

Abstract
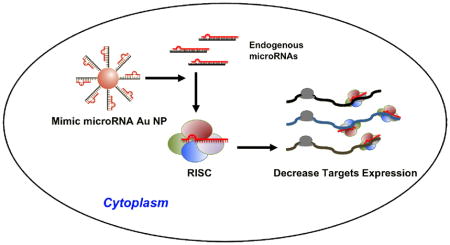
Novel conjugates of gold nanoparticles (13±1nm) functionalized with synthetic microRNAs can enter cells without the aid of cationic co-carriers and mimic the function of endogenous microRNAs. These conjugates can regulate multiple proteins through interactions with 3′ untranslated region of the target mRNA and control cell behavior. The conjugates are a promising new tool for studying miRNA function and new candidates for miRNA replacement therapies.
Keywords: Gold nanoparticle, siRNA, microRNA, gene regulation, cancer therapy
Recent advances in nanotechnology have led to promising constructs for drug delivery, disease detection, and molecular diagnostics. [1–8] Polyvalent oligonucleotide-functionalized gold nanoparticles have emerged as new and robust tools for drug delivery, analyte detection and gene regulation in biological systems. [9, 10] These unusual structures consist of gold nanoparticles (Au NP) that are functionalized with a dense shell of synthetic oligonucleotides, [11] and have several properties that make them ideal for biomedical applications. For example, they have the capacity to enter cultured cells or animal tissues without the aid of lipid- or polymer-based co-carriers. [12, 13] The oligonucleotides on their surfaces are resistant to nuclease degradation and, therefore, more stable than the same sequences free in solution. [14, 15] The innate immune response elicited by these conjugates is 25 fold lower than the same DNA delivered using commercial lipid co-carriers. [16, 17] Consequently, they have been used to develop a wide variety of molecular diagnostic and potential therapeutic systems. [12, 18, 19] Interestingly, to date, no methods have been developed for utilizing the unique properties of polyvalent nanoparticles to introduce microRNA (miRNAs) into cells and tissues, despite the fact that they might be very effective agents for stabilizing the miRNAs and regulating gene expression in multiple ways. Herein, we report the synthesis and characterization of novel miRNA-gold nanoparticle conjugates (miRNA-Au NPs) that are capable of entering cells without the aid of cationic co-carriers, regulating the expression of multiple genes and controlling cellular behavior.
MicroRNAs are small RNAs (approximately 22 nucleotides in length) of cellular origin that function as the endogenous regulators of gene expression in eukaryotes through the RNA interference pathway. [20–22] Individual miRNAs are able to regulate multiple target genes through interactions with sites of imperfect complementarity in the 3′-untranslated regions (UTRs) of target mRNAs, which cause either the destabilization of the targeted transcripts or translational repression. [20–23] The remarkable ability of individual miRNAs to regulate numerous transcripts allows miRNAs to control a variety of cellular processes, including cellular differentiation, proliferation and apoptosis. [24–26] Aberrant miRNA levels have been shown to contribute to a number of diseases, including several types of cancers, where miRNAs have the ability to act as both tumor suppressors and oncogenes. [27–30] Since dysregulation of a single miRNA can effectively contribute to tumorogenesis, correcting the abnormal miRNA activities by either antagonizing or restoring miRNA function may provide significant anti-oncogenic benefits. [27, 28, 31, 32] However, a major obstacle in miRNA functional studies and miRNA-directed therapy is the lack of viable delivery systems that achieve an effective regulation of gene expression and cell phenotype using miRNAs. Current methods of miRNA introduction include the use of virus-based cloning and expression vectors, complexation with auxiliary transfection agents, electroporation, and polymer- or liposome-based carrier systems. [33–36] These tools are challenged by several limitations, including an undesired immune response, cell toxicity, and inefficient delivery to target tissues. [37–39] We hypothesized that the polyvalent oligonucleotide-functionalized gold nanoparticles, which have been shown to enter cells without the use of a cationic co-carrier, could be functionalized with synthetic miRNA sequences and potentially overcome cellular transfection hurdles and mimic endogenous miRNA function.
