

Abstract
Ethanol exposure and withdrawal alter the generation of new neurons in the adult hippocampus. The endogenous opioid system, in particular the μ opioid receptor (MOR), can modulate neural progenitors and also plays a critical role in ethanol drinking and dependence. In the present study, we sought to determine whether MOR contributes to the effects of ethanol on the dentate gyrus (DG) neurogenic niche. MOR wild-type (WT), heterozygous (Het) and knockout (KO) littermates were subjected to voluntary ethanol drinking in repeated limited-access two-bottle choice (2BC) sessions. MOR deficiency did not alter progenitor proliferation, neuronal differentiation and maturation, apoptosis or microglia in ethanol-naïve mice. When exposed to five consecutive weeks of 2BC, MOR mutant mice exhibited a gene-dosage dependent reduction of ethanol consumption compared to WT mice. Introducing a week of ethanol deprivation between each week of 2BC increased ethanol consumption in all genotypes and produced equivalent intakes in WT, Het and KO mice. Under the latter paradigm, ethanol drinking decreased progenitor proliferation and neuronal differentiation in the DG of WT mice. Interestingly, WT mice exhibited a strong negative correlation between ethanol intake and proliferation, which was disrupted in Het and KO mice. Moreover, MOR deficiency blocked the effect of ethanol on neuronal differentiation. MOR deficiency also protected against the neuroimmune response to ethanol drinking. Finally, chronic binge drinking induced a paradoxical decrease in apoptosis, which was independent of MOR. Altogether our data suggest that MOR is implicated in some of the neuroplastic changes produced by chronic ethanol exposure in the DG.
Keywords: AC3, BrdU, DCX, Iba1, Ki-67, neurogenesis
Introduction
The hippocampus is implicated in the pathophysiology of alcohol use disorders. Alcohol dependence is linked to neuronal loss in the dentate gyrus (DG) (Bengochea and Gonzalo, 1990; Beresford et al., 2006), decreased hippocampal volume (Brandt et al., 1983), and deficits in hippocampus-dependent cognitive tasks (Glenn and Parsons, 1991; Sullivan et al., 2002; Sullivan et al., 1995; Sullivan et al., 2000a; Sullivan et al., 2000b). Importantly, the hippocampus is involved in ethanol reward and relapse of ethanol seeking (Koob and Volkow, 2010; Zarrindast et al., 2010). It therefore appears that maladaptive plasticity in the hippocampus may contribute to some of the behavioral impairments associated with chronic alcohol use.
Recent evidence demonstrates that ethanol exposure alters adult hippocampal neurogenesis (Mandyam and Koob, 2012; Nixon, 2006). Adult hippocampal neurogenesis is a multi-stage dynamic process, which includes birth, survival, and integration of granule cell neurons in the subgranular zone (SGZ) of the DG of the adult hippocampus (Altman and Das, 1965; Kempermann, 2002). Ethanol can exert an inhibitory effect on every stage of hippocampal neurogenesis, whose amplitude varies with the dose, duration and pattern of ethanol exposure (Bengochea and Gonzalo, 1990; Beresford et al., 2006; Crews et al., 2004; Hansson et al., 2010; He et al., 2005; Ieraci and Herrera, 2007; Nixon and Crews, 2002; Richardson et al., 2009; Stevenson et al., 2009; Taffe et al., 2010). Altogether, the inhibitory effect of ethanol on the regenerative capacity of the adult hippocampus is considered a precursor for ethanol-induced neurodegeneration in the hippocampus.
The endogenous opioid system is known to modulate hippocampal neurogenesis (Arguello et al., 2008; Drake et al., 2007; Eisch et al., 2000; Harburg et al., 2007; Hauser et al., 2000; Koehl et al., 2008). The μ opioid receptor (MOR) is highly expressed in the DG (Mansour et al., 1995), in particular by neural progenitors (Drake et al., 2007; Drake and Milner, 2002; Eisch and Harburg, 2006; Persson et al., 2003a; Persson et al., 2003b). Importantly, chronic ethanol consumption reduces the expression of hippocampal MORs (Saland et al., 2005) and their functional coupling to G proteins (Chen and Lawrence, 2000; Saland et al., 2004; Sim-Selley et al., 2002), potentially through increased phosphorylation by G-protein receptor kinase 2 (Saland et al., 2008), suggesting they are activated by endogenous ligands upon ethanol exposure. We therefore hypothesized that hippocampal MORs play a critical role in ethanol-induced inhibition of neural progenitors in the DG.
In the present study, we subjected MOR wild-type (WT), heterozygous (Het) and knockout (KO) littermates to voluntary ethanol exposure and determined whether chronic binge drinking differentially regulates the neurogenic niche in the presence and absence of MORs. We used exogenous (5-bromo-2′-deoxyuridine, BrdU, which labels cells during the synthesis (S) phase of the cell cycle) and endogenous (Kee et al., 2002) markers of proliferation, an endogenous marker of neuronal differentiation and maturation (Brown et al., 2003), an endogenous marker of apoptosis (Nicholson et al., 1995) and an endogenous marker of microglia (Ito et al., 2001) to test the hypothesis that MOR is implicated in some of the neuroplastic changes produced by chronic ethanol exposure in the DG.
Methods
Animals
MOR KO mice were generated by homologous recombination (Matthes et al., 1996) and fully backcrossed on C57BL/6J background. Male MOR WT, Het and KO littermates were bred at The Scripps Research Institute. C57Bl/6J male mice were imported from Jackson laboratories (stock #000664). Mice were group-housed and were acclimated for a minimum of 10 days to reverse light cycle before testing started. Behavioral testing and ethanol exposure were started at 2–5 months of age, and mice were 4–6 months old at the end of the experiments. Mice were maintained on a 12-h light/dark cycle (lights on at 09:00 pm) in a temperature (22°C) and humidity (50%) controlled vivarium. Water and food (standard rodent chow, Harlan Teklad, Frederick, MD, USA) were available ad libitum at all times. All procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by The Scripps Research Institute Institutional Animal Care and Use Committee.
Limited-access two-bottle choice (2BC) ethanol drinking
Mice were subjected to limited-access sessions of voluntary drinking, during which they had access to a bottle containing ethanol (15% w:v) and a bottle containing water, in individual cages. Control mice had access to two bottles of water. Sessions lasted 2 h, starting 3 h into the dark cycle, and were conducted five days per week (Monday-Friday) for five weeks. Mice were group-housed in their home cages the rest of the time. Positions of the ethanol and water bottles were alternated daily. Mice were not food or water deprived at any time. In a first group, drinking sessions were conducted for five consecutive weeks and mice were killed Friday of the fifth week (Figure 2a). In a second group, mice were also tested for a total of five weeks, but, starting after the second week of testing, a week of ethanol deprivation was intercalated every other week. In this group, mice were tested for an additional day (Monday of the sixth drinking week, following a fourth deprivation week) and killed the day after (Figure 2c). In both groups, perfusions were conducted 22–24 h after the last drinking session, at a time mice anticipated having free access to ethanol consumption. Brains from the first cohort were subjected to Ki-67 analysis, and brains from the second cohort were subjected to BrdU, Ki-67, DCX, AC3 and Iba1 analysis.
