

Abstract
Age, drugs, and noise are major causes of acquired hearing loss. The involvement of reactive oxygen species (ROS) in hair cell death has long been discussed, but there is considerably less information available as to the mechanisms underlying ROS formation. Most cellular ROS arise in mitochondria and this review will evaluate evidence for mitochondrial pathology in general and dysfunction of the mitochondrial respiratory chain in particular in acquired hearing loss. We will discuss evidence that different pathways can lead to the generation of ROS and that oxidative stress might not necessarily be causal to all three pathologies. Finally, we will detail recent advances in exploiting knowledge of aminoglycoside-mitochondria interactions for the development of non-ototoxic antibacterials.
Keywords: acquired hearing loss, age, noise, aminoglycosides, reactive oxygen species, mitochondria, ribosomes
1. Introduction
1.1 Mitochondria and ROS in health and disease
Mitochondria are the givers of aerobic life and the mediators of cell death. This dual role is intrinsic in their basic and most important function, namely to reduce oxygen in the electron transport chain and provide energy in the form of ATP. However, this process also produces potentially cell-damaging reactive oxygen species (ROS). Probably through this lifelong exposure to ROS as well as through noxious environmental influences, mitochondrial DNA (mtDNA) is at risk to sustain mutations that can impede the function of these organelles (Lee and Wei, 2012).
Dysfunction of mitochondria is not only a phenomenon associated with the aging process but also contributes to the pathogenesis of many diseases, including neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and amyotrophic lateral sclerosis (Martin, 2012). The central role for mitochondria in cellular life and death makes them suspects in many more disorders, including hearing loss. The genetic aspects of “mitochondrial deafness” are well-documented (Fischel-Ghodsian, 2003; Kokotas et al., 2007; Berrettini et al., 2008) and will not be addressed here. Instead, this review concerns itself with the involvement of mitochondria in acquired hearing loss. First, however, we want to consider some basic issues of mitochondrial energy metabolism.
1.1 Mitochondrial energy metabolism and ROS formation
When we discuss mitochondrial energy metabolism and the role of ROS, we should not overlook that ROS are essential molecules in cellular physiology. They are normal products of biochemical pathways and serve as regulatory messengers in a multitude of processes, ranging from proliferation and survival to gene expression and apoptosis, as well as being signaling molecules for homeostatic adaptation under stress conditions (Ray et al., 2012; Finkel, 2012). Not only do ROS serve multiple physiological roles, they also arise from multiple sources. ROS can be generated as radicals or pro-radicals by, among others, NADPH oxidase, nitric oxide synthase, and mitochondrial, peroxisomal, or microsomal pathways.
The process of mitochondrial ROS formation itself is complex (Adam-Vizi, 2005; Dröse and Brandt, 2012), potentially involving several of the four complexes of the respiratory chain. In brief, the production of ATP is linked to the reduction of oxygen (O2) to water, a process that requires the coordinated addition of two electrons. The respiratory chain is highly efficient in this coordination but not completely so. During regular metabolism 1–4% of oxygen is estimated to be incompletely reduced in a one-electron transfer, yielding superoxide as the primary radical. In addition to the electron transport chain, monoamine oxidase in the outer membrane and the α-ketoglutarate dehydrogenase complex in the matrix are potential contributors to mitochondrial ROS.
While mitochondria can generate ROS, their intrinsic antioxidants also guard against damaging ROS escaping into the cell. Superoxide has a very short half-life and is dismutated to hydrogen peroxide (H2O2) either by the mitochondrial superoxide dismutase (Mn-SOD; SOD2) or its cytosolic counterpart, Cu-Zn-SOD (SOD1). Hydrogen peroxide is not a radical by itself but a pro-radical capable of yielding the highly reactive hydroxyl radical (·OH) in the presence of transition metals such as iron (Fe2+) in a non-enzymatic Fenton-type reaction. Normally, however, H2O2 is rendered harmless by reacting with glutathione or converted to water by catalase or glutathione peroxidase.
Under basic metabolic conditions the intrinsic mitochondrial and cytosolic antioxidant machinery can maintain redox homeostasis, the steady state between oxidative and reductive forces. The situation, however, may change if ROS are being produced in excess creating oxidative stress that might affect various organelles and pathways in the cell, ultimately leading to apoptosis or other forms of cell death. In addition, ROS may damage the mitochondria themselves, affecting the mitochondrial membrane potential and energy metabolism.
1.3 Oxidative metabolism in the cochlea
From the very early investigations into cochlear respiration we learned that the stria vascularis has by far the highest metabolic rate of all inner ear structures (Hughes and Chou, 1964; Marcus et al., 1978) commensurate with its task of establishing the endocochlear potential and the driving force for transduction. Hair cells have a rather low metabolic rate, estimated to be even lower than that of supporting cells based on histochemical studies of enzymes involved in energy metabolism (Nakai and Hilding, 1968). This fact has led to speculations of a high glycolytic (“anaerobic”) metabolism in outer hair cells, but quantitative determination of the contributions of glycolysis and oxidative phosphorylation have clearly established that their metabolism is primarily aerobic (Puschner and Schacht, 1997) and, hence, their fate subject to mitochondrial function or dysfunction as much as other cell types. In fact, outer hair cells may be even more susceptible to oxidative stress due to a lower content of antioxidants (Sha et al., 2001).
1.4 Mitochondria and oxidative stress in acquired hearing loss
Oxidative stress, the failure to maintain redox homeostasis permitting cell death pathways to proceed, has been associated with many forms of disease as well as the aging process (Lenaz et al., 2006). Oxidative stress also figures prominently in the pathology of acquired hearing loss and several recent review articles have addressed ROS-based and other mechanisms of toxicity in drug-induced, noise-induced, and age-related hearing loss (Warchol, 2010; Huth et al., 2011; Op de Beeck et al., 2011: Xie et al., 2011). A causal relationship linking ROS to hearing loss has been inferred for both drug-induced and noise-induced hearing loss as the co-administration of antioxidants effectively prevents or attenuates morphological and functional damage in animal models. As we will discuss later, the case for ROS as the basis for age-related hearing loss is more tenuous, although oxidative stress can be documented in the aging cochlea (Jiang et al., 2007).