The prototype miRNA mimics (miRNA-Au NPs) consist of 13±1 nm gold nanoparticles, each functionalized with a monolayer of double stranded alkylthiol-modified RNA molecules (33±4 duplexes/NP, Scheme 1, Supporting Information Table 1). [15] The immobilization of the oligonucleotides on the surfaces of the gold nanoparticles has been shown to increase the stability of the bound RNA while retaining its ability to act in the native cellular RNAi pathways (See Supporting Information for Experimental Section). [15] Therefore, we hypothesized that miRNA-Au NPs could effectively introduce miRNA into cells without transfection agents, regulate target gene expression, and exert the miRNA specific phenotype.
Scheme 1.
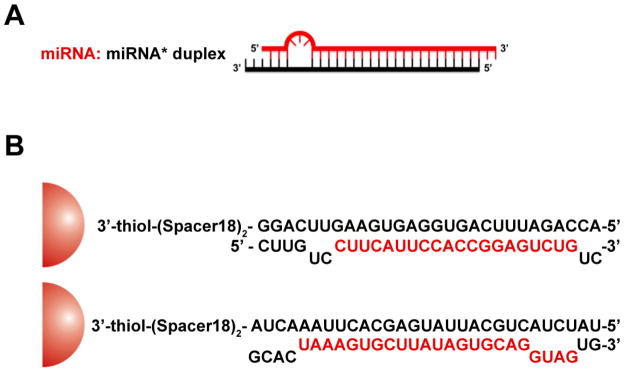
Schematic representation of the mimic miRNA Au NPs. In (A), the typical endogenous miRNA structure (miRNA: miRNA*) with one or more bulges is shown. Only the mature miRNA strand, in red, is known to be involved in downstream gene regulation pathways. In (B), the scheme and sequences of the mimic miRNA-205 Au NP (upper) and mimic miRNA-20a Au NP (lower) attached to 13 nm gold nanoparticle (red hemisphere) are depicted. In both cases, the sense strand is attached to the gold surface through the adsorption of alkyl thiol on gold. The red letters represent mature miRNA sequences.
To test this hypothesis, we investigated whether miRNA-Au NPs are able to control cellular processes by elevating the endogenous miRNA level in the human prostate cancer (PCa) cell line, PC-3. Human miR-205 was chosen as a proof-of-concept miRNA because it is markedly down-regulated in PCa cell lines and tissue specimens, and performs tumor-suppressive functions through the down-regulation of protein kinase C epsilon (PRKCε). [40] The PRKCε mRNA contains a 3′-UTR sequence that is partially complementary to miR-205 (Figure 1A). PC-3 cells were treated with a 2mL solution of 10 nM miRNA-Au NPs that mimic miR-205 or non-targeting sequences (NT Au NPs), respectively. After 96 hours, the expression of PRKCε was measured using immunoblotting. The results show that the treatment with the miRNA-Au NPs that mimic miR-205 decreases the expression of PRKCε by 52% compared to cells treated with control particles functionalized with non-targeting sequences (Figure 1C).
Figure 1.

Human protein kinase C epsilon (PRKCε) expression as a function of nanoparticle-conjugate treatment in PC-3 cells. (A) The construction of the pMIR-REPORT™ vector. The duplex formation demonstrates the partially complementary binding between miR-205 and PRKCε mRNA at position 2732–2738 of PRKCε 3′-UTR. (B) Bar graph shows that firefly luciferase activity is suppressed by transfection of mimic miR-205 Au NPs when compared with the non-targeting Au NPs. Commercially available mimic miR-205, transfected using DharmaFECT™, shows 12% reduction in firefly luciferase activity. The error bars indicate the standard deviation (SD) from three different experiments. (C) Immunoblotting of PC-3 cell lysates for PRKCε, showing an over 50% decrease in PRKCε expression in PC-3 cells following transfection with mimic miR-205 Au NPs. GAPDH serves as the loading control. The relative protein expression level was quantified by ImageJ.