Figure 2.

(a) Schematic of 2BC paradigm for the first group of mice. Mice were given free-choice access to ethanol drinking for 2 h per day, Monday through Friday, for five consecutive weeks. (b) Weekly average ethanol intake for the first group of mice (WT, n = 5; Het, n = 5; KO, n = 6). (c) Schematic of 2BC paradigm for the second group of mice. Mice were given free-choice access to ethanol drinking for 2 h per day, Monday through Friday, for a total of five weeks. After the second week of drinking, weeks of ethanol deprivation were intercalated. (d) Weekly average ethanol intake for the second group of mice (WT, n = 6; Het, n = 9; KO, n = 5). (e) Linear regression analysis of ethanol intake and resulting blood alcohol levels (BALs) in an independent cohort of C57Bl/6J mice subjected to limited-access 2BC on alternated weeks. BALs are plotted against the amount of ethanol consumed during the first hour (gray) and the two hours (black) of the drinking session.
An independent group of C57Bl/6J mice subjected to limited-access 2BC on alternated weeks was used to correlate ethanol intake with blood alcohol levels (BALs). Tail vein blood samples were collected 1 h into the session and at the end the 2-h session using heparinized capillary tubes, and centrifuged for 5 min at 13000 rpm. The supernatant was processed in a GM7 analyser (Analox Instruments, London, UK).
Bromodeoxyuridine (BrdU) injections
BrdU (Boehringer, Mannheim) was dissolved in 0.9% saline and 0.007 N NaOH at 20 mg/mL and administered i.p. at 150 mg/kg. BrdU was injected 2 h prior to perfusion in ethanol-naïve and ethanol-drinking mice from the second group.
Perfusions
Mice from all groups were anaesthetized using chloral hydrate 35% (0.15 mL/10 g body weight, i.p.) and perfused transcardially with 10 mL phosphate buffered saline, followed by 50 mL of 4% paraformaldehyde. The brains were postfixed overnight at 4°C with 4% paraformaldehyde and stored in 30% sucrose solution. Brains were cut through the hippocampus from bregma −0.82 to −4.24 (Paxinos and Franklin, 2001) at 40 μm in the coronal plane on a freezing microtome as described previously (Mandyam et al., 2004). Sections were collected in nine serial sets and were stored in vials containing 0.1% sodium azide in 1x PBS at 4°C.
Antibodies
The following primary antibodies were used for immunohistochemistry: rat anti-BrdU (MCA2060,1:100; Serotec), rabbit anti-Ki-67 (RM-9106-S, 1:1000; LabVision), goat anti-DCX (SC-8066, 1:700; Santa Cruz Biotechnology), rabbit anti-AC3 (9661, 1:500; Cell Signaling Technology) and rabbit anti-Iba1 (019–19741, 1:1000; Wako). The following biotinylated secondary antibodies were used: rabbit anti-rat (BA-4001, 1:200, Vector Laboratories), goat anti-rabbit (BA-1000, 1:200, Vector Laboratories) and horse anti-goat (BA-9500, 1:200, Vector Laboratories).
Immunohistochemistry
Every 9th section through the hippocampus was slide mounted and dried overnight. Slides were coded and the code was not broken until after analysis was complete. All incubations were carried out at room temperature unless otherwise indicated. Slide-mounted sections were subjected to three pretreatment steps: antigen unmasking, permeabilization, and denaturation as described previously (Mandyam et al., 2004). Slides were incubated with 0.3% H2O2 to remove any endogenous peroxidase activity. Nonspecific binding was then blocked with 5% serum and 0.5% Triton-X in 1x PBS for 30 min and incubated with the primary antibody (in 5% serum and 0.5% Tween-20) for 18–20 h. After washing with 1x PBS, the sections were exposed to biotin-tagged secondary antibodies for 60 min. Slides were then incubated in avidin-biotin complex (ABC, PK-6100, Vector Laboratories) for 1 h and staining was visualized with 3,3′-diaminobenzidine (DAB, SK-4100, Vector Laboratories). After immunostaining, sections were counterstained with Nuclear Fast Red (Vector Laboratories), dehydrated, and coverslipped.
Microscopic Analysis and Quantification
All microscopic quantification and analysis were made at 600x magnification by an observer blind to the study using a Zeiss PrimoStar microscope. Staining was examined and quantified in the subgranular zone (SGZ: 2-h BrdU, Ki-67 and DCX), granule cell layer (GCL: AC3 and Iba1) or hilus (Iba1) of the DG of the hippocampus (from bregma −0.82 to −4.24). The SGZ was defined as a region straddling the border of the GCL and the hilus: three GCL cell widths into the hilus, and the half of the GCL adjacent to the hilus (Mandyam et al., 2004). Immunoreactive cells were visually counted in every 9th section through the DG. Absolute cell counting of heterogeneously distributed cells (for AC3) and cells in clusters (for BrdU, Ki-67 and DCX) was performed rather than unbiased stereological analysis, because unbiased stereological analysis has been demonstrated to produce lower efficiency in cell quantification for heterogeneously distributed cell staining commonly visualized in the adult neurogenesis field (Noori and Fornal, 2011). The number of immunoreactive cells obtained by visual, absolute quantification method in the SGZ and GCL, were summed and multiplied by 9 to give the total number of cells (Eisch et al., 2000).
For DCX cell quantification, additional morphological analysis was performed as DCX is a marker for young neurons, and the developmental stages of DCX cells can be further delineated using morphological and co-labeling strategies (Mandyam et al., 2008a; Mandyam et al., 2008b). Specifically immature (early phase) DCX-labeled cells were differentiated from the mature (late phase) DCX-labeled cell types with careful morphological analysis; immature cells having short processes and mature cells having long processes that extend into the molecular layer of the DG (Figure 1d). Immature DCX cells are an indicator of neuronal differentiation, while mature DCX cells are an indicator of neuronal maturation (Brown et al., 2003; Couillard-Despres et al., 2005; Kempermann et al., 2003; Kuhn et al., 2005; Rao and Shetty, 2004; Seri et al., 2004).
Figure 1.