While ROS are formed by several cellular reactions, most ROS production is due to mitochondrial metabolism. Therefore, this review will explore the evidence for mitochondrial involvement in acquired hearing loss and, more specifically, mitochondria as primary targets of ototoxic actions. Mitochondria also harbor the mediators of cell death pathways that have been implicated in acquired hearing loss, for example the release of cytochrome c and activation of Bcl-2 family proteins and caspases, leading to apoptosis. While such pathways are important in an overall consideration of mitochondrial involvement in disease or acquired hearing loss, this review will bypass these downstream consequences of mitochondrial injury in favor of a close look into presumed primary events.
2. Noise-induced hearing loss
ROS emerge following noise exposure in cochlear tissues (Yamane et al., 1995; Ohinata et al., 2000) and spill into inner ear fluids (Ohlemiller et al., 1999), suggesting that they can leave the site of their original production to impact surrounding structures. Although their formation is almost immediate, there is also delayed ROS production that persists for extended periods of time (days) after cessation of noise exposure, spreading from the base of the cochlea to the apex (Yamashita et al., 2004) and widening the area of morphological damage. Antioxidants, when given prior to or shortly after noise exposure, can attenuate both hair cell death and threshold shifts (see Oishi and Schacht, 2011), a fact that suggests a causal relationship between oxidant stress and noise-induced hearing loss.
2.1 Mitochondria and noise-induced hearing loss
Mitochondria have long been speculated as being involved in noise trauma and the source of ROS. Spoendlin (1971) observed mitochondrial damage in outer hair cells of guinea pigs following exposure to noise, but such gross pathology may depend very well on the conditions of the trauma. If severe enough, noise exposure may lead to major intracellular derangements as well as mechanical destruction of cochlear structures; metabolic effects, although possibly leading to cell death, will be more subtle and not necessarily morphologically obvious. Metabolic effects signifying mitochondrial involvement in the initial events after noise exposure might include, for example, a rapid depletion of ATP levels (Chen et al., 2012). Genetic evidence also points to a link between mitochondria, oxidative stress, and noise. Factory workers exposed to occupational noise were at higher risk for hearing loss when they carried a specific single nucleotide polymorphism in the mitochondrial Mn-superoxide dismutase gene (Liu et al., 2010) compromising their ability to effectively inactivate mitochondria-generated superoxide.
It has been argued (Henderson et al., 2006) that the exposure to noise induces a higher metabolic rate in the affected cochlear cells, “overstimulates” mitochondrial energy metabolism, and thereby increases the rate of leakage of ROS. However, it appears unlikely that an enhanced metabolic rate per se should lead to a surge in ROS sufficient to overwhelm the cells’ considerable antioxidant defenses. An enhanced metabolism might even lower ROS formation by making better use of energy-supplying substrates. In fact, it is not stimulation but selective inhibition of the electron transport chain (in particular, of complex I) that produces the largest excess of ROS under stress situations (Sipos et al., 2003). Therefore, we need to look for alternative explanations of the elevation of ROS in noise trauma.
2.2 A mechanism for noise-induced ROS generation
The most likely trigger for elevated ROS is calcium. Free Ca2+ increases in outer hair cells immediately after acoustic overstimulation (Fridberger et al., 1998), probably both via entry through ion channels and liberation from intracellular stores. In agreement with calcium as a decisive factor in noise trauma is the success of attenuating noise-induced hearing loss through a blockade of L-type (Heinrich et al., 1999) or T-type voltage-gated calcium channels (Shen et al., 2007). Another suggestive link between calcium and noise trauma is the observation that sound conditioning prior to noise exposure reduces both the anticipated threshold shift and the calcium load of outer hair cells (Zuo et al., 2005). If we add to this scenario the well-established fact that elevated calcium significantly stimulates mitochondrial ROS production, leading to the degeneration of neurons or other afflicted cells (Peng and Jou, 2010), we can construct a reasonable scenario from noise-induced calcium elevation to mitochondrial ROS formation to cell death.
This is an appealing hypothesis, but calcium does not directly affect the respiratory chain. Other mechanisms must come into play in order to increase mitochondrial ROS production and these might include calcium-induced lipid peroxidation, protein kinase activation, or changes in mitochondrial membrane permeability. However, there is another potential source of ROS production that we had briefly mentioned earlier and that seems a likely target of calcium: α-ketoglutarate dehydrogenase, a calcium-regulated enzyme of the Krebs cycle able to form significant amounts of superoxide and H2O2 (Tretter and Adam-Vizi, 2004). Dysregulation of this enzyme would not only increase oxidative stress but have additional consequences for mitochondrial function: ROS generation by α-ketoglutarate dehydrogenase might lead to free-radical damage within mitochondria and inhibition of the respiratory chain, resulting in a further elevation of ROS and the initiation of apoptotic pathways.
An even stronger case for a crucial role of mitochondria and ROS in noise trauma can be made by invoking a secondary set of reactions. Noise-induced (and calcium-mediated) mitochondrial ROS produce vasoactive lipid peroxidation products such as isoprostanes (Ohinata et al., 2000) which may contribute to the decrease in cochlear blood flow observed after noise (Thorne and Nuttall, 1987). The resulting ischemia and subsequent reperfusion would add to increased mitochondrial ROS formation and oxidative stress in a self-perpetuating (or at least, self-enhancing) cycle.
One caveat applies. Calcium is a multifaceted ion in cellular homeostasis and pathology and its overload can trigger apoptotic and necrotic cell death pathways in addition to or independent of ROS formation. For example, calcineurin, a calcium/calmodulin-dependent protein phosphatase, is activated after noise exposure (Minami et al., 2004) and can, in turn, activate mitochondria-mediated cell death pathways via the Bcl-2-associated death promoter (BAD) in outer hair cells (Vicente-Torres and Schacht, 2006).
3. Mitochondria and age-related hearing impairment
Due to the central role of mitochondria for cellular homeostasis, it seems self-evident that their proper function is vital to maintain auditory function, and many pathways indeed cooperate in assuring mitochondrial integrity. This homeostatic capacity appears limited, however, and studies from essentially all tissues and organs demonstrate that certain pathological changes in mitochondria are intimately linked to the aging process. Thirteen of the mitochondrial proteins are encoded by mtDNA, rendering them particularly vulnerable to mutations. The reason for this vulnerability is that, in contrast to the situation for nuclear DNA, mitochondria possess a less efficient machinery for DNA damage repair, and spontaneous or induced mutations may remain uncorrected. As mutations accumulate, the integrity of mitochondrial protein synthesis and hence mitochondrial function is compromised, and phenotypic pathology becomes evident, varying with cell type and the type of mutation (Lenaz et al., 2006; Cline, 2012; Lee and Wei, 2012). Given this fact, we should expect plenty of information on mitochondrial changes in presbycusis. This is indeed the case, and herein lies a challenge: which of the observed changes are related to the aging process in general and which are related to and determine the progression and severity of presbycusis?
