CSCs may be responsible for tumor metastasis and radiation resistance. It has been reported that cells with surface expression of CD44, CD24 and ESA, or CD133 exhibit features of CSC in pancreatic cancer (46–48). To assess whether metastatic tumors in our orthotopic model are associated with elevated expression of CSC markers, we performed two types of assays. First, we compared primary pancreatic tumors with metastatic tumors for cell surface expression of CD44, ESA and CD24 by flow cytometry. By RT-PCR, we detected CD24 expression in AsPC1 cells (Supplementary Figure 1, as Fig. S1). In the AsPC1-based orthotopic mouse model of pancreatic cancer metastasis, we first gated ESA positive cells and then examined the percentage of CD24+CD44+ cells. We found that the percentage of CD24+CD44+ cells in metastatic tumors was several fold higher than that of pancreatic tumors (Fig. 3A), indicating an increase of CSC population during metastasis.
Fig. 3. Expression of CSC cell surface markers and Hh target genes in pancreatic cancer metastases.

Single cells were isolated from pancreatic cancer or lymph node metastases. Expression of CSC cell surface markers were detected by flow cytometry using specific antibodies to CD24, CD44 and ESA (Fig. 3A). ESA positive cells were first gated to further detect CD24+CD44+ cells. Expression of Hh target genes GLI and PTCH1 of human origin was detected by real-time PCR using total RNAs from AsPC1-derived pancreatic tumor (indicated as Panc) and different metastatic tumors. LN indicates lymph node metastatic nodules, and liver indicates liver metastatic nodules. *p value<0.05 indicates significant difference from pancreatic tumor.
Next, we examined the transcripts of Hh target genes GLI, PTCH1, and found an increase of GLI and PTCH1 in lymph node (LN) and liver metastatic tumors (Fig. 3B). We also noticed that expression of Hh target genes in primary pancreatic cancer was higher than that of cultured cells, suggesting an effect of tumor microenvironment on Hh signaling. This phenotype was not limited to AsPC1 cells because models derived from Panc02, MIA PaCa2 and MMC16 cells all had elevated Hh signaling in metastatic tumors (Fig. S2). The exact reason for the pathway activation was not entirely clear but we found elevated expression of SHH and IHH, major ligands for Hh signaling activation, in metastatic tumors in comparison with the primary tumor (Fig. S3). In addition, we detected stromal Hh target gene expression in models derived from AsPC1 and MIA PaCa2 (Fig. S4), which was consistent with previous reports (49, 50). Taken together, these results suggest that metastasis of pancreatic cancer is associated with elevated signaling of the Hh pathway and an increase of the CSC population.
Based on our data, we hypothesized that residual cells of the pancreatic cancer surviving the radiation are enriched in CSC-like cell population which can be regulated by Hh signaling. We propose that in the primary pancreatic cancer, the majority of cancer cells are destroyed by radiation, but CSC-like cells continue to grow and metastasize. Because Hh signaling is highly activated in metastatic tumors, we predicted that radiation in combination with Hh signaling inhibition would be more effective in reducing tumor metastasis. To test our hypothesis, we first evaluated the combined effect of radiation and Hh signaling inhibition in CSC-enriched cells. Although cell sorting using specific cell surface markers can yield a more pure CSC population, it is difficult to yield sufficient cell numbers for further experiments. Alternatively, tumor spheres, which are known to be enriched in CSC, can be used for such in vitro functional studies.
Combination of Hh inhibition with radiation reduces tumor sphere regeneration
After obtaining tumor spheres (see Fig. S5 for morphology), we used two approaches to test whether the CSC population was indeed enriched: expression of cell surface markers ESA, CD24 and CD44, and the percentage of ALDH+ cell population. By comparing tumor sphere cells with the parental cells, we found about 1% CD24+CD44+ESA+ cells in parental cells but over 8% triple positive cells in tumor spheres (Fig. 4A). At the transcript level, CD24 was detectable in parental AsPC1 cells and was increased in the tumor spheres (Fig. S1).
Fig. 4. Characterization of tumor spheres.

