

Abstract
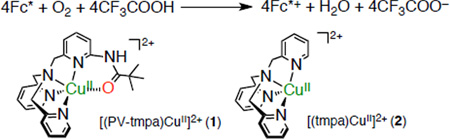
A copper complex, [(PV-tmpa)CuII](ClO4)2 (1) [PV-tmpa = bis(pyrid-2-ylmethyl){[6-(pivalamido)pyrid-2-yl]methyl}-amine], acts as a more efficient catalyst for the four-electron reduction of O2 by decamethylferrocene (Fc*) in the presence of trifluoroacetic acid (CF3COOH) in acetone as compared with the corresponding copper complex without a pivalamido group, [(tmpa)CuII](ClO4)2 (2) (tmpa = tris(2-pyridylmethyl)amine). The rate constant (kobs) of formation of decamethylferrocenium ion (Fc*+) in the catalytic four-electron reduction of O2 by Fc* in the presence large excess CF3COOH and O2 obeyed first-order kinetics. The kobs value was proportional to the catalyst concentrations, 1 or 2 whereas the kobs value remained constant irrespective of the concentrations of CF3COOH or O2. This indicates that electron transfer from Fc* to 1 or 2 is the rate-determining step in the catalytic cycle of the four-electron reduction of O2 by Fc* in the presence of CF3COOH. The second-order catalytic rate constant (kcat) for 1 is four times larger than the corresponding value determined for 2. Separate studies reveal that electron transfer from Fc* to the CuII complex is the rate-determining step in the case of PV-tmpa ligand, because the carbonyl oxygen of the pivalamido group inhibits the coordination of CF3COO− to copper, in direct contrast to the case for the tmpa ligand-complex. 1 is also an excellent catalyst for the two-electron two-proton reduction of H2O2 to water, and is also more efficient than is 2. For both complexes, reaction rates are greater than for overall four-electron O2-reduction to water, an important asset in the design of catalysts for the latter.
Introduction
Cytochrome c oxidases (CcOs), with a bimetallic active-site consisting of a heme a and Cu (Fea3/CuB), catalyze the four-electron reduction of dioxygen (O2) to water by the soluble electron carrier, cytochrome c.1–4 Extensive efforts have been devoted to develop synthetic Fea3/CuB analogs, because the four-electron four-proton reduction of O2 has attracted much attention from the viewpoint not only of great biological interest,5–11 but also of technological significance such as in fuel cells.12–15 Multicopper oxidases such as laccase (possessing four copper ions centers per functional unit) can also catalyzed the four-electron four-proton reduction of O2, (the “ORR”) at potentials approaching 1.2 V (vs RHE).16–22 Thus, it has become of interest to study discrete coordination complexes with copper ion for such studies. In fact, certain Cu complexes have recently been reported to exhibit electrocatalytic activity for the four-electron four-proton reduction of O2.14,23–29 In contrast to such heterogeneous systems, investigations on the catalytic reduction of O2 by metal complexes in homogeneous systems have provided valuable mechanistic insight into the role of metal-dioxygen intermediates in the catalytic cycle in the two-electron and four-electron reduction of O2.30–34 With regard to copper-dioxygen intermediates, trans–μ–1,2–peroxo-, μ-η2:η2-peroxo- and bis-μ-oxo-dinuclear copper complexes (Chart 1), have been extensively studied in reactions of low-valent metal complexes and O2.35–49
Chart 1.