3.1 Animal models of age-related hearing impairment
Human presbycusis is difficult to characterize because of its mix of genetic and environmental influences and its complexity of structural changes (Schuknecht and Gacek, 1993). Different animal models have been proposed and each of them may reflect different aspects of presbycusis (Spicer and Schulte, 2002; Ohlemiller, 2006), but some models may be so confounded that their value is debatable. C57 mouse strains, for example, carry a mutation in cadherin 23 that predisposes to accelerated hearing loss. In humans, Cdh23 mutations are present in type I Usher syndrome, but a relation of this mutation to presbycusis is tenuous and perhaps non-existent: a recent population study found no association between Cdh23 mutations and presbycusis (Hwang et al., 2012). Mouse mutants deficient in mitochondrial proofreading (Kujoth et al., 2005) present features of accelerated aging, including accelerated hearing loss. These animals accumulate excessive mtDNA mutations and show dramatic degenerative changes in phenotype and disastrous consequences for the animals’ health that are not seen in late-life wild-type mice. Therefore the model has been criticized for introducing catastrophic mutations that do not represent a normal aging process (Miller, 2005; Gershon, 2005). Finally, inbred animal strains in general may not be good models for the heterogeneity of the human population. Outbred—and hence genetically heterogeneous—strains might provide results that more likely can be reproduced in other populations of animals and perhaps lend themselves more easily to translational applications. The National Institute on Aging recognized this problem and provided guidance when it adopted a four-way cross mouse population as its standard stock for aging research (Miller et al., 1999).
Studies on age-related hearing loss and mitochondrial involvement mostly take one of two approaches, comparisons of the young and the old cochlea or comparisons of good and bad hearing cohorts at the same age. Comparing young and old cochleae gives information on age-related changes in metabolism (Jiang et al., 2007), but such changes constitute a combination of general age-related pathology and pathology potentially specific to presbycusis. The fact that the aging process and presbycusis can be dissociated is seen in the human population where subjects in the same (old) age group may maintain excellent hearing (“golden ears”) while others do not. The direct comparison of good and bad ears in the same age-group therefore seems essential and such results are available from some animal studies (e.g., Schacht et al., 2012) and from analyses of human temporal bones. Interestingly, the latter are probably yet the best source of information into the role of “mitochondrial deletions”.
3.2 Mitochondrial deletions and age-related hearing impairment
The spectrum of age-related mitochondrial DNA mutations is similar to that seen in nuclear DNA, comprising point mutations, insertions, and deletions. As might be expected, some mtDNA deletions found in human temporal bone material were generally age-related and shared between individuals with good hearing and hearing loss (Markaryan et al., 2008). In contrast, a specific “common deletion” (mtDNA4977) was apparent in temporal bone material from 14 out of 17 patients with presbycusis while its presence was somewhat lower (eight of 17) in individuals with normal audiograms (Bai et al., 1997). This observation was confirmed and expanded by subsequent studies evaluating temporal bone materials from presbycusic individuals against age-matched controls and young and middle-aged groups (Dai et al., 2004). Quantification of archival human mtDNA by real-time polymerase chain reaction and comparison to individual audiometric thresholds established a significant correlation between levels of the mtDNA4977 deletion in human cochlear tissue and the severity of presbycusis (Markaryan et al., 2009). Mitochondrial dysfunction as one cause of presbycusis is, therefore, likely and in agreement with observations that mtDNA mutations resulting in mitochondrial dysfunction and apoptosis are frequently associated with age-related disorders (Reeve et al., 2008). On the other hand, such a conclusion must be drawn with caution because mitochondrial variants per se do not determine the rate of age-related deterioration of hearing. An analysis of the entire mitochondrial genome of 400 individuals (200 with good hearing, 200 with bad hearing) found no association between the mutation load of inherited mitochondrial variants and age-related hearing impairment (Bonneux et al., 2011).
3.3 Oxidative stress in age-related hearing impairment
Oxidative stress is frequently associated with mitochondrial mutations and dysfunction (Lee and Wei, 2012) and has been documented in aging cochlear tissue (Jiang et al., 2007). The speculation that mitochondrial deletions give rise to ROS which, in turn, lead to tissue damage and hearing loss therefore seems well founded (Seidman et al., 2004). However, the issue is not resolved. Strategies to ameliorate oxidative stress by antioxidant supplementation have been convincingly successful in drug- and noise-induced hearing loss but have met with variable success in age-related hearing loss.
Rats on a life-long diet supplemented with antioxidants maintained better auditory sensitivity than their cohort on a placebo (Seidman, 2000). However, different feeding regimens with other vitamins, energy metabolites, or antioxidants have yielded inconsistent results in various animal models and recent studies have outright questioned whether oxidative stress is causal to presbycusis. Dosing aging rats with L-carnitine, an enhancer of mitochondrial bioenergetics, was unable to alter the course of presbycusis (Bielefeld et al., 2008). Likewise, a long-term intervention from 10 to 24 months of age in CBA/J mice with an antioxidant cocktail (α-lipoic acid and vitamins A, C, and E) and L-carnitine failed to attenuate or delay age-related hearing loss (Sha et al., 2012). Another independent line of aging research is adding to the uncertainty to what extent mtDNA deletions and oxidative stress contribute to presbycusis. Calorically restricted diets can extend longevity and slow age-related physiological declines in a variety of species (Bordone and Guarente, 2005). A prominent effect of such a regimen is the reduction of mitochondrial ROS production and oxidative damage, preventing the formation of mtDNA mutations (Lee and Wei, 2012). However, in terms of age-related hearing loss, the results are mixed, ranging from attenuation to aggravation of auditory damage in different animal models (Henry, 1986; Park et al., 1990; Sweet et al., 1988; Willott et al. (1995).
Combining evidence from human and animal studies, we may argue that mtDNA deletions might relate to presbycusis but that the metabolic effects must be more complex than simply generating excess ROS. Mutations in mtDNA have multiple consequences on mitochondrial gene expression (Cline, 2012) and might lead to ROS-independent dysregulation of oxidative phosphorylation and energy production or mitochondria-mediated pathways of apoptosis. Notwithstanding the case for a mitochondrial involvement, we should keep in mind that the aging process is multifaceted and that non-mitochondrial cellular pathways might also contribute to age-related hearing impairment (Kidd and Bao, 2012).