It is known that a single miRNA can regulate multiple targets through interactions with the 3′-UTRs of the target mRNA. To test whether the novel miRNA-Au NPs could function similarly, we cloned and inserted the cDNA fragment containing the 3′-UTR sequence of PRKCε downstream of firefly luciferase into the pMIR-REPORT™ miRNA expression reporter vector (Applied Biosystems) and then transfected this plasmid into HeLa cells. When the ectopic miR-205 is successfully delivered to the cells by miRNA-Au NPs, it interacts with the miR-205 binding site in the PRKCε 3′-UTR fragment inserted into the reporter vector and causes translational repression of luciferase. The pRL-SV40 Renilla luciferase vector that did not contain a miR-205 binding site and could not be targeted by miR-205 was co-transfected as a control. Luciferase expressing HeLa cells were treated with 5 nM mimic miRNA-Au NPs targeting the 3′-UTR sequence of PRKCε. After 72 hours, firefly luciferase signal was measured in cell lysates and normalized to the Renilla luciferase signal using the Dual-Glo™ Luciferase Assay System (Promega). The results show that the firefly luciferase signal in cells treated with mimic miR-205 Au NPs was reduced by 38% compared with cells transfected with Au NPs functionalized with non-targeting sequences (Figure 1B). Importantly, the same concentration of commercially available miR-205 mimic molecules delivered using DharmaFECT™ only provided a 12% reduction in firefly luciferase activity (Figure 1B). The decrease in firefly luciferase signal confirms that the miRNA-Au NPs have the capacity to recognize and interact with the 3′-UTR of the target gene. Interestingly, the mimic miR-205 Au NPs are approximately three times more potent than the analogous molecular system delivered with DharmaFECT™. Furthermore, while the protein expression levels were reduced, no significant changes in the mRNA levels were observed suggesting that the mimic miRNA-Au NPs block the translation of corresponding mRNA target rather than cause degradation of the target mRNA (Supporting Information,Figure 1). This observation is in agreement with literature precedent, which shows that most mammalian miRNAs regulate their targets through translational repression rather than mRNA degradation. [23]
Having confirmed the biochemical activity of the miRNA-Au NPs, we proceeded to examine the phenotypic effects of mimic miRNA-Au NPs in tissue culture. A previous report demonstrated that re-expression of miR-205 in prostate cancer cells induces apoptosis and impairs the cells’ invasive properties. [41] Consistent with these results, we observed an approximately 50% decrease in cell survival for PC-3 cells transfected with miRNA-Au NPs compared with control cells transfected with the non-targeting Au NPs (Figure 2A; Supporting Information, Figure 2). Thus, mimic miR-205 functionalized Au NPs not only inhibit PRKCε expression, but also induce PCa cell apoptosis within one week after transfection in a sequence-specific manner.
Figure 2.
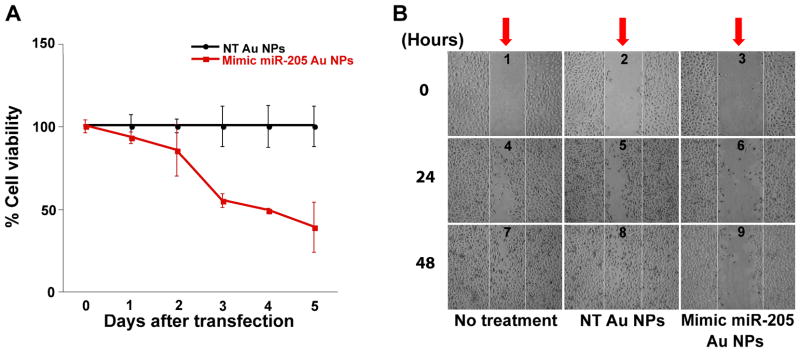
The proliferation and migration of cancer cells as a function of nanoparticle-conjugate treatment. (A) miR-205 Au NPs (red) significantly reduce PC-3 cell viability over 5 days of treatment. Non-targeting Au NPs (black) do not affect PC-3 cell viability over the same time period. (B) Mimic miR-205 Au NPs restrict PC-3 cell migration. The scratch wounds (red arrows) made in the untreated or non-targeting Au NPs treated cells (center of panels 1 and 2) closed completely within 48 hours (panels 7 and 8). By contrast, miR-205 Au NPs treated cells were not able to close a similar scratch wound (panel 9).