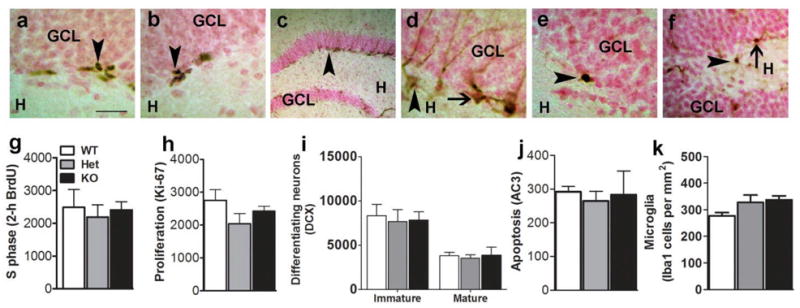
Representative images of immunolabeling in the SGZ (a–d) and GCL (e–f) of the DG from an ethanol-naive WT mouse. (a–b) Representative images of proliferating BrdU-labeled cells (a) and Ki-67- labeled cells (b), which were almost always seen in clusters of many cells. (c–d) Representative images of DCX-labeled cells, at low (c) and higher magnification (d), demonstrating distinct morphology of the cell types (arrowhead in (d) points to a mature DCX cell and thin arrow in (d) points to an immature DCX cell). (e) Representative image of an AC3-labeled cell in the GCL (arrowhead points to a positive cell). (f) Representative image of Iba1-labeled cells in the GCL and hilus (arrowhead points to a cell in the hilus and arrow points to a cell in the GCL). Scale bar in (a) is 20 μm for (a–b), 50 μm for (c), 10 μm for (d–e) and 30 μm for (f). Orientation of the DG is indicated by hilus (H) and GCL. Quantitative analysis of labeled cells in the DG from serial coronal sections of WT, Het and KO ethanol-naive mice (g–k). 2-h BrdU-labeled S-phase cells (g), Ki-67 proliferating cells (h), DCX-labeled immature and mature cells (i), AC3-labeled apoptotic cells (j) and Iba1-labeled microglia (k), n = 4 in each genotype. One-way ANOVAs did not reveal any significant effect of genotype for any of the cell markers analyzed.
For Iba1 analysis, dorsal hippocampal sections (4 per brain) were examined with a Zeiss Axiophot photomicroscope equipped with a three axis Mac 5000 motorized stage, a digital Zeiss CCD ZVS video camera, PCI color frame grabber and PC workstation, and MicroBrightField Stereo-Investigator software. Live video images were used to draw contours delineating the GCL and the hilus, and each region was traced separately at 50x magnification. The contours were realigned at high magnification (200x). Following determination of mounted section thickness (cut section thickness 40 μm; measured mounted section thickness 11–15 μm), z plane values and selection of contours, an optical fractionator analysis was used to determine bilateral estimates of Iba1 cell number per GCL and hilus of each section. A counting frame of appropriate dimensions, denoting forbidden and nonforbidden boundaries, was superimposed on the video monitor, and the optical fractionator analysis was performed at 200x. Cells were identified as Iba1 immunoreactive cells based on standard morphology (McClain et al., 2011), and only cells with a focused nucleus within the nonforbidden regions of the counting frame were counted. Over 30 cells (microglia with cell bodies falling entirely within the borders of the frame) were counted using a 150 × 150 μm counting grid, and a 2 μm top and bottom guard zone.Total number of Iba1 cells per mm2 was calculated for each section.
Data analysis
Statview software (SAS Institute Inc.) was used for statistical analysis. Ethanol drinking data (weekly average intake) were analyzed using a two-way repeated-measures analysis of variance (ANOVA) with time as within-subject factor and genotype as a between-subject factor. For the BrdU, Ki-67, DCX, AC3, and Iba1 analyses, one- or two-way ANOVAs were used with genotype and/or ethanol exposure as between-subject factors. The Tukey-Kramer test was used for post hoc analysis when appropriate. Linear regression analyses were performed using Pearson’s correlation test. The data are expressed as mean ± SEM in all graphs.
Results
MOR deficiency does not affect the neurogenic niche in ethanol-naïve mice
The role of MOR in modulating basal cell proliferation (2-h BrdU, Figure 1a, and Ki-67, Figure 1b), neuronal differentiation and maturation (DCX, Figure 1c–d), apoptosis (AC3, Figure 1e) and microglia (Iba1, Figures 1f) in the DG was examined in ethanol-naïve MOR KO, Het and WT mice. Ki-67 cells were irregularly shaped, showed immature morphology consistent with proliferating cells and were often clustered (Mandyam et al., 2008b). Immature and mature DCX-labeled cells were differentiated based on their morphology and indicate neuronal differentiation and maturation respectively (Figure 1c–d).
Ethanol-naïve MOR Het and KO mice had a similar number of 2–h BrdU cells (F2,9 = 0.14; n.s., Figure 1g) and Ki-67 cells (F2,9 = 1.66; n.s., Figure 1h) in the SGZ compared to WT littermates. MOR WT, Het and KO mice also showed equivalent levels of immature (F2,9 = 0.07; n.s., Figure 1i) and mature (F2,9 = 0.11; n.s., Figure 1i) DCX- expressing cells. Finally, MOR WT, Het and KO mice showed comparable levels of apoptosis (F2,9 = 0.10; n.s.; Figure 1j) and microglia (F2,9 = 2.71; n.s.; Figure 1k) in the GCL.
The effect of MOR deficiency on voluntary ethanol drinking depends on the temporal pattern of access to ethanol
A first group of MOR WT, Het and KO mice had limited access to 2BC ethanol self-administration for five consecutive weeks (Figure 2a). A two-way repeated-measures ANOVA of ethanol intake (weekly average, Figure 2b) revealed an effect of genotype (F2,13 = 7.9, p < 0.01) and time (F4,52 = 7.7, p < 0.001), as well as a significant interaction between both factors (F8,52 = 2.5, p < 0.05). A gene-dosage effect was observed, with a virtually null consumption in KO mice and an intermediate intake in Het mice compared to WT littermates.
In a second group of MOR WT, Het and KO mice, 2BC drinking weeks were interspersed with ethanol deprivation weeks (Figure 2c). Intercalating a week of deprivation between drinking weeks enhanced ethanol self-administration across genotypes (Figure 2b versus Figure 2d, effect of group: F1,30 = 8.6, p < 0.01, no significant interaction between group and genotype: F2,30 = 0.2, n.s.). A two-way repeated-measures ANOVA of ethanol intake (weekly average, Figure 2d) revealed a significant effect of time (F4,68 = 26.1, p < 0.001), but did not detect an effect of genotype (F2,17 = 0.5, n.s.) nor an interaction between time and genotype (F8,68 = 1.3, n.s.).