To obtain tumor spheres, single cells were first isolated from pancreatic tumor or LN metastases. Tumor spheres were formed after 5 days of incubation with serum-free medium with defined growth factors (see Methods for details). Cell surface expression of CD24, CD44 and ESA was analyzed by flow cytometry (Fig. 4A shows CD24+CD44+ cells in ESA positive population). ALDH+ cell population of pancreatic cancer cells was detected by staining with ALDEFLUORR, and detected by flow cytometry (Fig. 4B). When a specific ALDH inhibitor DEAB was used, this population was nearly undetectable (<0.1%) (not shown). Fig. 4C shows real-time analysis of Hh target gene expression for AsPC-1 cells and tumor spheres from pancreatic tumors (PANC) or lymph node metastases (LN) in the AsPC1-based orthotopic model. Gene expression of GLI, PTCH1, BCL2, MYCN, SNAI1, JAG2 and FST in tumor spheres was significantly higher than that from the parental cells. CCND2 was not detectable in parental cells but was highly expressed in tumor spheres. Not all reported Hh target genes were elevated in tumor spheres. For example, expression of BMI1 and CCND1 was similar between parental cells and tumor spheres. Fig. 4D shows the diagram of Hh signaling and basic structures of two compounds used in this study (see Materials and Methods for references).
In addition, CSC population often has an increased level of ALDH activity (33). By comparing AsPC1 cells growing in monolayer with those in tumor spheres, we noticed that ALDH positive cells were higher in tumor sphere (1.2%) than the parental cells (0.159%) (Fig. 4B). These data indicate that tumor spheres indeed have elevated expression of CSC markers.
At the same time, we found increased expression of GLI/PTCH1 and other reported Hh target genes [reviewed in (51)] in tumor spheres in comparison with the parental cells (Fig. 4C), suggesting that Hh signaling may be important for tumor sphere maintenance and regeneration. Most notably, we found elevated expression of proliferation-related genes MYCN, CCND2, stem cell signaling molecules JAG2, FST, PDGFRα, WNT2, as well as genes involved in survival (BCL2) and in epithelial-to-mesenchymal transition (SNAI1).
Next, we tested whether combination of Hh signaling inhibition with radiation would be more effective in reducing tumor sphere formation efficiency. For the parental AsPC1 cells, irradiation at 2 Gy was sufficient to reduce cell number by half (Fig. 5A), suggesting that AsPC1 cells are quite sensitive to radiation. Formation of secondary tumor spheres is commonly used to test cell stemness in CSC functional studies (47). Because tumor spheres have more CSCs and express a high level of Hh target genes, we examined whether regeneration of tumor spheres is affected by Hh signaling inhibition and radiation. We first treated single cells from tumor spheres with radiation at different dosages (2, 4, 6, 8 Gy) or left untreated, and examined the efficiency of secondary tumor sphere formation. Effects of irradiation on tumor spheres were assessed by the number of spheres formed from 1,000 cells. As shown in Fig. 5B, we found that 2 Gy only reduced the number of secondary tumor spheres by 30%. We estimated the EC50 dose of radiation for inhibiting tumor sphere formation was over 5Gy.
Fig. 5. Responses of tumor spheres to radiation in the absence or presence of Hh signaling inhibition.

To test the sensitivity of AsPC1 cells and the tumor spheres, single cells were irradiated at different doses (see Methods), and were put back into appropriate media (RPMI with 10% FBS for AsPC1 culture in regular 96 well plates and tumor sphere medium in extra low adhesive plates (see Methods). Cell viability of AsPC1 was measured by measurements with Alama blue metabolism, and the number of visible tumor spheres was counted 5 days after radiation (5A). For tumor spheres, if single cells were seeded with regular RPMI 5%FBS medium in regular 96 well plates, the level of CSC markers will be reduced to the basal level, possibly due to differentiation. We kept all tumor spheres in the defined medium. 5B shows the effect of radiation on regeneration of tumor spheres. Sphere-L indicates tumor spheres from lymph node metastases, and sphere-P indicates tumor spheres from pancreatic tumors. Fig. 5C shows the effect of radiation and Hh signaling inhibition (Hh-I) on tumor sphere regeneration. We used BMS833932 or CycT to inhibit Hh signaling. Both drugs gave similar results, and Fig. 5C shows the data from CycT. Fig. 5D shows cell surface marker expression following radiation. ESA+ cells were first gated before analysis with CD24 and CD44 expression. Fig. 5E shows Hh target gene expression changes after radiation by real-time PCR analyses with no radiation group as the control.