A decrease in miR-205 expression is known to increase cellular motility and eventual metastasis of tumor cells. [40, 41] We postulated that an effective increase in cellular miR-205 through treatment with miRNA-Au NPs would have the opposite effect and decrease the migration rate of PCa cells. We tested this hypothesis by using a wound closing assay (commonly used for studying migration rates of cultured cells). [42] PC-3 cells were grown to confluency in a 6-well plate, and a wound was made using a sterile P-200 pipet tip. The cells were then incubated with mimic miRNA-Au NPs or non-targeting Au NPs and the wound closure was monitored using light microscopy. We found that the untreated cells, or the non-targeting Au NPs treated cells, were able to close the wound within 48 hours. However, the cells treated with miR-205 functionalized nanoparticles only showed a partial closure of the wound over this time period, indicating that the mimic miRNA-Au NPs are decreasing the motility of PC-3 cells (Figure 2B). Since miR-205 is reported to decrease both cellular motility and viability, it is likely that the inability of PC-3 cells to close the wound is due to a combination of these two reported functions of miR-205. [40, 41] These experiments confirm that miR-205 introduced via miRNA-Au NPs can function in the endogenous pathways to exert the expected phenotypic response from PC-3 cells and control cell migratory behavior.
To show the general applicability of these constructs, we tested a different sequence, miR-20a, which plays an oncogenic role in PCa carcinogenesis by reducing the expression of transcription factors E2F1 and the phosphatase and tensin homolog (PTEN). [43–45] We cloned the 3′ UTRs of E2F1 and PTEN downstream of firefly luciferase gene as described above (Figure 3A, 3B) and performed a luciferase activity assay to determine the effects of miRNA-Au NPs. Consistent with the known biological function of miR-20a, the mimic miR-20a Au NPs were able to regulate the expression of firefly luciferase gene modified to include 3′-UTRs of E2F1 and PTEN. The luciferase signal in cells treated with mimic miR-20a Au NPs was reduced by 32% (E2F1) and 41% (PTEN) compared with cells transfected with Au NPs functionalized with non-targeting sequences (Figure 3A–B). [44] Furthermore, oncogenic miR-20a delivered by miRNA-Au NPs protects PC-3 cells from apoptosis induced by the anti-cancer drug doxorubicin. [43] After treatment with doxorubicin, PC-3 cells incubated with Au NPs functionalized with non-targeting sequences showed a 3-fold increase in Caspase 3/7 activity, indicating the induction of cell apoptosis. However, PC-3 cells treated with mimic miR-20a Au NPs showed no significant change in Caspase 3/7 activity, suggesting that mimic miRNAs prevent the cells from undergoing doxorubicin induced apoptosis (Figure 3C).
Figure 3.

Mimic miR-20a Au NPs regulate multiple known miR-20a targets and promote cell proliferation. (A) Upper panel: the transcription factor E2F1 has two miR-20a binding sites at positions 386–392 and 979–985 in its 3′-UTR. Lower panel (bar graph): ectopic introduction of miR-20a by mimic miRNA Au NPs inhibits the activity of luciferase gene containing E3F1 3′-UTR. (B) The upper panel shows the binding site of miR-20a at position 272–278 of PTEN 3′-UTR. The lower panel (bar graph) indicates that the mimic miR-20a Au NPs cause reduction in the activity of luciferase gene containing PTEN 3′-UTR. (C) miR-20a Au NPs treatment suppresses doxorubicin induced apoptosis in PC-3 cells. After treatment with doxorubicin, PC-3 cells transfected with Au NPs functionalized with non-targeting sequences show a 3-fold increase in Caspase 3/7 activity, an indication of apoptosis. Caspase activity in miR-20a Au NPs treated PC-3 cells remains unchanged with or without doxorubicin indicating an inhibition of apoptosis induced by doxorubicin. The error bars indicate S.D. from 3 different experiments in (A), (B) and 6 different experiments in (C). The groups of NT-Au NPs and mimic miRNA-Au NPs in (A) and (B) have P values less than 0.05.