Copper-dioxygen intermediates
We recently reported that μ-η2:η2-peroxo- and bis-μ-oxo-dinuclear copper complexes were readily reduced by two equivalents of one-electron reductants such as decamethylferrocene (Fc*), leading to the catalytic four-electron reduction of O2 by Fc*.44 We also reported that [(tmpa)CuII](ClO4) (tmpa = tris(2-pyridylmethyl)amine), which afforded the trans–μ–1,2–peroxo-dinuclear copper complex ([(tmpa)CuII(O2)CuII(tmpa)]2+) as an intermediate formed from [(tmpa)CuI]+/O2 chemistry, also catalyzed the four-electron reduction of O2 by Fc* in the presence of perchloric acid (HClO4) acetone.46 In contrast to the case of μ-η2:η2-peroxo- and bis-μ-oxo-dinuclear copper complexes, the trans–μ–1,2–peroxo complex with TMPA ligand could not be reduced by Fc* without also the presence of an acid such as HClO4.46 However, the role of an acid in the catalytic four-electron reduction of O2 with the trans–μ–1,2–peroxo complex has yet to be clarified. Thus, it is interesting to study the homogeneours catalytic four-electron reduction of O2 by Cu complexes with a weaker acid.
We report herein that a copper complex, [(PV-tmpa) CuII](ClO4)2 (1) [PV-tmpa = bis(pyrid-2-ylmethyl){[6-(pivalamido)pyrid-2-yl]methyl}amine]48b which has a pivalamido group, acts as a more efficient catalyst for the four-electron reduction of dioxygen (O2) with Fc* in the presence of trifluoroacetic acid (CF3COOH) which is much weaker acid than HClO4 in acetone as compared with the corresponding copper complex without a pivalamido group, [(tmpa)CuII](ClO4)2 (2). The role of CF3COOH in the catalytic four-electron reduction of O2 with 1 is clarified based on kinetic and electrochemical studies and detection of trans–μ–1,2–peroxo and Cu(II) hydroperoxo intermediates at low temperature in the absence and presence of CF3COOH, respectively. Direct comparisons are made between the behavior of 1 and 2 with CF3COOH as acid source, to understand why 1 acts as a better O2-reduction catalyst.
The two-electron two-proton reduction of hydrogen peroxide to water is also a critically important reaction in societal energy concerns.50,51 If H2O2 or a metal-(hydro)peroxide species is an intermediate in the hoped-for four-electron four-proton reduction of O2 to water, it is desirable to design/develop catalysts that take H2O2-to-water faster than the O2-to-H2O reaction occurs, to insure efficiency. Another purpose in the study of hydrogen peroxide reduction (or oxidation) mechanism(s) is for future efforts in “H2O2 fuel cell” technology.50,51 For these reasons, we have also initiated a research program in hydrogen peroxide reduction chemistry. We also describe here experiments demonstrating that complexes [(PV-tmpa)CuII](ClO4)2 (1) and [(tmpa)CuII](ClO4)2 (2) are catalysts for this process, using Fc* as reductant and CF3COOH as proton source.
Experimental Section
Materials
Commercially available reagents, decamethylferrocene (Fc*), perchloric acid (70%), trifluoroacetic acid, hydrogen peroxide (30%) and NaI (Wako Pure Chemical Industries) were the best available purity and used without further purification. Acetone was dried according to the literature procedures52 and distilled under Ar prior to use. Copper complexes ([(PV-tmpa) CuII](ClO4)2 (1) 48b and [(tmpa)CuII](ClO4)2 (2)45,46) were prepared according to literature procedures.
Reaction Procedure
The catalytic reduction of O2 was observed by the spectral change in the presence of various concentrations of CF3COOH at 298 K using a Hewlett Packard 8453 photodiode-array spectrophotometer with a quartz cuvette (path length = 10 mm). Typically, an acetone solution of CF3COOH (0 – 5.0 × 10−2 M) was added by means of a microsyringe to an O2-saturated acetone solution containing [(PV-tmpa)CuII](ClO4)2 (1) or [(tmpa)CuII](ClO4)2 (2) (4.0 × 10−5 M) and Fc* (2.0 × 10−3 M). The concentration of Fc*+ was determined from the absorption band at λmax = 780 nm (ε = 500 M−1 cm−1 at 298 K and 600 M−1 cm−1 at 213 K). The ε value of Fc*+ was estimated by the electron-transfer oxidation of Fc* with [RuIII(bpy)3](PF6)3. The limiting concentration of O2 in an acetone solution was prepared by a mixed gas flow of O2 and N2. The mixed gas was controlled by using a gas mixer (Kofloc GB-3C, KOJIMA Instrument Inc.), which can mix two or more gases at a certain pressure and flow rate. The amount of H2O2 was determined by the titration by iodide ion. The diluted (× 10) acetone solution of the O2 reduction product was treated with an excess of NaI. The amount of I3− formed was then determined by its visible spectrum (λmax = 361 nm, ε = 2.5 × 104 M−1 cm−1).53 UV–vis absorption spectra and spectral changes at low temperature were recorded on a Hewlett Packard 8453A diode array spectrophotometer equipped with a liquid nitrogen chilled Unisoku USP-203-A cryostat.
Kinetic Measurements
Kinetic measurements for fast reactions with short half-lifetimes (within 10 s) were performed on a UNISOKU RSP-601 stopped flow spectrophotometer with a MOS-type high selective photodiode array at 298 K using a Unisoku thermostated cell holder. Rates of electron transfer from Fc* to 1 were monitored by the rise of absorption bands due to Fc*+. All kinetic measurements were carried out under pseudo-first-order conditions; concentrations of Fc* was maintained to be more than in 10-fold excess compared to the concentration of 1.
Electrochemistry
Cyclic voltammetry measurements of Cu(II) complexes were performed on an ALS 630B electrochemical analyzer and measured in the absence and presence of CF3COOH in deaerated acetone solutions containing 0.1 – 0.5 M [(n–butyl)4N]PF6 (TBAPF6) as an supporting electrolyte at room temperature or otherwise noted. A conventional three-electrode cell was used with a platinum working electrode (surface area of 0.3 mm2) and platinum wire as the counter electrode. The Pt working electrode (BAS) was routinely polished with BAS polishing alumina suspension and rinsed with acetone before use. The potentials were measured with respect to the Ag/AgNO3 (0.01 M) reference electrode. All potentials (vs Ag/AgNO3) were converted the values vs SCE by addition of 0.29 V.54 All electrochemical measurements were carried out under an atmospheric pressure of nitrogen or argon otherwise noted.
EPR Measurements
EPR spectra were recorded on a JEOL JES-RE1XE spectrometer. The magnitude of modulation was chosen to optimize the resolution and signal-to-noise (S/N) ratio of the observed spectra under non-saturating microwave power conditions. The g values and hyperfine coupling constants were calibrated using a Mn2+ marker.
ESI Mass Measurements
Electrospray ionization mass spectrometry (ESI-MS) data were obtained using an API 150EX quadrupole mass spectrometer (PE-Sciex), equipped with an ion spray interface. The sprayer was held at a potential of +5.0 kV or −4.4 kV for positive or negative ion detection modes, respectively, and compressed N2 was employed to assist liquid nebulization. The orifice potential was maintained at +30.0 V or −40.0 V for positive or negative modes, respectively.
Theoretical Calculations
DFT calculations of copper complexes were performed on a 32-processor QuantumCube™ using using Gaussian 09, revision A.02.55 The geometry optimization carried out at the UCAM-B3LYP/6-311G(d) level of theory.56–59 Graphical outputs of the computational results were generated with the GaussView software program (ver. 3.09) developed by Semichem, Inc.60
Results and Discussion
Four-Electron Reduction of O2 with Fc* Catalyzed by 1 in the Presence of CF3COOH
The addition of a catalytic amount of 1 to an acetone solution of Fc*, O2 and CF3COOH at 298 K results in the efficient oxidation of Fc* by O2 to afford Fc*+. When more than four equiv of Fc* relative to O2 (i.e., limiting [O2]) were employed, four equiv of Fc*+ were formed in the presence of excess CF3COOH (Figure 1a). It has also been confirmed that no H2O2 is detected, via iodometric titration experiments (Figure S1 in the Supporting Information (SI)).49 Thus, the stoichiometry of the catalytic reduction of O2 by Fc* is given by eq 1. The four-
| (1) |
 |
electron reduction of O2 by Fc* with catalyst (1) in the presence of CF3COOH was also confirmed at 193 K. (Figure S2 in SI). Time courses of formation of Fc*+ comparing results for [(PV-tmpa) CuII]2+ (1) and [(tmpa)CuII]2+ (2) under the same reaction conditions are shown in Figure 1b. Complex 2 is also a catalyst for four-electron O2-reduction (Figure 1b). It is also clearly seen from Figure 1b that the rate of reaction with [(PV-tmpa)CuII]2+ (1) is significantly greater than that with [(tmpa)CuII]2+ (2).
Figure 1.