4. Aminoglycoside ototoxicity
4.1 Mitochondria and ototoxicity
A mitochondrial “involvement” in aminoglycoside toxicity is well established through ample documentation of intrinsic pathways of cell death (Huth et al., 2011; Op de Beeck et al., 2011). Whether mitochondria are a direct and possibly even a first target for aminoglycosides is another question.
In morphological descriptions of ototoxicity, overt mitochondrial damage does not seem to play a major role. Gross mitochondrial pathology was seen, to our knowledge, in only one study dosing lizards with “consecutive large doses” of gentamicin (Bagger-Sjöbäck and Wersäll, 1978). In contrast, an elaborate day-by-day analysis of kanamycin ototoxicity in the guinea pig (8 days of injections and 14 days of follow-up; Darrouzet and Guilhaume, 1974) reports prominent intracellular changes in the Golgi apparatus and lysosomes but unambiguously concludes “Jusqu’au stade de l’eclatement, les mitochondries ne sont pas modifies” [Until the demise [of the hair cells], the mitochondria are not altered]. It is therefore reasonable to assume that any effects on mitochondria in chronic ototoxicity would be metabolic and morphologically subtle.
4.2 Mitochondrial energy metabolism in aminoglycoside ototoxicity
There appears to be general acceptance that ROS are involved in aminoglycoside ototoxicity, and the mitigating influence of antioxidants on ototoxicity is perhaps the strongest argument for a causal relationship (Rybak and Ramkumar, 2007; Poirrier et al., 2010; Xie et al., 2011). However, antioxidants do not distinguish between the origin of ROS and frequently nor do they between different species of ROS, leaving the question of the mechanism of ROS formation open. Among the options are non-enzymatic formation by an aminoglycoside-iron complex (Priuska and Schacht, 1995) and enzymatic reactions catalyzed by nitric oxide synthase (Hong et al., 2006) or NADPH oxidase (Jiang et al., 2006). In addition, attenuation of hair cell damage by stabilizing mitochondrial homeostasis with L-carnitine (Kalinec et al., 2005) points to a mitochondrial contribution.
Two recent studies report effects of aminoglycosides on mitochondrial metabolism (Jensen-Smith et al., 2012; Majumder et al., 2012). Both studies employ an acute exposure of early postnatal murine cochlear explants to relatively high drug concentrations (0.3 mM gentamicin and 1.0 mM neomycin) so that some caveats might apply to the applicability of the results to an in-vivo situation. Jensen-Smith et al. (2012) implicate compromised energy metabolism from the measurement of a gentamicin-induced decrease of the formation of NADH, interpreted as an overall indication of mitochondrial dysfunction. Given that mitochondrial dysfunction ought to have the capacity to elevate ROS, some level of gentamicin mediated mitochondrial ROS formation can be inferred. The effect is most pronounced in outer hair cells at the base of the cochlea, the region most sensitive to aminoglycosides—a suggestive link between this observation and hair cell death. Majumder et al. (2012) take a more direct approach to the question of oxidative stress and image the fluorescence of dihydroethidium, a marker of ROS formation, as well as of intracellular free calcium. Their pharmacological manipulations suggest sources of ROS that depend on mitochondrial respiratory complex I but also sources that appear independent.
These studies make a compelling case for a mitochondrial dysfunction induced by aminoglycosides but leave us with the question as to where exactly the drugs attack. A disturbance of energy metabolism could result from a direct action on proteins regulating the mitochondrial membrane potential or enzymes of the respiratory complexes; or it could be indirect via, for example, an aberrant synthesis of mitochondrial proteins. While acute experiments tend to direct speculation towards the first type of action, the slow development of ROS during chronic injections of drugs in vivo (Jiang et al., 2005) argues for a mechanism of the latter type. In fact, there is evidence for such a more complex sequence of events.
4.3 The mitochondrial ribosome in aminoglycoside ototoxicity
Common to most aminoglycosides is the neamine core composed of a 2-deoxystreptamine linked to a glucopyranosyl moiety. Additional sugars are attached to position 5 or 6 of the 2-deoxystreptamine to give rise to disubstituted 4,5- or 4,6-aminoglycosides (fig. 1). In their antibacterial action, aminoglycosides bind to ribosomal RNA and affect protein synthesis by inducing codon misreading and inhibiting tRNA translocation (Wilson, 2009). The binding site is located within a conserved loop of 16S rRNA helix 44 including the small subunit’s aminoacyl-tRNA acceptor site (A site), the decoding site of the ribosome. The molecular details of aminoglycoside-rRNA complexes have been determined by X-ray crystallography at atomic resolution (Carter et al., 2000; Francois et al., 2005).
Figure 1. Structures of drugs and complexes with ribosomal RNA.
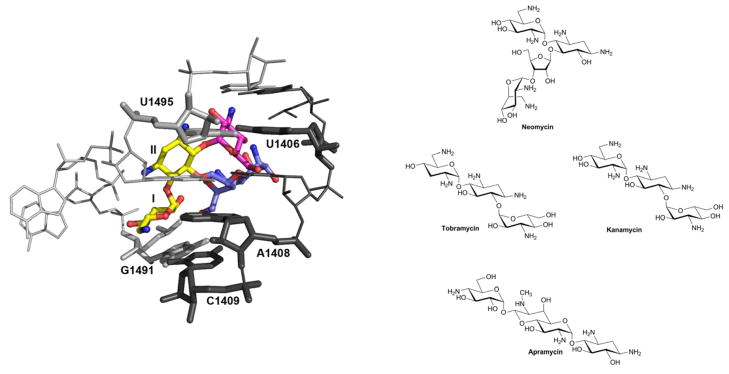
Left: The three-dimensional structure of the ribosomal A site in complex with 4,5- and 4,6-disubstituted 2-deoxystreptamines. The common neamine core is denoted in yellow; ring III of the 4,6-aminoglycosides (tobramycin) in red; rings III and IV of the 4,5-aminoglycosides (paromomycin) in blue.
Right: Neomycin is a 4,5-disubstituted 2-deoxystreptamine, tobramycin and kanamycin are 4,6-disubstituted 2-deoxystreptamines, and apramycin is a 4-monosubstituted 2-deoxystreptamine.