Analysis of BALs in an independent cohort of C57Bl/6J mice subjected to limited-access 2BC ethanol drinking on alternated weeks indicated that BALs were significantly correlated with ethanol intake (Figure 2e), both when BALs were analyzed 1 h into the session (R2 = 0.96, F1,9 = 216.4, p < 0.001) and at the end of the 2-h session (R2 = 0.74, F1,9 = 25.9, p < 0.001).
The effect of chronic binge drinking on the hippocampal neurogenic niche (2-h BrdU, Ki-67, DCX, AC3 and Iba1 cell counts) in the presence and absence of MOR was examined in the second group of mice, in which ethanol exposure was equivalent across genotypes (Figure 2d). In addition, Ki-67 cell counts from the first and second groups were combined to analyze the relationship between ethanol intake and cell proliferation using linear regression.
Chronic binge drinking dose-dependently reduces cell proliferation in the SGZ of WT mice
Limited-access ethanol drinking for five alternated weeks reduced cell proliferation in the SGZ, as assessed both by 2-h BrdU labeling (Figure 3a, significant effect of ethanol: F1,26 = 20.73, p < 0.001; no effect of genotype: F2,26 = 0.88, n.s.; no interaction: F2,26 = 2.44, n.s.) and by Ki-67 expression (Figure 3b, significant effect of ethanol: F1,26 = 22.29, p < 0.001; no effect of genotype: F2,26 = 0.37, n.s.; trend for an interaction: F2,26 = 3.23, p = 0.056). Post hoc analysis indicated a significant reduction in proliferating cells (using both markers) in ethanol-drinking WT and KO mice compared to their water-drinking counterparts (Figures 3a–b). Linear regression analysis revealed a significant correlation between ethanol intake (average intake over the last five drinking sessions) and proliferation (number of Ki-67-expressing cells) in WT mice (Figure 3c, R2 = 0.77, p < 0.001). In contrast, no correlation was detected in MOR Het (R2 = 0.10, n.s.) and KO mice (R2 = 0.04, n.s.).
Figure 3.
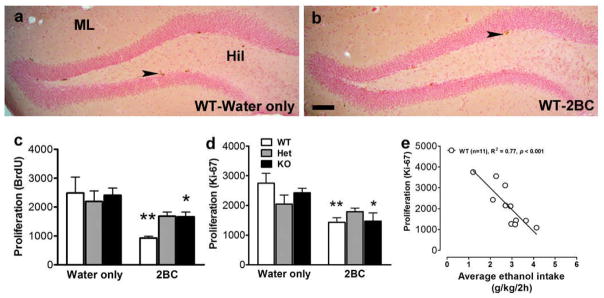
Photomicrographs of BrdU labeling from (a) WT-water only mouse and (b) WT-2BC mouse. Arrowhead points to BrdU clusters in the subgranular zone of the DG. Scale bar in (b) is 100 μm, applies to (a–b). (c–d) Quantitative analysis (cell counts) of 2-h BrdU-labeled S-phase progenitors (c) and Ki-67 proliferating cells (d) in the DG from the second group of mice (water only: WT, n = 4; Het, n = 4; KO, n = 4; 2BC: WT, n = 6; Het, n = 9; KO, n = 5). Post hoc analysis revealed significant differences between water-and ethanol-drinking mice of a given genotype (*, p < 0.05; **, p < 0.01). (e) Linear regression analysis of the effect of chronic binge drinking on progenitor proliferation in the DG. The total number of Ki-67-labeled cells in the hippocampus is plotted against the average ethanol intake over the last five drinking sessions for each mouse from the first and second groups.
Chronic binge drinking inhibits neuronal differentiation through a MOR- dependent mechanism
Chronic binge drinking significantly decreased the number of immature DCX- labeled cells in the SGZ (F1,26 = 10.51, p < 0.01, Figure 4a). Although there was no main effect of genotype (F2,26 = 0.50, n.s.) and no significant genotype x ethanol interaction (F2,26 = 1.07, n.s.), post hoc analysis indicated that ethanol self- administration significantly decreased the number of immature DCX cells in WT mice, without producing any effects in Het and KO mice. In contrast, chronic binge drinking did not affect the number of mature DCX-labeled cells (F1,26 = 0.11, n.s., Figure 4b). There was no main effect of genotype (F2,26 = 0.66, n.s.) and no significant genotype x ethanol interaction (F2,26 = 0.28, n.s.) either.
Figure 4.
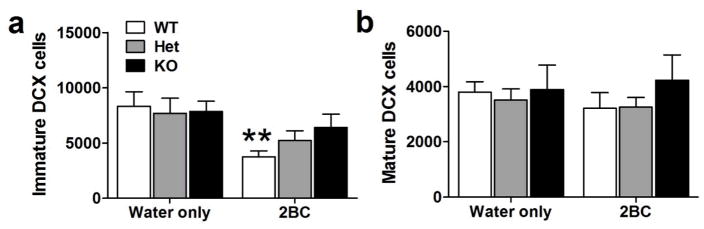
(a–b) Quantitative analysis of DCX-labeled cells of immature (a) and mature (b) morphology in the DG from the second group of mice (water only: WT, n = 4; Het, n = 4; KO, n = 4; 2BC: WT, n = 6; Het, n = 9; KO, n = 5). Post hoc analysis revealed significant differences between water- and ethanol-drinking mice of a given genotype (**, p < 0.01).
Chronic binge drinking reduces apoptosis independently of MOR signaling
The number of AC3-labeled apoptotic cells was reduced in the GCL of ethanol- drinking mice (F1,26 = 29.10, p < 0.001, Figure 5), and there was no effect of genotype (F2,26 = 0.28, n.s.) and no interaction between both factors (F2,26 = 0.01, n.s.). Post hoc analysis indicated a significantly decreased number of AC3 cells in ethanol-exposed WT and Het mice and a trend for reduction in KO mice compared to their respective water-drinking counterparts.
Figure 5.