The effect of Hh signaling inhibition on tumor sphere formation was tested by addition of Hh signaling inhibitor CycT in the tumor sphere formation medium. We found that Hh signaling inhibition by CycT at 5μM reduced tumor sphere formation by 50% (Fig. S6). We therefore determined whether combination of the two will be more effective. After irradiation of tumor sphere cells in the presence or absence of Hh inhibitor CycT, we examined the tumor sphere forming efficiency. As shown in Fig. 5C, tumor sphere formation was reduced by 30–50% after 4 Gy, by 50% after Hh inhibition, but was completely eliminated when radiation was combined with Hh inhibition. By Bliss independence analysis (see Methods), we found that combining focal irradiation with Hh signaling inhibition had more than additive effects (the response fraction for irradiation was 0.08; the response fraction for Hh signaling inhibition was 0.42 whereas the combined treatment had a response fraction of 0.62)[0.62 > (0.42+0.08)− 0.42 X 0.080] (Fig. 5C). We also found that radiation of tumor spheres further increased the level of Hh target genes (GLI and PTCH1), which were inhibited by BMS833923 (Fig. 5D). Consistent with Hh signaling changes, we discovered that the level of CSC cell surface markers was induced by radiation, but reduced to the basal level after radiation plus Hh inhibition (Fig. 5E). Taken together, our results indicate that the combination of radiation with Hh signaling inhibition will be more effective in treatment of pancreatic cancer metastasis in our mouse model.
To further support our data derived from AsPC1 cells, we used another cell line, MMC16, for tumor sphere studies. MMC16 cells were derived from a mouse model of pancreatic cancer (36), with a high efficiency in tumor sphere formation. For this mouse cell line, we used side population as a marker for CSC population because the mouse cell surface markers are not widely used. The side population is the cells with high expression of ABC transporter protein family members, rendering these cells less effective in uptake of fluorescent dyes (52). Like normal tissue stem cells, CSCs are also reported to possess this property (24). It has been reported that the side population of pancreatic cancer cells has some features of CSCs, such as high invasive and metastatic potential, and susceptibility to EMT (24). After staining with the fluorescent dye Hoechst 33342, we can detect side population (SP) by flow cytometry. For MMC16 cells, the side population was less than 2% whereas radiation increased the percentage of the population to >6% (Fig. S7A). This increase was specific because addition of the ABC transporter inhibitor fumitremorgin C reduced the side population (data not shown). Consistent with an increase of the side population, we observed elevated Hh target gene expression in tumor spheres after radiation (Fig. S7B). Further analysis indicate that while radiation and Hh signaling inhibition both reduced tumor sphere formation efficiency, combination of Hh signaling inhibition with radiation at 8Gy resulted in no visible tumor spheres (Fig. S7C). Similar to AsPC1-derived tumor spheres, Bliss independence analysis showed that the combination had greater than additive effects when irradiation was over 2Gy (Fig. S7C).
Once placed in regular culture medium, however, tumor sphere cells began to lose expression of the cell surface markers as well as Hh target genes (not shown), suggesting a unique microenvironment is required to keep the stemness of CSCs. Consistent with the high expression of CSC markers, single cells from tumor spheres were also more efficient in tumor sphere formation (20–30 spheres per 1,000 cells from tumor sphere cells vs. 2–3 spheres per 1,000 cells from parental AsPC1 cells). These results suggest that the CSC-enriched tumor spheres require a tumor environment for CSC to be functional.
Our results from tumor sphere studies indicate that Hh target gene expression is associated with CSC markers in two independent cell lines, and radiation plus a Hh signaling inhibitor have more than additive effects in tumor sphere formation. Because tumor metastasis is associated with high Hh target gene expression and increased expression of CSC markers, our data support that combining Hh signaling inhibition with radiation can be more effective in suppressing tumor metastasis in pancreatic cancer.