Early speculation of mitochondrial ribosomes as targets of aminoglycosides pointed to the structural relationship of mitochondrial to bacterial ribosomes, since mitochondrial ribosomes (mitoribosomes) are remnants of previous symbiotic bacteria. Speculation was further fuelled by familial cases of aminoglycoside-induced deafness which are maternally transmitted and linked to mutations in mtDNA. Transition mutations in the mitochondrial small ribosomal RNA gene, particularly A1555G and, less frequently, C1494T are primary genetic traits in a predisposition towards aminoglycoside-induced deafness. A1555G and C1494T both map to the decoding A site, and would replace a non-canonical RNA base-pair with a perfect base-pairing. By themselves, the A1555G and C1494T alterations produce a clinical phenotype that may range from normal hearing through moderate progressive hearing loss of later onset to severe congenital deafness (Fischel-Ghodsian, 2003). In addition to the 1555 A→G transition, a plethora of other mitochondrial RNA mutations have been suggested to influence aminoglycoside ototoxicity (Guan, 2004; Lu et al., 2010). However, the roles of some of these variants in aminoglycoside-induced hearing loss remain unclear because of potentially confounding factors in analysis or interpretation of the data (Yao et al., 2006).
The mechanistic link between mitochondrial rRNA mutation, mitoribosomal function, deafness, and aminoglycoside ototoxicity has remained largely elusive because of the absence of suitable experimental models. The presence of hundreds to thousands of mitochondria in a eukaryotic cell, each organelle carrying multiple copies of its genome, renders genetic manipulations of mitochondrial rRNA in higher eukaryotes impossible. To establish an experimental and genetically tractable model for the study of human A site function, we replaced A-site residues in bacterial 16S rRNA helix 44 with that of various eukaryotic homologs (Hobbie et al., 2007; Hobbie et al. 2008a). Replacement of a 34-nucleotide part of bacterial 16S rRNA helix 44 with human homologs resulted in rRNA decoding A sites corresponding to those in cytosolic and mitochondrial (wild-type and mutant) human ribosomes, essentially constructing a bacterial ribosomal backbone with functional eukaryotic decoding sites. These hybrid ribosomes provided an opportunity to study aminoglycoside susceptibility of the eukaryotic cytoplasmic, mitochondrial, and mitochondrial C1494U and A1555G mutant ribosomes (Hobbie et al., 2008b). In contrast to the cytohybrid ribosomes, the mitohybrid ribosomes show remarkable aminoglycoside susceptibility (see fig. 2A) with malfunction of protein synthesis in-vitro correlating with the ototoxicity of the drugs (fig. 2B). In addition, mutations A1555G and C1494U in mitohybrid ribosomes conferred increased levels of spontaneous misreading and hypersusceptibility to aminoglycoside action (fig. 2C). Mitochondrial dysfunction may thus represent a unifying mechanism, integrating both sporadic dose-dependent and genetically inherited susceptibility to aminoglycoside ototoxicity.
Figure 2. Drug effects on protein synthesis and recombinant microorganisms.

A. Susceptibility to aminoglycosides of the mitochondrial and the cytosolic decoding site as assessed by MIC assays (minimal inhibitory concentrations) on recombinant microorganisms with hybrid ribosomes.
B. Relationship between inhibition of protein synthesis in mitochondrial hybrid ribosomes and relative cochleotoxicity of aminoglycoside antibiotics. The potencies of a series of cochleotoxic aminoglycosides (Ak, amikacin; Gm, gentamicin; Ne, neamine; Nm, neomycin) in inhibiting protein synthesis in hybrid mitochondrial ribosomes (see Table 1) correlates with their previously reported relative cochleotoxicity (Kotecha and Richardson, 1994).
C. Susceptibility to aminoglycosides of mutant and wild-type mitochondrial hybrid ribosomes. Dose-response curves with wild-type mitochondrial (red squares), mutant A1555G (green triangles), and mutant C1494T (blue inverted triangles) hybrid ribosomes; bacterial ribosomes (black circles) are included for comparison. Left: Gentamicin-induced increase in mis-incorporation of the near-cognate [3H]-leucine using polyU as template relative to the drug-free control (mean ± SD; n ≥ 3). Right: Gentamicin-induced inhibition of luciferase synthesis relative to the drug-free control (mean ± SD; n ≥ 3). Modified from Proc. Natl. Acad. Sci. USA 2008, 105: 20888–20893 with permission from the publisher.
The hypothesis that aminoglycoside ototoxicity is related to the compounds’ anti-ribosomal activity can in principle be tested, provided that it is possible to separate the antibacterial activity of aminoglycosides from the compounds’ activity toward the eukaryotic ribosome. We recently observed (Matt et al., 2012) that apramycin, a unique 4-monosubstituted 2-deoxystreptamine (see fig. 1) shows a dissociation of its anti-bacterioribosomal and anti-mitoribosomal activities, as assessed by the interaction with hybrid ribosomes (table 1; Matt et al., 2012). The commonly used disubstituted 4,5- and 4,6-aminoglycosides significantly affect the mitoribosome and this effect is aggravated by the A1555G mutation. In contrast, apramycin affects all three eukaryotic drug binding pockets in an equally poor fashion and does not induce misreading on mitohybrid or mitohybrid-A1555G ribosomes. Studies in murine organ of Corti explants and in the guinea pig in vivo testified to a much lower hair cell toxicity of apramycin compared to gentamicin. Furthermore, in accordance with its lesser toxicity, apramycin elicited only little ROS formation. From these data, an interplay of mitochondrial dysfunction and ROS formation resulting in cell death becomes a logical postulate for a mechanism of ototoxicity. In addition, the dissociation of antibacterial and anti-mitoribosomal activity resulting in greatly alleviated ototoxicity provides a long-sought rationale for hypothesis-driven modifications of aminoglycosides to improve further this important class of antibacterials.
Table 1.