In summary, we have synthesized and characterized novel miRNA-Au NPs, which mimic native miRNAs. We have shown that the nanoparticles carrying the mimics of tumor suppressive microRNA miR-205 repress the expression of miR-205 target protein by interaction with the 3′ untranslated region of the target mRNA. Significantly, phenotypic assays have shown that these conjugates can act to inhibit cancer cell proliferation and migration. Nanoparticles functionalized with mimics of oncogenic microRNA miR-20a promote cell survival by down-regulating known miR-20a target proteins. Importantly, these constructs regulate gene expression up to three fold more effectively than analogous molecular miRNAs delivered using a commercial co-carrier DharmaFECT™. Taken together, the ability of polyvalent nucleic acid-functionalized nanoparticles to enter cells without the aid of cationic lipids and polymers, coupled with their demonstrated function as endogenous miRNAs, makes them a promising new tool for studying miRNA function as well as important new candidates for miRNA replacement therapies.
Supplementary Material
Acknowledgments
C.A.M. acknowledges a Cancer Center for Nanotechnology Excellence (NCI-CCNE) Award and the AFOSR for support of this research. P.C.P. was supported by a Ryan Fellowship. A.H.A. acknowledges the K.S.A. King Abdullah Scholarship.
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
References
- 1.Rotello V. Cell Cycle. 2009;8:3615–3616. doi: 10.4161/cc.8.22.9915. [DOI] [PubMed] [Google Scholar]
- 2.Sandhu K, McIntosh C, Simard J, Smith S, Rotello V. Bioconjug Chem. 2002;13:3–6. doi: 10.1021/bc015545c. [DOI] [PubMed] [Google Scholar]
- 3.You C, Miranda O, Gider B, Ghosh P, Kim I, Erdogan B, Krovi S, Bunz U, Rotello V. Nat Nanotechnol. 2007;2:318–323. doi: 10.1038/nnano.2007.99. [DOI] [PubMed] [Google Scholar]
- 4.Agbasi-Porter C, Ryman-Rasmussen J, Franzen S, Feldheim D. Bioconjug Chem. 2006;17:1178–1183. doi: 10.1021/bc060100f. [DOI] [PubMed] [Google Scholar]
- 5.Tkachenko A, Xie H, Coleman D, Glomm W, Ryan J, Anderson M, Franzen S, Feldheim D. J Am Chem Soc. 2003;125:4700–4701. doi: 10.1021/ja0296935. [DOI] [PubMed] [Google Scholar]
- 6.Huang CC, Huang YF, Cao Z, Tan W, Chang HT. Anal Chem. 2005;77:5735–5741. doi: 10.1021/ac050957q. [DOI] [PubMed] [Google Scholar]
- 7.Huang YF, Chang HT, Tan W. Anal Chem. 2008;80:567–572. doi: 10.1021/ac702322j. [DOI] [PubMed] [Google Scholar]
- 8.Medley C, Smith J, Tang Z, Wu Y, Bamrungsap S, Tan W. Anal Chem. 2008;80:1067–1072. doi: 10.1021/ac702037y. [DOI] [PubMed] [Google Scholar]
- 9.Giljohann D, Seferos D, Daniel W, Massich M, Patel P, Mirkin C. Angew Chem-Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhar S, Daniel W, Giljohann D, Mirkin C, Lippard S. J Am Chem Soc. 2009;131:14652–14653. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirkin C, Letsinger R, Mucic R, Storhoff J. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 12.Rosi N, Giljohann D, Thaxton C, Lytton-Jean A, Han M, Mirkin C. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 13.Patel P, Giljohann D, Daniel W, Zheng D, Prigodich A, Mirkin C. Bioconjug Chem. 2010:2250–2256. doi: 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seferos D, Prigodich A, Giljohann D, Patel P, Mirkin C. Nano Lett. 2009;9:308–311. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giljohann D, Seferos D, Prigodich A, Patel P, Mirkin C. J Am Chem Soc. 2009;131:2072–2073. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massich M, Giljohann D, Schmucker A, Patel P, Mirkin C. ACS Nano. 2010;4:5641–5646. doi: 10.1021/nn102228s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massich M, Giljohann D, Seferos D, Ludlow L, Horvath C, Mirkin C. Mol Pharm. 