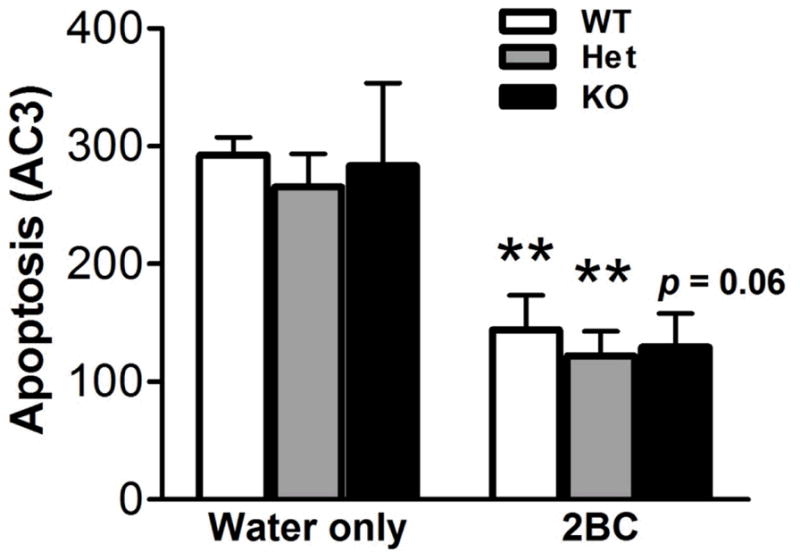
Quantitative analysis of AC3-labeled apoptotic cells in the DG from the second group of mice (water only: WT, n = 4; Het, n = 4; KO, n = 4; 2BC: WT, n = 6; Het, n = 9; KO, n = 5). Post hoc analysis revealed significant differences between water-and ethanol-drinking mice of a given genotype (**, p < 0.01).
MOR deficiency protects against the neuroimmune response to chronic binge drinking
There was a trend for an effect of ethanol (F1,26 = 3.42, p = 0.08, Figure 6) and no effect of genotype (F2,26 = 0.28, n.s.) on the number of Iba1-labeled cells in the GCL. There was, however, a strong interaction between ethanol and genotype (F2,26 = 5.68, p < 0.01), which reflected that chronic binge drinking had opposite effects in WT (increased number of Iba1 cells, indicative of a neuroimmune response) and MOR deficient mice (decreased number of Iba1 cells). The same analysis in the hilus led to similar results (significant ethanol x genotype interaction, F2,26 = 5.83, p < 0.01, data not shown).
Figure 6.

Photomicrograph of Iba1 staining from (a) WT-water only mouse and (b) WT-2BC mouse. Insets in (a) and (b) show an Iba1 immunoreactive cell at 600x magnification. Scale bar in (b) is 100 μm, applies to (a–b). (c) Quantitative analysis of Iba1-labeled microglia in the DG from the second group of mice (water only: WT, n = 4; Het, n = 4; KO, n = 4; 2BC: WT, n = 6; Het, n = 9; KO, n = 5). Two-way ANOVA revealed a significant interaction between ethanol drinking and genotype (##, p < 0.01).
Discussion
Ongoing research in the neurogenesis field is focusing on the identification and functional study of cell-intrinsic and extrinsic regulatory factors modulating the process of hippocampal adult neurogenesis (Dhaliwal and Lagace, 2011; Duan et al., 2008; Gage, 2010; Hsieh and Eisch, 2010; Johnson et al., 2009; Shi et al., 2010). Neurotransmitters have been proposed to act upstream from intracellular mechanisms and other extracellular cues by influencing the secretion of growth factors and the activation of transcription factors (Pathania et al., 2010). Importantly, identification of specific molecular pathways governing neurogenesis would potentially enable us to reverse the neuroplasticity induced by ethanol exposure, thereby aiding recovery and reducing the vulnerability to relapse (Canales, 2007; DeCarolis and Eisch, 2010; Mandyam and Koob, 2012).
Of particular interest to the ethanol abuse field, the endogenous opioid system was shown to regulate the development of SGZ neural progenitors (Harburg et al., 2007; Persson et al., 2004; Persson et al., 2003a; Persson et al., 2003b). Our data obtained in ethanol-naive mice however suggest that the hippocampal neurogenic niche is not significantly affected by the absence of MOR signaling under basal conditions. In particular, we found that MOR Het and KO mice demonstrate normal levels of progenitor proliferation, neuronal differentiation and maturation, and apoptosis compared to WT littermates, which is in agreement with the literature (Harburg et al., 2007). We also found that levels of microglia were not altered by MOR deficiency. The enhanced survival of adult-generated hippocampal cells previously reported in MOR deficient mice (Harburg et al., 2007) may therefore arise from ‘stabilizing’ (anti-cell death) factors that still remain to be identified.
Several lines of evidence demonstrate that the endogenous opioid system is involved in ethanol drinking. In particular, nonselective opioid receptor antagonists reduce ethanol consumption in humans, and are used in the treatment of alcoholism (O’Malley et al., 1992; Rubio et al., 2001; Volpicelli et al., 1992). A specific role of MORs is supported by preclinical studies demonstrating that MOR-selective antagonists decrease ethanol self-administration in genetically heterogenous Wistar rats (Stromberg et al., 1998) and in rats bred for high ethanol intake (Krishnan-Sarin et al., 1998). In agreement with these pharmacological studies, genetic deletion of MOR in KO mice also reduced ethanol consumption in paradigms of home-cage drinking and operant self-administration (Becker et al., 2002; Ghozland et al., 2005; Hall et al., 2001; Roberts et al., 2000). It should be noted, however, that under certain experimental conditions, MOR KO mice voluntarily consumed ethanol to a similar extent as WT mice (e.g., continuous 2BC drinking prior to operant training in Roberts et al., 2000, and continuous 2BC drinking at high concentrations of ethanol in Hall et al., 2001). Accordingly, in our study, we initially found a strong gene-dosage effect of MOR deletion on ethanol intake in a limited-access 2BC paradigm when drinking sessions were conducted on consecutive weeks, but the effect of genotype vanished upon insertion of deprivation weeks between drinking weeks. The escalation of voluntary ethanol consumption following periods of deprivation has been previously shown under various experimental conditions (Heyser et al., 1997; Melendez et al., 2006; Spanagel and Holter, 1999). This approach enabled us to normalize the ethanol intake of MOR deficient mice to that of WT mice, and to study the effect of chronic binge drinking on the hippocampal neurogenic niche in the presence or absence of MOR without the bias of variable ethanol exposure.
We found that chronic ethanol binge drinking in WT mice reduces proliferation and neuronal differentiation (immature DCX cells) of hippocampal progenitors, thereby confirming an earlier report (Crews et al., 2004). Importantly, we also provide striking new evidence for a negative correlation between the amount of ethanol consumed and the number of dividing progenitors in the SGZ of WT mice, thereby highlighting the significance of ethanol-induced inhibition of proliferation in the DG in the context of excessive drinking. The existence of a direct relationship between ethanol intake and the extent of proliferation inhibition may help explain some of the discrepancies found in the literature. For example, our findings in WT mice are consistent with a previous report (Crews et al., 2004) that demonstrated reduced progenitors in the DG after chronic ethanol consumption. However, other studies showed increased or unchanged progenitor proliferation and survival of newly born neurons after weeks of ethanol drinking (Aberg et al., 2005; Stevenson et al., 2009). There are important methodological differences between the 2BC paradigms used by us and others. In the studies by Aberg et al. and Stevenson et al., mice were single housed, and had continuous access to ethanol (10% v/v) in their home cage (24 h/day, 7 days/week). In our study, mice were group-housed except during 2BC sessions, and had access to ethanol (15% w/v) only for 2 h/day, 5 days/week in a different testing cage. The limited-access 2BC paradigm in our study produced a binge drinking pattern with BALs approximating 150 mg/dl (based on the linear relationship established in C57Bl/6J mice), which is higher than the BALs reported in the other studies. Therefore differences in drinking pattern and degree of ethanol exposure may have contributed to the differential regulation of neural progenitors in WT mice.