Aminoglycoside interaction with drug binding pockets
| Aminoglycoside | Bacterial wild-type and hybrid ribosome A site rRNAa
|
|||
|---|---|---|---|---|
| Bacterial | Mitochondrial hybrid | Mitochondrial A1555G hybrid | Cytosolic hybrid | |
| Apramycin | 0.08 ± 0.02 | 115.6 ± 27.8 | 47.5 ± 10.7 | 89.4 ± 21.2 |
| Gentamicin | 0.02 ± 0.01 | 11.1 ± 1.6 | 0.69 ± 0.12 | 40.4 ± 10.4 |
| Tobramycin | 0.03 ± 0.01 | 32.6 ± 2.1 | 1.07 ± 0.19 | 58.5 ± 11.8 |
| Kanamycin | 0.04 ± 0.02 | 31.1 ± 14.3 | 1.12 ± 0.16 | 91.2 ± 12.3 |
| Neomycin | 0.02 ± 0.01 | 2.64 ± 0.58 | 0.35 ± 0.08 | 37.9 ± 11.2 |
Aminoglycoside-induced inhibition of protein synthesis (IC50 μM). IC50 values represent the drug concentrations in μM required to inhibit in-vitro synthesis of functional firefly luciferase to 50% (mean ± SD; n = 3).
5. Conclusion
The sum of the evidence strongly implies mitochondria as primary targets in cochlear dysfunction induced by noise, ototoxic drugs, and the aging process. This, however, is also where the commonality seems to end. While we still need to explore more details, the three otopathologies cause mitochondrial failure apparently by different mechanisms, including aberrant calcium-regulation, mitochondrial DNA deletions, and the targeting of mitochondrial ribosomes. Commensurate with the multifaceted roles of mitochondria in cell physiology and pathology, the three stimuli invoke different sequels of this failure, ROS formation being only one of the options. Consequently, a “silver bullet” to ameliorate acquired hearing loss may not exist, and each of these pathologies might require its own consideration in terms of mechanisms and prevention.
Highlights.
Mitochondria play a major role in cochlear function and dysfunction.
Noise may affect mitochondrial metabolism by calcium influx.
Presbycusis correlates with distinct mitochondrial lesions but not necessarily with oxidative stress.
Aminoglycosides inhibit the respiratory chain via targeting mitochondrial function.
Non-ototoxic aminoglycosides can be developed by eliminating their activity against the mitochondrial ribosome.
Acknowledgments
Dr. Schacht’s research is supported by research grant R01 DC-03685 and core center grant P30 DC05188 from the National Institute on Deafness and Other Communication Disorders, National Institutes of Health.
Abbreviations
- ROS
reactive oxygen species
- mtDNA
mitochondrial DNA
- SOD
superoxide dismutase
- rRNA
ribosomal RNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antiox Redox Signal. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- Bagger-Sjöbäck D, Wersäll J. Gentamicin-induced mitochondrial damage in inner ear sensory cells of the lizard Calotes versicolor. Acta Otolaryngol. 1978;86:35–51. doi: 10.3109/00016487809124718. [DOI] [PubMed] [Google Scholar]
- Bai U, Seidman MD, Hinojosa R, Quirk WS. Mitochondrial DNA deletions associated with aging and possibly presbycusis: a human archival temporal bone study. Am J Otolaryngol. 1997;18:449–453. [PubMed] [Google Scholar]
- Berrettini S, Forli F, Passetti S, Rocchi A, Pollina L, Cecchetti D, Mancuso M, Siciliano G. Mitochondrial non-syndromic sensorineural hearing loss: a clinical, audiological and pathological study from Italy, and revision of the literature. Biosci Rep. 2008;28:49–59. doi: 10.1042/BSR20070027. [DOI] [PubMed] [Google Scholar]
- Bielefeld EC, Coling D, Chen GD, Henderson D. Multiple dosing strategies with acetyl L-carnitine (ALCAR) fail to alter age-related hearing loss in the Fischer 344/NHsd rat. J Negat Results Biomed. 2008;7:4. doi: 10.1186/1477-5751-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneux S, Fransen E, Van Eyken E, Van Laer L, Huyghe J, Van de Heyning P, Voets A, Gerards M, Stassen AP, Hendrickx AT, Smeets HJ, Van Camp G. Inherited mitochondrial variants are not a major cause of age-related hearing impairment in the European population. Mitochondrion. 2011;11:729–734. doi: 10.1016/j.mito.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- Chen FQ, Zheng HW, Hill K, Sha SH. Traumatic Noise Activates Rho-Family GTPases through Transient Cellular Energy Depletion. J Neurosci. 2012;32:12421–12430. doi: 10.1523/JNEUROSCI.6381-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline SD. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim Biophys Acta. 2012;1819:979–991. doi: 10.1016/j.bbagrm.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Yang W, Jiang S, Gu R, Yuan H, Han D, Guo W, Cao J. Correlation of cochlear blood supply with mitochondrial DNA common deletion in presbyacusis. Acta Otolaryngol. 2004;124:130–136. doi: 10.1080/00016480410016586. [DOI] [PubMed] [Google Scholar]
- Darrouzet J, Guilhaume A. Ototoxicité de la kanamycine au jour le jour. Étude expérimentale en microscopie électronique. Rev Laryng (Bordeaux) 1974;95:601–621. [PubMed] [Google Scholar]
- Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischel-Ghodsian N. Mitochondrial deafness. Ear Hear. 2003;24:303–313. doi: 10.1097/01.AUD.0000079802.82344.B5. [DOI] [PubMed] [Google Scholar]
- Francois B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci U S A. 1998;95:7127–7132. doi: 10.1073/pnas.95.12.7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MX. Molecular pathogenetic mechanism of maternally inherited deafness. Ann N Y Acad Sci. 2004;1011:259–271. doi: 10.1007/978-3-662-41088-2_25. [DOI] [PubMed] [Google Scholar]
- Gershon D. Evaluating evidence for aging. Science. 2005;310:441–443. [PubMed] [Google Scholar]
- Heinrich UR, Maurer J, Mann W. Ultrastructural evidence for protection of the outer hair cells of the inner ear during intense noise exposure by application of the organic calcium channel blocker diltiazem. ORL J Otorhinolaryngol Relat Spec. 1999;61:321–327. doi: 10.1159/000027693. [DOI] [PubMed] [Google Scholar]
- Henderson D, Bielefeld EC, Harris KC, Hu BH. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006;27:1–19. doi: 10.1097/01.aud.0000191942.36672.f3. [DOI] [PubMed] [Google Scholar]
- Henry KR. Effects of dietary restriction on presbyacusis in the mouse. Audiology. 1986;25:329–337. doi: 10.3109/00206098609078397. [DOI] [PubMed] [Google Scholar]