Radiation with Hh signaling inhibition on pancreatic cancer metastasis in mouse model
To test our hypothesis derived from tumor sphere studies, mice with orthotopic pancreatic tumors were treated with radiation (6Gy) with or without Hh inhibitor BMS833932 (as Hh-I in the figure), or left untreated. The mice were imaged weekly, and were sacrificed 5 weeks after pancreatic injection. Hh signaling inhibition by BMS833932 was shown to reduce expression of Hh target genes (Fig. S8). Typical images from each group were shown in Fig. 6A. We found that the combined treatment group gave the best response. Tumor growth was monitored by bioluminescence imaging. Hh signaling inhibition had no significant effect on primary tumor growth whereas tumors from mice with radiation did not grow (Fig. 6B). Despite lack of any significant effects of Hh inhibitor BMS833932 for primary tumor growth, the number of metastatic tumor nodules in lymph nodes was significantly reduced (Fig. 6C). We observed that combining Hh inhibition with focal irradiation gave a greater than additive effect in reducing lymph node metastases based on Bliss independence analysis. Combination of IR and Hh inhibition was also more effective than either single treatment alone on suppressing lung metastases (Fig. S9). Thus, our data demonstrated more therapeutic efficacy when irradiation is combined with Hh inhibition.
Fig. 6. Treatment of pancreatic cancer by combination of focal radiation with Hh signaling inhibition.

Twelve mice in each group [four groups: control; irradiation alone (IR); Hh inhibition alone (Hh-I); and combination of irradiation with Hh signaling inhibition (IR+Hh-I)] received appropriate treatments 3 days after pancreatic injection of luciferase-GFP expressing AsPC1 cells. Mice were monitored for tumor growth by bioluminescence imaging, and the tumor metastases were examined by the appearance of GFP at the end of the study (5 weeks after pancreatic injection). A shows the typical images of GFP expression from each group, and the pancreatic tumors were circled in orange. Metastatic nodules were indicated by *. B shows tumor growth curves based on the bioluminescence imaging. C shows the average number of lymph node metastases in each group of mice. * indicates statistical different from the control group. + indicates a greater than additive effect based on Bliss independence analysis.
To investigate the molecular mechanisms by which Hh signaling regulates CSC and tumor metastases, we examined putative Hh target genes in our mouse models. As shown in Supplementary Fig. 10, we found elevated expression of PDGFRα, WNT2, OPG and IGFBP4 (Fig. S10A), and BMS833923 inhibited expression of PDGFRα, WNT2 and OPG (Fig. S10B). As described earlier, PDGFRα is a known Hh target gene (53–56), and may be responsible for Hh signaling-mediated cell proliferation and tumor metastasis. Similarly, WNT2 is reported to be a specific marker for circulating pancreatic cancer cells in the pancreatic cancer patients with metastatic disease (57), and was shown to be regulated by Hh signaling in another study (50). OPG is known to be involved in cancer metastasis, and is regulated by Indian hedgehog. We believe that PDGFRα and WNT2 are good candidates for the underlying molecular mechanisms by which Hh signaling mediates cell proliferation and maintenance of CSC population.
DISCUSSION
Hh signaling during pancreatic carcinogenesis and metastasis
During tumor development of pancreatic cancer, increasing evidence suggests two major changes: a cell-autonomous non-canonical Gli regulation and Hh ligand-dependent paracrine Hh signaling (12, 49, 50, 58–61). It has been shown that Gli1 or Gli2 activation cooperates with KRAS to promote pancreatic cancer formation (12, 61). However, Hh signaling during pancreatic cancer metastasis is less clear although it is generally believed that similar signaling events also occur. Variable rates of metastasis in different GEM models, and the dependence of the phenotypes on the mouse genetic background make the study of metastasis more challenging. Using AsPC1 and MIA PaCa2 cells in the orthotopic mouse models of pancreatic cancer metastasis, we were able to specifically examine Hh signaling in the tumor compartment (human Hh target genes) and the stroma (mouse Hh target genes). Our data revealed that Hh signaling is activated in the stroma and in the tumor compartment. Our data are consistent with previous studies in E3LZ10.7 cell-derived orthotopic model (33). We further indicated that SHH and IHH were highly expressed in metastatic tumors (Fig. S3). We believe that ligand-mediated signaling is at least partially responsible for Hh signaling activation because SMO signaling inhibitors (CycT and BMS833923) did reduce the levels of human and mouse Hh target genes. Similar results were also observed in isolated tumor spheres where only human Hh target genes are detectable (Fig. 5D). The highly association between human Hh target gene expression and CSC markers in tumor spheres and in mouse models indicate that tumor specific Hh signaling may be important for maintenance of CSC population during pancreatic cancer metastasis.