2009;6:1934–1940. doi: 10.1021/mp900172m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prigodich A, Seferos D, Massich M, Giljohann D, Lane B, Mirkin C. ACS Nano. 2009;3:2147–2152. doi: 10.1021/nn9003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng D, Seferos D, Giljohann D, Patel P, Mirkin C. Nano Lett. 2009;9:3258–3261. doi: 10.1021/nl901517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He L, Hannon G. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 21.Bartel D. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 22.Bartel D. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bushati N, Cohen S. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 24.Brennecke J, Hipfner D, Stark A, Russell R, Cohen S. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 25.Dostie J, Mourelatos Z, Yang M, Sharma A, Dreyfuss G. RNA. 2003;9:180–186. doi: 10.1261/rna.2141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Li L, Lodish H, Bartel D. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 27.Kota J, Chivukula R, O’Donnell K, Wentzel E, Montgomery C, Hwang H, Chang T, Vivekanandan P, Torbenson M, Clark K, Mendell J, Mendell J. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calin G, Croce C. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 29.He L, Thomson J, Hemann M, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe S, Hannon G, Hammond S. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meltzer P. Nature. 2005;435:745–746. doi: 10.1038/435745a. [DOI] [PubMed] [Google Scholar]
- 31.Bader A, Brown D, Winkler M. Cancer Res. 2010;70:7027–7030. doi: 10.1158/0008-5472.CAN-10-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeshita F, Patrawala L, Osaki M, Takahashi R, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader A, Brown D, Ochiya T. Mol Ther. 2010;18:181–187. doi: 10.1038/mt.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anand S, Majeti B, Acevedo L, Murphy E, Mukthavaram R, Scheppke L, Huang M, Shields D, Lindquist J, Lapinski P, King P, Weis S, Cheresh D. Nat Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen M, Larsen L, Kauppinen S, Schratt G. Front Neural Circuits. 2010:3. doi: 10.3389/neuro.04.016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Paula D, Bentley M, Mahato R. RNA. 2007;13:431–456. doi: 10.1261/rna.459807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh N, Agrawal A, Leung A, Sharp P, Bhatia S. J Am Chem Soc. 2010;132:8241–8243. doi: 10.1021/ja102132e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song W, Du J, Sun T, Zhang P, Wang J. Small. 2010;6:239–246. doi: 10.1002/smll.200901513. [DOI] [PubMed] [Google Scholar]
- 38.Lee S, Han M, Asokan S, Tung C. Small. 2010:364–370. doi: 10.1002/smll.201001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryou S, Kim S, Jang H, Kim J, Yeom J, Eom M, Bae J, Han M, Lee K. Biochem Biophys Res Commun. 2010;398:542–546. doi: 10.1016/j.bbrc.2010.06.115. [DOI] [PubMed] [Google Scholar]
- 40.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, Salvioni R, Supino R, Moretti R, Limonta P, Valdagni R, Daidone M, Zaffaroni N. Cancer Res. 2009;69:2287–2295. doi: 10.1158/0008-5472.CAN-08-2894. [DOI] [PubMed] [Google Scholar]
- 41.Majid S, Dar A, Saini S, Yamamura S, Hirata H, Tanaka Y, Deng G, Dahiya R. Cancer. 2010:5637–5649. doi: 10.1002/cncr.25488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Peng H, Ruan Q, Fatima A, Getsios S, Lavker R. FASEB J. 2010;24:3950–3959. doi: 10.1096/fj.10-157404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay U, Bourdeau V, Major F, Ferbeyre G, Chartrand P. J Biol Chem. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 44.O’Donnell K, Wentzel E, Zeller K, Dang C, Mendell J. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 45.Pesta M, Klecka J, Kulda V, Topolcan O, Hora M, Eret V, Ludvikova M, Babjuk M, Novak K, Stolz J, Holubec L. Anticancer Res. 2010;30:3579–3583. [PubMed] [Google Scholar]
- 46.Pitsch S, Weiss P, Jenny L, Stutz A, Wu X. Helvetica Chimica Acta. 2001;84:3773–3795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.