- Hobbie SN, Kalapala SK, Akshay S, Bruell CM, Dabow S, Vasella A, Sander P, Böttger EC. Engineering the rRNA decoding site of eukaryotic cytosolic ribosomes in bacteria. Nucleic Acids Res. 2007;35:6086–6093. doi: 10.1093/nar/gkm658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie SN, Bruell CM, Akshay S, Kalapala SK, Shcherbakov D, Böttger EC. Mitochondrial deafness alleles confer misreading of the genetic code. Proc Natl Acad Sci U S A. 2008a;105:3244–3249. doi: 10.1073/pnas.0707265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Böttger EC. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A. 2008b;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Park SK, Cho YS, Lee HS, Kim KR, Kim MG, Chung WH. Gentamicin induced nitric oxide-related oxidative damages on vestibular afferents in the guinea pig. Hear Res. 2006;211:46–53. doi: 10.1016/j.heares.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hughes DE, Chou JT-Y. Respirometrie. In: Rauch S, editor. Biochemie des Hörorgans. Georg Thieme; Stuttgart: 1964. pp. 446–457. [Google Scholar]
- Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of prevention. Int J Otolaryngol. 2011:937861. doi: 10.1155/2011/937861. Epub 2011 Oct 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JH, Liu KS, Wu CC, Liu TC. Association of cadherin23 single nucleotide polymorphism with age-related hearing impairment in han chinese. Otolaryngology Head Neck Surg. 2012;147:531–534. doi: 10.1177/0194599812446904. [DOI] [PubMed] [Google Scholar]
- Jensen-Smith HC, Hallworth R, Nichols MG. Gentamicin rapidly inhibits mitochondrial metabolism in high-frequency cochlear outer hair cells. PLoS One. 2012;7(6):e38471. doi: 10.1371/journal.pone.0038471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. The NF-κB pathway protects cochlear hair cells from aminoglycoside-induced ototoxicity. J Neurosci Res. 2005;79:644–651. doi: 10.1002/jnr.20392. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. Rac/Rho pathway regulates actin depolymerization induced by aminoglycoside antibiotics. J Neurosci Res. 2006;83:1544–1551. doi: 10.1002/jnr.20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol Aging. 2007;28:1605–1612. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinec GM, Fernandez-Zapico ME, Urrutia R, Esteban-Cruciani N, Chen S, Kalinec F. Pivotal role of Harakiri in the induction and prevention of gentamicin-induced hearing loss. Proc Natl Acad Sci U S A. 2005;102:16019–21604. doi: 10.1073/pnas.0508053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd AR, Iii, Bao J. Recent Advances in the Study of Age-Related Hearing Loss: A Mini-Review. Gerontology. 2012 Jun 15; doi: 10.1159/000338588. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clin Genet. 2007;71:379–391. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- Kotecha B, Richardson GP. Ototoxicity in vitro: effects of neomycin, gentamicin, dihydrostreptomycin, amikacin, spectinomycin, neamine, spermine and poly-L-lysine. Hear Res. 1994;73:173–184. doi: 10.1016/0378-5955(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–327. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Baracca A, Fato R, Genova ML, Solaini G. New insights into structure and function of mitochondria and their role in aging and disease. Antioxid Redox Signal. 2006;8:417–437. doi: 10.1089/ars.2006.8.417. [DOI] [PubMed] [Google Scholar]
- Liu YM, Li XD, Guo X, Liu B, Lin AH, Ding YL, Rao SQ. SOD2 V16A SNP in the mitochondrial targeting sequence is associated with noise induced hearing loss in Chinese workers. Dis Markers. 2010;28:137–147. doi: 10.3233/DMA-2010-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Li Z, Zhu Y, Yang A, Li R, Zheng J, Cai Q, Peng G, Zheng W, Tang X, Chen B, Chen J, Liao Z, Yang L, Li Y, You J, Ding Y, Yu H, Wang J, Sun D, Zhao J, Xue L, Wang J, Guan MX. Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion. 2010;10(4):380–390. doi: 10.1016/j.mito.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder P, Duchen M, Gale J. Investigating free radical production in the cochlea during ototoxicity. Abstr Inner Ear Biol. 2012;49:169. [Google Scholar]
- Marcus DC, Thalmann R, Marcus NY. Respiratory rate and ATP content of stria vascularis of guinea pig in vitro. Laryngoscope. 1978;88:1825–1835. doi: 10.1288/00005537-197811000-00011. [DOI] [PubMed] [Google Scholar]
- Markaryan A, Nelson EG, Hinojosa R. Detection of mitochondrial DNA deletions in the cochlea and its structural elements from archival human temporal bone tissue. Mutat Res. 2008;640:38–45. doi: 10.1016/j.mrfmmm.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Markaryan A, Nelson EG, Hinojosa R. Quantification of the mitochondrial DNA common deletion in presbycusis. Laryngoscope. 2009;119:1184–1189. doi: 10.1002/lary.20218. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci. 2012;107:355–415. doi: 10.1016/B978-0-12-385883-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Böttger EC. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci U S A. 2012;109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA. Evaluating evidence for aging. Science. 2005;310:441–443. doi: 10.1126/science.310.5747.441. [DOI] [PubMed] [Google Scholar]
- Miller RA, Burke D, Nadon N. Announcement: four-way cross mouse stocks: a new, genetically heterogeneous resource for aging research. J Gerontol A Biol Sci Med Sci. 1999;54A:B358. doi: 10.1093/gerona/54.8.b358. [DOI] [PubMed] [Google Scholar]
- Minami SB, Yamashita D, Schacht J, Miller JM. Calcineurin activation contributes to noise-induced hearing loss. J Neurosci Res. 2004;78:383–392. doi: 10.1002/jnr.20267. [DOI] [PubMed] [Google Scholar]
- Nakai Y, Hilding D. Oxidative enzymes in the cochlea. Acta Oto-laryngol. 1968;65:459–467. doi: 10.3109/00016486809120988. [DOI] [PubMed] [Google Scholar]