Hh signaling and CSCs
The cancer stem cell theory has explained several phenotypes of human cancer, including high metastatic potential, tumor recurrence and therapeutic resistance. Accumulating evidence indicates the existence of cancer stem cells in pancreatic cancer (45). Using cell surface markers, Li et al demonstrated that a subset of pancreatic cancer cells with CD24+CD44+ESA+ or CD44+cMEThigh is more potent in generating tumors (29), and inhibition of cMET reduces tumor metastasis (47). Furthermore, pancreatic cancer cells with high ALDH activity more readily form tumors in immune deficient mice (33). Additionally, the side population of pancreatic cancer cells is more metastatic when implanted in immune deficient mice (62). Our studies showed that tumor spheres are enriched in CD24+CD44+ESA+ cells and ALDH+ cells (Fig. 4). Functional analyses using tumor sphere regeneration indicate that inhibition of Hh signaling can sensitize tumor spheres to radiation (Fig. 5). The data were further confirmed in a mouse orthotopic model of pancreatic cancer metastasis, in which Hh signaling inhibition significantly reduced tumor metastasis (Fig. 6). Similar studies from other groups also indicate a role of Hh signaling in pancreatic cancer stem cells (15, 27, 33). Understanding the molecular mechanisms by which Hh signaling regulates CSCs require additional studies. For example, we do not know whether CSCs with genetic depletion of Hh signaling can still form tumor metastases. We know very little of the interaction between CSC and the tumor microenvironment. It is known that Hh signaling activation is a major factor for the development of desmoplastic stroma (63), which provides a physical barrier for drug delivery to the tumor.
In consistent with the role of Hh signaling for CSC, we detected elevated expression of several stem cell signaling molecules in tumor spheres, including JAG2, FST, WNT2 and PDGFRα. The CSC-enriched tumor spheres also have elevated expression of proliferation-related genes such as MYCN, CCND2. In addition, survival gene BCL2 and EMT regulatory gene SNAI1 were also up-regulated. Further understanding of the roles of Hh signaling for maintenance of CSCs and for the tumor microenvironment may help generate new therapeutic options for pancreatic cancer.
Is combining Hh inhibitor with radiation more effective for pancreatic cancer treatment?
Surgery, radiation, and chemotherapy have been the mainstays of treatment for human malignancies for several decades. Technical advances in irradiation have made the treatment very focused on cancer tissues with limited radiation exposure to normal adjacent tissues. The use of a combination of radiation and chemotherapy, chemoradiation, has been proven to be effective in many tumor types. In treatment of pancreatic cancer, combination of gemcitabine with irradiation is effective only in a subset of patients (64). Thus, there exists the need to develop better combinations of radiation with chemotherapy or targeted therapy.
Our data provide evidence to support that the combination of radiation with Hh signaling inhibitor results in greater than additive inhibition of tumor metastasis in mouse orthotopic model of pancreatic cancer. We found elevated expression of CSC markers following irradiation as well as increased Hh signaling. We showed that Hh signaling regulates expression of WNT2 and PDGFRα (Fig. S10), molecules known to regulate cell proliferation and cell stemness (53, 54, 57). This CSC-enriched tumor spheres are also more resistant to radiation, and contain activated Hh signaling. They became sensitive to radiation after Hh signaling inhibition (Fig. 5C and Fig. S7C). The relevance of our data in vitro to pancreatic cancer treatment was also shown in our animal model (Fig. 6). Since Hh signaling activation in tumor metastases has been observed in the two models tested, we believe that this mechanism is common for most pancreatic cancer.
Inhibitors of Hh signaling
Several Hh signaling inhibitors are in clinical trial, and all are targeted at smoothened. Off-target effects have been reported when the inhibitors are over-dosed (10mM for cyclopamine) (65). To avoid off-target effects of CycT and BMS833923, we first tested their biological effects in Hh-dependent tumor models (34) to obtain the EC50. In tumor sphere assays, BMS833923 (EC50= 50nM) is more potent than CycT (EC50= 5 μM) (Fig. S6). Furthermore, we correlate the change of metastatic tumor nodules with Hh target gene expression.
Taken together, our studies suggest that combining focal irradiation with Hh signaling inhibition may be more effective in future pancreatic cancer treatment. Focal irradiation reduces the size of pancreatic cancer but has little effects on tumor metastases, which are associated with elevated expression of CSC cell surface markers and Hh target gene expression. Combination of irradiation with Hh signaling inhibition gives more than additive effects than single treatment in the tumor sphere assay and in an orthotopic mouse model for pancreatic cancer metastasis.