(a) UV–vis spectral changes in four-electron reduction of O2 (11 mM) by Fc* (2.0 mM) with [(PV-tmpa)CuII]2+ (1) (0.040 mM) in the presence of CF3COOH (25 mM) in acetone at 298 K. (b) Time courses of absorbance at 780 nm due to Fc*+ with [(PV-tmpa)CuII]2+ (1) (red) and [(tmpa)CuII]2+ (2) (blue).
Time profiles for the absorbance at 780 nm due to Fc*+ formed by this four-electron O2-reduction chemistry by Fc* with various concentrations of [(PV-tmpa)CuII]2+ (1) in the presence of CF3COOH in O2-saturated acetone at 298 K are shown in Figure 2a, obeying pseudo-first-order kinetics (see inset of Figure 2a). The observed pseudo-first-order rate constants (kobs) are proportional to concentrations of 1 and 2 as shown in Figure 3b. The kobs values at a fixed concentration of 1 (0.040 mM) are constant with changes in the concentrations of O2 (Figure 3a) and CF3COOH (Figure 3b). Thus, the rate of formation of Fc*+ is given by eq 2,
| (2) |
where kcat is the second-order rate constant of the catalytic four-electron reduction of O2 by Fc* with 1 in the presence of CF3COOH in acetone at 298 K. The kcat value for 1 was thus determined to 8.6 × 102 M−1 s−1.
Figure 2.