- Ohinata Y, Miller JM, Altschuler RA, Schacht J. Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res. 2000;878:163–173. doi: 10.1016/s0006-8993(00)02733-5. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK. Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res. 2006;1091:89–102. doi: 10.1016/j.brainres.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol Neurootol. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- Oishi N, Schacht J. Emerging treatments for noise-induced hearing loss. Expert Opin Emerg Drugs. 2011;16:235–245. doi: 10.1517/14728214.2011.552427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op de Beeck K, Schacht J, Van Camp G. Apoptosis in acquired and genetic hearing impairment: The programmed death of the hair cell. Hear Res. 2011;281:18–27. doi: 10.1016/j.heares.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, Cook KC, Verde EA. Dietary restriction slows the abnormally rapid loss of spiral ganglion neurons in C57BL/6 mice. Hear Res. 1990;48:275–279. doi: 10.1016/0378-5955(90)90067-y. [DOI] [PubMed] [Google Scholar]
- Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Poirrier AL, Pincemail J, Van Den Ackerveken P, Lefebvre PP, Malgrange B. Oxidative stress in the cochlea: an update. Curr Med Chem. 2010;17:3591–3604. doi: 10.2174/092986710792927895. [DOI] [PubMed] [Google Scholar]
- Priuska E, Schacht J. Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem Pharmacol. 1995;50:1749–1752. doi: 10.1016/0006-2952(95)02160-4. [DOI] [PubMed] [Google Scholar]
- Puschner B, Schacht J. Energy metabolism in cochlear outer hair cells in vitro. Hear Res. 1997;114:102–106. doi: 10.1016/s0378-5955(97)00163-9. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann N Y Acad Sci. 2008;1147:21–29. doi: 10.1196/annals.1427.016. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Ramkumar V. Ototoxicity. Kidney Int. 2007;72:931–935. doi: 10.1038/sj.ki.5002434. [DOI] [PubMed] [Google Scholar]
- Schacht J, Altschuler RA, Burke DT, Chen S, Dolan D, Galecki AT, Kohrman D, Miller RA. Alleles that modulate late life hearing in genetically heterogeneous mice. Neurobiol Aging. 2012;33:1842.e15–1842.e29. doi: 10.1016/j.neurobiolaging.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol. 1993;102:1–16. doi: 10.1177/00034894931020S101. [DOI] [PubMed] [Google Scholar]
- Seidman MD. Effects of dietary restriction and antioxidants on presbyacusis. Laryngoscope. 2000;110:727–738. doi: 10.1097/00005537-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Seidman MD, Ahmad N, Joshi D, Seidman J, Thawani S, Quirk WS. Age-related hearing loss and its association with reactive oxygen species and mitochondrial DNA damage. Acta Otolaryngol Suppl. 2004;552:16–24. doi: 10.1080/03655230410017823. [DOI] [PubMed] [Google Scholar]
- Sha S-H, Kanicki A, Halsey K, Wearne KA, Schacht J. Antioxidant-enriched diet does not delay the progression of age-related hearing loss. Neurobiol Aging. 2012;33:1010.e15–1010.e16. doi: 10.1016/j.neurobiolaging.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha S-H, Taylor R, Forge A, Schacht J. Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear Res. 2001;155:1–8. doi: 10.1016/s0378-5955(01)00224-6. [DOI] [PubMed] [Google Scholar]
- Shen H, Zhang B, Shin JH, Lei D, Du Y, Gao X, Wang Q, Ohlemiller KK, Piccirillo J, Bao J. Prophylactic and therapeutic functions of T-type calcium blockers against noise-induced hearing loss. Hear Res. 2007;226:52–60. doi: 10.1016/j.heares.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos I, Tretter L, Adam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. 2003;84:112–118. doi: 10.1046/j.1471-4159.2003.01513.x. [DOI] [PubMed] [Google Scholar]
- Spoendlin H. Primary structural changes in the organ of Corti after acoustic overstimulation. Acta Otolaryngol. 1971;71:166–176. doi: 10.3109/00016487109125346. [DOI] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA. Spiral ligament pathology in quiet-aged gerbils. Hear Res. 2002;172:172–185. doi: 10.1016/s0378-5955(02)00581-6. [DOI] [PubMed] [Google Scholar]
- Sweet RJ, Price JM, Henry KR. Dietary restriction and presbyacusis: periods of restriction and auditory threshold losses in the CBA/J mouse. Audiology. 1988;27:305–312. doi: 10.3109/00206098809081601. [DOI] [PubMed] [Google Scholar]
- Thorne PR, Nuttall AL. Laser Doppler measurements of cochlear blood flow during loud sound exposure in the guinea pig. Hear Res. 1987;27:1–10. doi: 10.1016/0378-5955(87)90021-9. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Torres MA, Schacht J. A BAD link to mitochondrial cell death in the cochlea of mice with noise-induced hearing loss. J Neurosci Res. 2006;83:1564–1572. doi: 10.1002/jnr.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warchol ME. Cellular mechanisms of aminoglycoside ototoxicity. Curr Opin Otolaryngol Head Neck Surg. 2010;18:454–458. doi: 10.1097/MOO.0b013e32833e05ec. [DOI] [PubMed] [Google Scholar]
- Willott JF, Erway LC, Archer JR, Harrison D. Genetics of age related hearing loss in mice: II. Strain differences and effects of caloric restriction on cochlear pathology and evoked response thresholds. Hear Res. 1995;88:143–155. doi: 10.1016/0378-5955(95)00107-f. [DOI] [PubMed] [Google Scholar]
- Wilson DN. The A-Z of bacterial translation inhibitors. Crit Rev Biochem Mol Biol. 2009;44:393–433. doi: 10.3109/10409230903307311. [DOI] [PubMed] [Google Scholar]
- Xie J, Talaska AE, Schacht J. New developments in aminoglycoside therapy and ototoxicity. Hear Res. 2011;281:28–37. doi: 10.1016/j.heares.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane H, Nakai Y, Takayama M, Iguchi H, Nakagawa T, Kojima A. Appearance of free radicals in the guinea pig inner ear after noise-induced acoustic trauma. Eur Arch Otorhinolaryngol. 1995;252:504–508. doi: 10.1007/BF02114761. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Jiang HY, Schacht J, Miller JM. Delayed production of free radicals following noise exposure. Brain Res. 2004;1019:201–209. doi: 10.1016/j.brainres.2004.05.104. [DOI] [PubMed] [Google Scholar]
- Yao YG, Salas A, Bravi CM, Bandelt HJ. A reappraisal of complete mtDNA variation in East Asian families with hearing impairment. Hum Genet. 2006;119:505–515. doi: 10.1007/s00439-006-0154-9. [DOI] [PubMed] [Google Scholar]
- Zuo H, Cui B, She X, Wu M. Changes in Guinea pig cochlear hair cells after sound conditioning and noise exposure. J Occup Health. 2008;50:373–379. doi: 10.1539/joh.l8032. [DOI] [PubMed] [Google Scholar]