Supplementary Material
Acknowledgments
This work was supported by NCI, the Wells Center for Pediatric Research, IU Simon Cancer Center and The Signature Center for Pancreatic Cancer in Indiana University. We thank Anthony L. Sinn, Brian P. McCarthy, Drs. Karen Pollock and Paul R. Territo for technical support as well as the core facilities (In Vivo Therapeutics, and In Vivo Biomedical Imaging) for services.
Financial Support:
This work was supported by grants from National Cancer Institute (R01CA94160, R01CA155086) to J. Xie, from The Wells Center for Pediatric Research to J. Xie, from IU Simon Cancer Center and The Signature Center for Pancreatic Cancer in Indiana University to J.Xie.
Footnotes
Disclosure of Potential Conflict of Interest: Authors of this manuscript have no conflict of interests to disclose.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. Jama. 2007;297:267–77. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 3.Herman JM, Swartz MJ, Hsu CC, Winter J, Pawlik TM, Sugar E, et al. Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. J Clin Oncol. 2008;26:3503–10. doi: 10.1200/JCO.2007.15.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YJ, Lin CP, Hsu ML, Shieh HR, Chao NK, Chao KS. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. International journal of radiation oncology, biology, physics. 2011;80:851–9. doi: 10.1016/j.ijrobp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Parkin CA, Ingham PW. The adventures of Sonic Hedgehog in development and repair. I. Hedgehog signaling in gastrointestinal development and disease. Am J Physiol Gastrointest Liver Physiol. 2008;294:G363–7. doi: 10.1152/ajpgi.00457.2007. [DOI] [PubMed] [Google Scholar]
- 6.Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–34. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–22. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- 8.Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10:634–42. doi: 10.1128/mcb.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 10.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Tian X, Xie X, Zhuang Y, Wu W, Wang W. Expression and regulation of hedgehog signaling pathway in pancreatic cancer. Langenbecks Arch Surg. 2010;395:515–25. doi: 10.1007/s00423-009-0493-9. [DOI] [PubMed] [Google Scholar]
- 12.Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:E1038–47. doi: 10.1073/pnas.1114168109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–53. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012;4:218–33. doi: 10.1002/emmm.201100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodova M, Fu J, Watkins DN, Srivastava RK, Shankar S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One. 2012;7:e46083. doi: 10.1371/journal.pone.0046083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–9. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048–53. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobune M, Takimoto R, Murase K, Iyama S, Sato T, Kikuchi S, et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009;100:948–55. doi: 10.1111/j.1349-7006.2009.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. The EMBO journal. 2010;29:2646–58. doi: 10.1038/emboj.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature. 2011;472:110–4. doi: 10.1038/nature09851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song Z, Yue W, Wei B, Wang N, Li T, Guan L, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One. 2011;6:e17687. doi: 10.1371/journal.pone.0017687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takezaki T, Hide T, Takanaga H, Nakamura H, Kuratsu J, Kondo T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer science. 2011;102:1306–12. doi: 10.1111/j.1349-7006.2011.01943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su W, Meng F, Huang L, Zheng M, Liu W, Sun H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through beta-catenin signaling. Experimental hematology. 2012 doi: 10.1016/j.exphem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(+) glioma stem cells to temozolomide therapy. Mol Med. 2011;17:103–12. doi: 10.2119/molmed.2010.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer. 2012;131:30–40. doi: 10.1002/ijc.26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller MT, Hermann PC, Witthauer J, Rubio-Viqueira B, Leicht SF, Huber S, et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137:1102–13. doi: 10.1053/j.gastro.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 29.Ischenko I, Seeliger H, Schaffer M, Jauch KW, Bruns CJ. Cancer stem cells: how can we target them? Current medicinal chemistry. 2008;15:3171–84. doi: 10.2174/092986708786848541. [DOI] [PubMed] [Google Scholar]