(a) Time profiles of formation of Fc*+ monitored by absorbance at 780 nm (ε = 500 M−1 cm−1) in electron transfer oxidation of Fc* (2.0 mM) by O2 (11 mM), catalyzed by [(PV-tmpa)CuII]2+ (1) (0.020–0.12 mM) in the presence of CF3COOH (25 mM) in acetone at 298 K. Inset: First-order plots. (b) Plots of kobs vs [(PV-tmpa)CuII]2+ (1) (red) and [(tmpa)CuII]2+ (2) (blue).
Figure 3.

Plots of kobs vs (a) [O2] and (b) [CF3COOH] in electron-transfer oxidation of Fc* (2.0 mM) by O2, catalyzed by [(PV-tmpa)CuII]2+ (1)(red) and [(tmpa)CuII]2+ (2) (blue) (0.040 mM) in the presence of CF3COOH in acetone at 298 K.
The same kinetic equation (eq 1) was obtained when similar experiments and analyses were carried out for the catalyst [(tmpa)CuII]2+ (2) (see blue plots in Figures 2 and 3), leading to a kcat value for 2 determined to be 2.3 × 102 M−1 s−1, which is about one-fourth of the kcat value determined for 1. Because the catalytic rate is proportional to [1] and [Fc*] but it is constant with changes in concentrations of [CF3COOH] and [O2], the rate-determining step in the catalytic cycle can be concluded to be electron transfer from Fc* to 1 and 2, i.e., reduction of the catalysts from the CuII to CuI state, whereupon O2-binding and reaction could occur. In order to understand the reason why the catalytic reactivity of 1 is four times larger than that of 2, the one-electron reduction potentials of 1 and 2 were examined using cyclic voltammetry (vide infra).
Effects of CF3COOH on One-Electron Reduction Potentials of 1 and 2
Cyclic voltammograms of [(PV-tmpa) CuII]2+ (1) and [(tmpa)CuII]2+ (2) show reversible redox couples at −0.23 and −0.05 V vs SCE, respectively (Figure 4). Thus, 1 is more difficult to be reduced as compared with 2. Addition of CF3COONa to an acetone solution of 1 resulted in only slight change in the one-electron reduction potential. In the case of 2, however, the one-electron reduction potential is shifted from −0.05 V to −0.35 V vs SCE in the presence of CF3COONa (Figure 4b). This indicates that CF3COO− coordinates to 2 and increases the metal ion’s overall electron density (Scheme 1b), accounting for the large negative shift in reduction potential.61 A similar negative potential shift was observed for 2 in the presence of CF3COOH whereas no potential shift was observed for 1 under the same experimental conditions (Figure S4 in SI).
Figure 4.

Cyclic voltammograms of (a) [(PV-tmpa)CuII]2+ (1) and (b) [(tmpa)CuII]2+ (2) (1.0 mM) with CF3COONa (0 – 2.0 mM) in deaerated acetone containing TBAPF6 (0.10 M); scan rate 100 mV s−1.
Scheme 1.