Our data also indicate that chronic binge drinking selectively disrupts neuronal differentiation (immature DCX cells) without impacting neuronal maturation (mature DCX cells) in WT mice, in accordance with our previous findings in rats (Richardson 2009). Despite equivalent levels of ethanol exposure, chronic binge drinking in MOR Het and KO mice did not alter the differentiation of neural progenitors as it did in WT mice. Furthermore, the amount of ethanol consumed in MOR Het and KO mice did not correlate with the number of dividing progenitors, indicating that MOR deficiency switched off a mechanism linking ethanol exposure to cell proliferation.
Surprisingly, while an abundance of studies has shown that ethanol exposure increases cell death (Crews and Nixon, 2009), we found that chronic binge drinking reduced apoptosis in the GCL. Combined with our findings that cell proliferation was reduced under the same conditions, we propose that decreased apoptosis may represent a compensatory adaptation to conserve neuronal population in the DG. The mechanisms mediating this unexpected decrease in apoptosis still remain to be elucidated, but our study shows that MOR signaling is not involved since reduced apoptosis was observed in MOR deficient mice as well. It should be noted that alternative pathways of cell death, such as necrosis, have not been investigated in the present study and could possibly offset the effect of reduced apoptosis.
Lastly, we determined the effects of binge ethanol consumption on microglia, as neuroimmune factors can be triggered by ethanol exposure (He and Crews, 2008) and can in turn modulate neurogenesis (Zou and Crews, 2012). In our study, chronic binge drinking slightly increased the number of microglial cells in the GCL and hilus of WT mice, but had the opposite effect in MOR deficient mice, thereby suggesting that MOR directly contributes to ethanol-induced activation of microglia. Combined with our data on progenitor proliferation and neuronal differentiation, this striking interaction between ethanol exposure and MOR genotype on hippocampal microglia raises the possibility that ethanol-induced inhibition of neurogenesis may be partly mediated by MOR-dependent neuroimmune signaling within the neurogenic niche (McClain et al., 2011; Nixon et al., 2008).
In conclusion, MOR deficiency does not alter the hippocampal neurogenic niche under basal conditions, but reduces the inhibitory effect of chronic binge drinking on neuronal differentiation in the SGZ, and disrupts the effect of ethanol on progenitor proliferation. MOR deficiency also protects against the neuroimmune response triggered in the DG by chronic binge drinking. On the other hand, MOR is not involved in the paradoxical reduction of apoptosis we observed in ethanol-drinking mice.
A potential pitfall of our study is that constitutive KO mice were used and some of the observed phenotypic changes may therefore reflect compensatory changes occurring in the brain during development and adulthood due to the absence of MORs, or downstream consequences of MOR deficiency outside of the DG. In particular, the lack of correlation between ethanol intake and the number of dividing progenitors in KO mice, despite a preserved net effect of chronic binge drinking on cell proliferation, may reflect the recruitment of alternative inhibitory mechanisms. Importantly, in Het mice, which should be less prone than KO mice to the development of compensatory adaptations, chronic binge drinking had no effect on cell proliferation, as assessed using both exogenous and endogenous markers. Future studies aiming to specifically knockdown MORs in the hippocampus versus other brain regions, or to knockdown MORs in DG cells undergoing distinct stages of neuronal development (progenitor cells versus immature neurons) will help delineate the role of MOR activation in the regulatory effects of ethanol on DG neurogenesis.
Acknowledgments
Research was supported by National Institutes of Health grants DA022473 (CDM) from the National Institute on Drug Abuse and AA020913 (CC) from the National Institute on Alcohol Abuse and Alcoholism. We acknowledge the excellent technical assistance of Elena Crawford for stereological analysis, and Vinay Biligiri and Wednesday R. Bushong from the Life Sciences Summer Internship Program at The Scripps Research Institute, Kelly Ostertag, Joelle Harwin, Connie Choi and Kelly Svoboda from the independent study program at the University of California, San Diego, for assistance with immunohistochemistry and animal behavior. We appreciate the editorial assistance of Michael Arends. This is publication number 21633 from The Scripps Research Institute.
Footnotes
The authors have no conflict of interest to declare.