- 30.Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol. 2008;26:2813–20. doi: 10.1200/JCO.2008.16.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mimeault M, Hauke R, Mehta PP, Batra SK. Recent advances in cancer stem/progenitor cell research: therapeutic implications for overcoming resistance to the most aggressive cancers. Journal of cellular and molecular medicine. 2007;11:981–1011. doi: 10.1111/j.1582-4934.2007.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 33.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan Q, Gu D, He M, Liu H, Sheng T, Xie G, et al. Tumor shrinkage by cyclopamine tartrate through inhibiting hedgehog signaling. Chin J Cancer. 2011;30:472–81. doi: 10.5732/cjc.011.10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tremblay MR, Nesler M, Weatherhead R, Castro AC. Recent patents for Hedgehog pathway inhibitors for the treatment of malignancy. Expert Opin Ther Pat. 2009;19:1039–56. doi: 10.1517/13543770903008551. [DOI] [PubMed] [Google Scholar]
- 36.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–73. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Estabrook NC, Chin-Sinex H, Borgmann AJ, Dhaemers RM, Shapiro RH, Gilley D, et al. Inhibition of NF-kappaB and DNA double-strand break repair by DMAPT sensitizes non-small-cell lung cancers to X-rays. Free radical biology & medicine. 2011;51:2249–58. doi: 10.1016/j.freeradbiomed.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 39.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–46. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 40.Goodell MA. Stem cell identification and sorting using the Hoechst 33342 side population (SP) In: Paul Robinson J, et al., editors. Current protocols in cytometry / editorial board. Unit9. Chapter 9. 2005. p. 18. [DOI] [PubMed] [Google Scholar]
- 41.Athar M, Li C, Tang X, Chi S, Zhang X, Kim AL, et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004;64:7545–52. doi: 10.1158/0008-5472.CAN-04-1393. [DOI] [PubMed] [Google Scholar]
- 42.Huang S, He J, Zhang X, Bian Y, Yang L, Xie G, et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis. 2006;27:1334–40. doi: 10.1093/carcin/bgi378. [DOI] [PubMed] [Google Scholar]
- 43.Ma X, Chen K, Huang S, Zhang X, Adegboyega PA, Evers BM, et al. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005;26:1698–705. doi: 10.1093/carcin/bgi130. [DOI] [PubMed] [Google Scholar]
- 44.Berenbaum MC. A method for testing for synergy with any number of agents. The Journal of infectious diseases. 1978;137:122–30. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- 45.Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:5646–8. doi: 10.1158/1078-0432.CCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 46.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 47.Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141:2218–27. e5. doi: 10.1053/j.gastro.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–9. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 51.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–86. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 52.Lin KK, Goodell MA. Detection of hematopoietic stem cells by flow cytometry. Methods in cell biology. 2011;103:21–30. doi: 10.1016/B978-0-12-385493-3.00002-4. [DOI] [PubMed] [Google Scholar]
- 53.Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, Epstein E, et al. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc Natl Acad Sci U S A. 2001;98:9255–9. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelczar P, Zibat A, van Dop WA, Heijmans J, Bleckmann A, Gruber W, et al. Inactivation of patched1 in mice leads to development of gastrointestinal stromal-like tumors that express pdgfralpha but not kit. Gastroenterology. 2013;144:134–44. e6. doi: 10.1053/j.gastro.2012.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walton KD, Kolterud A, Czerwinski MJ, Bell MJ, Prakash A, Kushwaha J, et al. Hedgehog-responsive mesenchymal clusters direct patterning and emergence of intestinal villi. Proc Natl Acad Sci U S A. 2012;109:15817–22. doi: 10.1073/pnas.1205669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–8. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510–3. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem. 2007;282:14048–55. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- 59.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20:3161–73. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabashima A, Higuchi H, Takaishi H, Matsuzaki Y, Suzuki S, Izumiya M, et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. International journal of cancer Journal international du cancer. 2009;124:2771–9. doi: 10.1002/ijc.24349. [DOI] [PubMed] [Google Scholar]
- 63.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cardenes HR, Chiorean EG, Dewitt J, Schmidt M, Loehrer P. Locally advanced pancreatic cancer: current therapeutic approach. The oncologist. 2006;11:612–23. doi: 10.1634/theoncologist.11-6-612. [DOI] [PubMed] [Google Scholar]
- 65.Meyers-Needham M, Lewis JA, Gencer S, Sentelle RD, Saddoughi SA, Clarke CJ, et al. Off-target function of the Sonic hedgehog inhibitor cyclopamine in mediating apoptosis via nitric oxide-dependent neutral sphingomyelinase 2/ceramide induction. Mol Cancer Ther. 2012;11:1092–102. doi: 10.1158/1535-7163.MCT-11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.