ESI-MS analysis exhibits the strong corrdination of 2 with CF3COO− as detection of mass peak at m/z = 466.1 due to [(tmpa)CuII(CF3COO−)]+ (see Figure S5 in SI). The amido oxygen atom of 1 already coordinates to the Cu(II) center, disfavoring CF3COO− coordination (Scheme 1a). In contrast to the case of 2, no MS peak shift was observed by the addition of CF3COO−. Thus, the one-electron reduction potential of 1 becomes more positive in the presence of CF3COO− as compared with that of 2. This suggests that 1 can act as a stronger electron acceptor than 2 in the presence of CF3COO−.
The binding of the amido oxygen to the Cu(II) center of 1 is supported by the optimized structure by DFT calculations at the UCAM-B3LYP/6-311G(d) level of theory in Figure 6. The SOMO orbital is delocalized to the amide moiety. The SOMO level of 1 (−0.275 eV) is significantly higher than those of [(PV-tmpa) CuII]2+ isomers, where substituents are installed in positions 4 and 5 of the pyridine ring (−0.327 eV for 4-substituted isomer, −0.334 eV for 5-substituted isomer), and pyvalamido-unsubstituted complex (2) (−0.327 eV). These results indicate the stabilization by the coordination of the amide oxygen to the Cu(II) center of 1. It should be noted that the X-ray crystal structure of [(PV-tmpa) CuI]+ clearly shows the coordination of the amido O-atom.48b
Figure 6.

Difference absorption spectra observed in electron transfer from Fc* (2.0 mM) to [(PV-tmpa)CuII]2+ (1) (0.10 mM) (a) in deaerated and (b) O2-saturated acetone at 298 K. Black; 1st difference specturm. Red; final diffeernce spectrum. The spectra shown were obtained by subtraction of the final spectrum (as reference) from the observed spectra; the recovery of bleaching at 780 nm thus indicates formation of Fc*+.
Then, electron transfer from Fc* to [(PV-tmpa)CuII]2+ (1) was examined using the stopped flow technique as shown in Figure 6a, where the difference spectra obtained after the reaction indicates the formation of Fc*+ (λmax = 780 nm) as the recovery of bleaching by electron transfer from a large excess Fc* to 1 occurs. The rate of formation of Fc*+ obeyed first-order kinetics (see Figure S6 in SI) and the pseudo-first-order rate constant increased lienarly with increasing concentration of Fc*+ as shown in Figure 7. From the slope of the linear plot, the second-order rate constant for electron transfer from Fc* to 1 was determined to be (1.2 ± 0.1) × 103 M−1 s−1 in acetone at 298 K.
Figure 7.