References
- Aberg E, Hofstetter CP, Olson L, Brene S. Moderate ethanol consumption increases hippocampal cell proliferation and neurogenesis in the adult mouse. Int J Neuropsychopharmacol. 2005;8:557–567. doi: 10.1017/S1461145705005286. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- Arguello AA, Harburg GC, Schonborn JR, Mandyam CD, Yamaguchi M, Eisch AJ. Time course of morphine’s effects on adult hippocampal subgranular zone reveals preferential inhibition of cells in S phase of the cell cycle and a subpopulation of immature neurons. Neuroscience. 2008;157:70–79. doi: 10.1016/j.neuroscience.2008.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:296–302. doi: 10.1007/s00210-002-0533-2. [DOI] [PubMed] [Google Scholar]
- Bengochea O, Gonzalo LM. Effect of chronic alcoholism on the human hippocampus. Histol Histopathol. 1990;5:349–357. [PubMed] [Google Scholar]
- Beresford TP, Arciniegas DB, Alfers J, Clapp L, Martin B, Du Y, Liu D, Shen D, Davatzikos C. Hippocampus volume loss due to chronic heavy drinking. Alcohol Clin Exp Res. 2006;30:1866–1870. doi: 10.1111/j.1530-0277.2006.00223.x. [DOI] [PubMed] [Google Scholar]
- Brandt J, Butters N, Ryan C, Bayog R. Cognitive loss and recovery in long-term alcohol abusers. Arch Gen Psychiatry. 1983;40:435–442. doi: 10.1001/archpsyc.1983.01790040089012. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Canales JJ. Adult neurogenesis and the memories of drug addiction. Eur Arch Psychiatry Clin Neurosci. 2007;257:261–270. doi: 10.1007/s00406-007-0730-6. [DOI] [PubMed] [Google Scholar]
- Chen F, Lawrence AJ. Effect of chronic ethanol and withdrawal on the mu-opioid receptor- and 5-Hydroxytryptamine(1A) receptor-stimulated binding of [(35)S]Guanosine-5′-O-(3-thio)triphosphate in the fawn-hooded rat brain: A quantitative autoradiography study. J Pharmacol Exp Ther. 2000;293:159–165. [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME. Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol. 2004;33:63–71. doi: 10.1016/j.alcohol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- DeCarolis NA, Eisch AJ. Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology. 2010;58:884–893. doi: 10.1016/j.neuropharm.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaliwal J, Lagace DC. Visualization and genetic manipulation of adult neurogenesis using transgenic mice. Eur J Neurosci. 2011;33:1025–1036. doi: 10.1111/j.1460-9568.2011.07600.x. [DOI] [PubMed] [Google Scholar]
- Drake CT, Chavkin C, Milner TA. Opioid systems in the dentate gyrus. Prog Brain Res. 2007;163:245–263. doi: 10.1016/S0079-6123(07)63015-5. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA. Mu opioid receptors are in discrete hippocampal interneuron subpopulations. Hippocampus. 2002;12:119–136. doi: 10.1002/hipo.1107. [DOI] [PubMed] [Google Scholar]
- Duan X, Kang E, Liu CY, Ming GL, Song H. Development of neural stem cell in the adult brain. Curr Opin Neurobiol. 2008;18:108–115. doi: 10.1016/j.conb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisch AJ, Harburg GC. Opiates, psychostimulants, and adult hippocampal neurogenesis: Insights for addiction and stem cell biology. Hippocampus. 2006;16:271–286. doi: 10.1002/hipo.20161. [DOI] [PubMed] [Google Scholar]
- Gage FH. Molecular and cellular mechanisms contributing to the regulation, proliferation and differentiation of neural stem cells in the adult dentate gyrus. Keio J Med. 2010;59:79–83. doi: 10.2302/kjm.59.79. [DOI] [PubMed] [Google Scholar]
- Ghozland S, Chu K, Kieffer BL, Roberts AJ. Lack of stimulant and anxiolytic-like effects of ethanol and accelerated development of ethanol dependence in mu-opioid receptor knockout mice. Neuropharmacology. 2005;49:493–501. doi: 10.1016/j.neuropharm.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Glenn SW, Parsons OA. Impaired efficiency in female alcoholics’ neuropsychological performance. J Clin Exp Neuropsychol. 1991;13:895–908. doi: 10.1080/01688639108405106. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Nixon K, Rimondini R, Damadzic R, Sommer WH, Eskay R, Crews FT, Heilig M. Long-term suppression of forebrain neurogenesis and loss of neuronal progenitor cells following prolonged alcohol dependence in rats. Int J Neuropsychopharmacol. 2010;13:583–593. doi: 10.1017/S1461145710000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harburg GC, Hall FS, Harrist AV, Sora I, Uhl GR, Eisch AJ. Knockout of the mu opioid receptor enhances the survival of adult-generated hippocampal granule cell neurons. Neuroscience. 2007;144:77–87. doi: 10.1016/j.neuroscience.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Houdi AA, Turbek CS, Elde RP, Maxson W., 3rd Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: differential impact of mu and delta receptor activation on proliferation and neurite elongation. Eur J Neurosci. 2000;12:1281–1293. doi: 10.1046/j.1460-9568.2000.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Nixon K, Shetty AK, Crews FT. Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur J Neurosci. 2005;21:2711–2720. doi: 10.1111/j.1460-9568.2005.04120.x. [DOI] [PubMed] [Google Scholar]
- Heyser CJ, Schulteis G, Koob GF. Increased ethanol self-administration after a period of imposed ethanol deprivation in rats trained in a limited access paradigm. Alcohol Clin Exp Res. 1997;21:784–791. [PubMed] [Google Scholar]
- Hsieh J, Eisch AJ. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol Dis. 2010;39:73–84. doi: 10.1016/j.nbd.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieraci A, Herrera DG. Single alcohol exposure in early life damages hippocampal stem/progenitor cells and reduces adult neurogenesis. Neurobiol Dis. 2007;26:597–605. doi: 10.1016/j.nbd.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Ables JL, Eisch AJ. Cell-intrinsic signals that regulate adult neurogenesis in vivo: insights from inducible approaches. BMB Rep. 2009;42:245–259. doi: 10.5483/bmbrep.2009.42.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115:97–105. doi: 10.1016/s0165-0270(02)00007-9. [DOI] [PubMed] [Google Scholar]
- Kempermann G. Why new neurons? Possible functions for adult hippocampal neurogenesis. J Neurosci. 2002;22:635–638. doi: 10.1523/JNEUROSCI.22-03-00635.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130:391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Koehl M, Meerlo P, Gonzales D, Rontal A, Turek FW, Abrous DN. Exercise-induced promotion of hippocampal cell proliferation requires beta-endorphin. FASEB J. 2008;22:2253–2262. doi: 10.1096/fj.07-099101. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Wand GS, Li XW, Portoghese PS, Froehlich JC. Effect of mu opioid receptor blockade on alcohol intake in rats bred for high alcohol drinking. Pharmacol Biochem Behav. 1998;59:627–635. doi: 10.1016/s0091-3057(97)00474-7. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Biebl M, Wilhelm D, Li M, Friedlander RM, Winkler J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: a role for cell death during continued hippocampal neurogenesis. Eur J Neurosci. 2005;22:1907–1915. doi: 10.1111/j.1460-9568.2005.04377.x. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Crawford EF, Eisch AJ, Rivier CL, Richardson HN. Stress experienced in utero reduces sexual dichotomies in neurogenesis, microenvironment, and cell death in the adult rat hippocampus. Dev Neurobiol. 2008a;68:575–589. doi: 10.1002/dneu.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Koob GF. The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci. 2012 doi: 10.1016/j.tins.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Norris RD, Eisch AJ. Chronic morphine induces premature mitosis of proliferating cells in the adult mouse subgranular zone. Journal of Neuroscience Research. 2004;76:783–794. doi: 10.1002/jnr.20090. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Wee S, Crawford EF, Eisch AJ, Richardson HN, Koob GF. Varied access to intravenous methamphetamine self-administration differentially alters adult hippocampal neurogenesis. Biol Psychiatry. 2008b;64:958–965. doi: 10.1016/j.biopsych.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Akil H, Watson SJ. Immunohistochemical localization of the cloned mu opioid receptor in the rat CNS. J Chem Neuroanat. 1995;8:283–305. doi: 10.1016/0891-0618(95)00055-c. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM, Nixon K. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav Immun. 2011 doi: 10.1016/j.bbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Middaugh LD, Kalivas PW. Development of an alcohol deprivation and escalation effect in C57BL/6J mice. Alcohol Clin Exp Res. 2006;30:2017–2025. doi: 10.1111/j.1530-0277.2006.00248.x. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nixon K. Alcohol and adult neurogenesis: roles in neurodegeneration and recovery in chronic alcoholism. Hippocampus. 2006;16:287–295. doi: 10.1002/hipo.20162. [DOI] [PubMed] [Google Scholar]
- Nixon K, Crews FT. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem. 2002;83:1087–1093. doi: 10.1046/j.1471-4159.2002.01214.x. [DOI] [PubMed] [Google Scholar]
- Nixon K, Kim DH, Potts EN, He J, Crews FT. Distinct cell proliferation events during abstinence after alcohol dependence: microglia proliferation precedes neurogenesis. Neurobiol Dis. 2008;31:218–229. doi: 10.1016/j.nbd.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noori HR, Fornal CA. The appropriateness of unbiased optical fractionators to assess cell proliferation in the adult hippocampus. Front Neurosci. 2011;5:140. doi: 10.3389/fnins.2011.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence. A controlled study. Arch Gen Psychiatry. 1992;49:881–887. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- Pathania M, Yan LD, Bordey A. A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology. 2010;58:865–876. doi: 10.1016/j.neuropharm.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2°. Academic Press; San Diego: 2001. [Google Scholar]
- Persson AI, Naylor AS, Jonsdottir IH, Nyberg F, Eriksson PS, Thorlin T. Differential regulation of hippocampal progenitor proliferation by opioid receptor antagonists in running and non-running spontaneously hypertensive rats. Eur J Neurosci. 2004;19:1847–1855. doi: 10.1111/j.1460-9568.2004.03268.x. [DOI] [PubMed] [Google Scholar]
- Persson AI, Thorlin T, Bull C, Eriksson PS. Opioid-induced proliferation through the MAPK pathway in cultures of adult hippocampal progenitors. Mol Cell Neurosci. 2003a;23:360–372. doi: 10.1016/s1044-7431(03)00061-7. [DOI] [PubMed] [Google Scholar]
- Persson AI, Thorlin T, Bull C, Zarnegar P, Ekman R, Terenius L, Eriksson PS. Mu- and delta-opioid receptor antagonists decrease proliferation and increase neurogenesis in cultures of rat adult hippocampal progenitors. Eur J Neurosci. 2003b;17:1159–1172. doi: 10.1046/j.1460-9568.2003.02538.x. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Richardson HN, Chan SH, Crawford EF, Lee YK, Funk CK, Koob GF, Mandyam CD. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis. 2009;36:1–10. doi: 10.1016/j.nbd.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Rubio G, Jimenez-Arriero MA, Ponce G, Palomo T. Naltrexone versus acamprosate: one year follow-up of alcohol dependence treatment. Alcohol Alcohol. 2001;36:419–425. doi: 10.1093/alcalc/36.5.419. [DOI] [PubMed] [Google Scholar]
- Saland LC, Abeyta A, Frausto S, Raymond-Stintz M, Hastings CM, Carta M, Valenzuela CF, Savage DD. Chronic ethanol consumption reduces delta-and mu-opioid receptor-stimulated G-protein coupling in rat brain. Alcohol Clin Exp Res. 2004;28:98–104. doi: 10.1097/01.ALC.0000108658.00243.BF. [DOI] [PubMed] [Google Scholar]
- Saland LC, Chavez JB, Lee DC, Garcia RR, Caldwell KK. Chronic ethanol exposure increases the association of hippocampal mu-opioid receptors with G-protein receptor kinase 2. Alcohol. 2008;42:493–497. doi: 10.1016/j.alcohol.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saland LC, Hastings CM, Abeyta A, Chavez JB. Chronic ethanol modulates delta and mu-opioid receptor expression in rat CNS: immunohistochemical analysis with quantitiative confocal microscopy. Neurosci Lett. 2005;381:163–168. doi: 10.1016/j.neulet.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, Collado-Morente L, McEwen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol. 2004;478:359–378. doi: 10.1002/cne.20288. [DOI] [PubMed] [Google Scholar]
- Shi Y, Zhao X, Hsieh J, Wichterle H, Impey S, Banerjee S, Neveu P, Kosik KS. MicroRNA regulation of neural stem cells and neurogenesis. J Neurosci. 2010;30:14931–14936. doi: 10.1523/JNEUROSCI.4280-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ, Sharpe AL, Vogt LJ, Brunk LK, Selley DE, Samson HH. Effect of ethanol self-administration on mu- and delta-opioid receptor-mediated G-protein activity. Alcohol Clin Exp Res. 2002;26:688–694. [PubMed] [Google Scholar]
- Spanagel R, Holter SM. Long-term alcohol self-administration with repeated alcohol deprivation phases: an animal model of alcoholism? Alcohol Alcohol. 1999;34:231–243. doi: 10.1093/alcalc/34.2.231. [DOI] [PubMed] [Google Scholar]
- Stevenson JR, Schroeder JP, Nixon K, Besheer J, Crews FT, Hodge CW. Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology. 2009;34:1209–1222. doi: 10.1038/npp.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O’Brien CP. A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol. 1998;15:281–289. doi: 10.1016/s0741-8329(97)00131-6. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Fama R, Rosenbloom MJ, Pfefferbaum A. A profile of neuropsychological deficits in alcoholic women. Neuropsychology. 2002;16:74–83. doi: 10.1037//0894-4105.16.1.74. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Marsh L, Mathalon DH, Lim KO, Pfefferbaum A. Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol Clin Exp Res. 1995;19:110–122. doi: 10.1111/j.1530-0277.1995.tb01478.x. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Lim KO, Pfefferbaum A. Longitudinal changes in cognition, gait, and balance in abstinent and relapsed alcoholic men: relationships to changes in brain structure. Neuropsychology. 2000a;14:178–188. [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Pfefferbaum A. Pattern of motor and cognitive deficits in detoxified alcoholic men. Alcohol Clin Exp Res. 2000b;24:611–621. [PubMed] [Google Scholar]
- Taffe MA, Kotzebue RW, Crean RD, Crawford EF, Edwards S, Mandyam CD. Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. Proc Natl Acad Sci U S A. 2010;107:11104–11109. doi: 10.1073/pnas.0912810107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Meshkani J, Rezayof A, Beigzadeh R, Rostami P. Nicotinic acetylcholine receptors of the dorsal hippocampus and the basolateral amygdala are involved in ethanol-induced conditioned place preference. Neuroscience. 2010;168:505–513. doi: 10.1016/j.neuroscience.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews FT. Inflammasome-IL-1beta Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front Neurosci. 2012;6:77. doi: 10.3389/fnins.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]