Plots of psuedo-first-order rate constants vs [Fc*] in electron transfer from Fc* to 1 in deaerated (closed circles) and O2-saturated (open circles) acetone at 298 K.
In the presence of O2, electron transfer from Fc* to [(PV-tmpa) CuII]2+ (1) also occurs as shown in Figure 6b, where the formation of Fc*+ is accompanied by formation of the μ-1,2-peroxodicopper(II) complex, [{(PV-tmpa)CuII}2(O22−)]2+ (λmax = 515 nm).48 The second-order rate constant of electron transfer was determined from a linear plot of the psuedo-first-order rate constant vs [Fc*] shown in Figure 7 to be (1.3 ± 0.1) × 103 M−1 s−1, which agrees with the rate constant in the absence of O2 (vide supra).62 Because electron transfer from Fc* to 1 coincides with formation of the peroxo complex (Figure S6 in SI), as soon as the Cu(II) complex is reduced to the Cu(I) complex, this is rapidly converted to the peroxo dicopper(II) complex (vide infra).
The ket vlaue of electron transfer from Fc* to [(PV-tmpa) CuII]2+ (1) [(1.2 ± 0.1) × 103 M−1 s−1] is significantly smaller than the value found for 2 (1.1 × 105 M−1 s−1),46 because the one-electron reduction potential of 1 (−0.23 V vs SCE) is more negative than that of 2 (−0.05 V) as shown in Figure 4. In the presence of CF3COO−, the significant deceleration effect on the electron-transfer reduction of 2 with Fc* was observed by the addition of CF3COOH (ket = 2.3 × 102 M−1 s−1) (Figure 2b) and CF3COONa (ket = 2.2 × 102 s−1) (Figure S7 in SI).
Detection of Intermediates in Catalytic Four-Electron Reduction of O2 by Fc* with [(PV-tmpa)CuII]2+ (1)
Intermediates in the catalytic four-electron reduction of O2 by Fc* with 1 were examined step by step at low temperature (vide infra). Addition of Fc* to an O2-saturated acetone solution of 1 resulted in formation of the μ-1,2-peroxodicopper(II) complex, [{(PV-tmpa) CuII}2(O22−)]2+ (λmax = 515 nm)48 and Fc*+ (λmax = 780 nm) as shown in Figure 8a.63 The peroxo complex is formed via electron transfer from Fc* to [(PV-tmpa)CuII]2+ (1) (eq 1) as shown in eqs 4 and 5 as reported previously.48 The electron transfer is fast enough to observe the peroxo complex, because no further reaction of the peroxo complex occurred without CF3COOH at 213 K. The CuI complex ([(PV-tmpa)CuI]+) reacts with O2 to produce the superoxo complex, [(PV-tmpa)CuII(O2•−)]+ (eq 4), which was previously detected at very low temperature (148 K) in 2-methyltetrahdrofuran.48b However, we could not detect the copper(II)-superoxide adduct, because, as monitored by UV-Vis spectroscopy,48b formation of μ-1,2-peroxodicopper(II) complex, [{(PV-tmpa)CuII}2(O22−)]2+ is faster than the addition of O2 to the copper(I) species under the present conditions at 213 K.
| (4) |
Figure 8.

(a) Absorption spectra of 1 (0.10 mM) in O2-saturated acetone before (blue) and after (black) addition of Fc* (0.50 mM) at 213 K. (b) Absorption spectra of 1 (0.10 mM) in the presence of CF3COOH (0.10 mM) in O2-saturated acetone before (blue) and after (black) addition of Fc* (0.50 mM) at 213 K.
The superoxo complex is converted to the peroxo complex by further reaction with CuI complex (eq 5).48 In the presence of
| (5) |
one-equivalent of CF3COOH added to 1, however, no absorption band (λmax = 515 nm)48 due to [{(PV-tmpa)CuII}2(O22−)]2+ was observed although the formation of Fc*+ occurred, as shown in Figure 8b. This indicates that the peroxo complex is protonated by CF3COOH to produce the hydroperoxo complex ([(PV-tmpa) CuII(OOH)]+) and [(PV-tmpa)CuII]2+ (eq 6). The
| (6) |
hydroperoxo complex was not reduced by Fc* in the absence of excess CF3COOH, whereas [(PV-tmpa)CuII]2+ was reduced by Fc* to [(PV-tmpa)CuI]+, which is converted to [{(PV-tmpa) CuII}2(O22−)]2+ (eq 7) via the reactions in eqs 4 and 5. Then the overall reaction is given by eq 8.
| (7) |
| (8) |
In the presence of excess CF3COOH (10 mM), Fc* was fully converted to Fc*+ as shown in Figure 9. This indicates that proton-coupled electron transfer from Fc* to [(PV-tmpa) CuII(OOH)]+ may occur to regenerate [(PV-tmpa)CuII]2+ (1) (eq 9), providing for the overall catalytic oxidation of Fc* by excess O2 with 1.
| (9) |
Figure 9.

(a) UV-vis spectral changes observed in electron transfer from Fc* (0.50 mM) to [(PV-tmpa)CuII]2+ (1) (0.10 mM) in O2-saturated acetone at 213 K (black to orange). UV-vis spectral changes observed by addition of CF3COOH (10 mM) (orange to red). UV-vis spectral changes observed in catalytic reduction of O2 monitored by the formation of Fc*+ at 780 nm (red to purple). (b) Absorption time profiles at 515 nm due to the μ-1,2-peroxo complex and at 780 nm due to Fc*+.
Copper Hydroperoxo Complexes
The hydroperoxo complex [(PV-tmpa)CuII(OOH)]+ can also be produced by the reaction of [(PV-tmpa)CuII]2+ (1) with H2O2 in the presence of base (Me4NOH) in acetone at 213 K (eq 10) as shown in Figure 10a. The absorption maxima at 398, 760 nm shoulder at 640 nm are assigned to [(PV-tmpa)CuII(OOH)]+. The ligand-to-metal charge-transfer absorption at 398 nm is typical for what is observed for other ligand-CuII-hydroperoxo complexes.47,64–67 Overall, these absorption bands are somewhat blue-shifted as compared with those known already known for [(tmpa)CuII(OOH)]+,47 and also observed here, see Figure 10b.
| (10) |
Figure 10.

(a) UV-vis spectral changes observed (blue to green) in the addition of H2O2 and Me4NOH to an acetone solution of (a) [(PV-tmpa) CuII]2+ (1) (0.50 mM) and (b) [(tmpa)CuII]2+ (2) (0.50 mM) in deaerated acetone at 213 K.
The formation of [(PV-tmpa)CuII(OOH)]+ was also confirmed by EPR as shown in Figure 11. The EPR spectrum of [(PV-tmpa)CuII(OOH)]+ produced by the reaction of [(PV-tmpa) CuII]2+ with HO2− in acetone (part c) with g⊥ = 2.21, |A⊥| = 135 G, g// = 2.02, |A//| = 60 G is virtually the same as that produced by the reaction of Fc* with [(PV-tmpa)CuII]2+ in the presence of CF3COOH in O2-saturated acetone (part b), but it is different from that of [(PV-tmpa)CuII]2+ itself (part a) (g⊥ = 2.21, |A⊥| = 128 G, g// = 2.03, |A//| = 86 G).64 The EPR spectrum of [(tmpa)CuII(OOH)]+ is also different from that observed for [(tmpa)CuII]+ (Figure S8 in SI).
Figure 11.

EPR spectra of (a) [(PV-tmpa)CuII]2+ (1)(0.50 mM) observed at 77 K, (b) [(PV-tmpa)CuII(OOH)]+ produced by reaction of Fc* (0.50 mM) with 1 (0.50 mM) in the presence of CF3COOH (0.50 mM) in O2-saturated acetone at 213 K and observed at 77 K. (c) [(PV-tmpa)CuII(OOH)]+ produced by the reaction of 1 (0.50 mM) with H2O2 (1.0 mM) and Me4NOH in deareated acetone at 213 K, observed at 77 K.47c
When Fc* was added to an acetone solution of [(PV-tmpa) CuII(OOH)]+, no electron transfer from Fc* to [(PV-tmpa) CuII(OOH)]+ occurred, as shown in Figure 12. In the presence of excess CF3COOH, however, proton coupled electron transfer (PCET) from Fc* to [(PV-tmpa)CuII(OOH)]+ occurred, leading to the catalytic two-electron reduction of H2O2 by Fc*. Similar UV-vis spectral changes were observed with [(tmpa)CuII(OOH)]+ as shown in Figure S9, however where the rate of PCET reduction of H2O2 by Fc* with 2 was much slower than the rate with 1.
Figure 12.

(a) UV-vis spectral changes and (b) time profile of absorbance at 780 nm observed upon addition of hydrogen peroxide (5.0 mM) to [(PV-tmpa) CuII]2+ (1) (0.50 mM) and Me4NOH (0.50 mM) (blue to red) (first arrow), followed by addition of Fc* (0.50 mM) (red to purple) (second arrow) and then CF3COOH (25 mM) (purple to orange) (third arrow) in deareated acetone at 233 K.
Catalytic Two-Electron Reduction of H2O2 by Fc* with [(PV-tmpa)CuII]2+ (1) and CF3COOH
As mentioned in the Introduction, it is of considerable interest to also design or develop catalysts and elucidate mechanisms for hydrogen peroxide to water reaction chemistry. The two-electron reduction of H2O2 by Fc* ocurred with 1 in the presence of CF3COOH in acetone at 298 K. The stoichiometry of the reaction was confirmed as given by eq 11. The rate of formation of Fc*+ for the
| (11) |
H2O2 reduction to water in the presence of a large excess Fc* and CF3COOH obeyed first-order kinetics (Figure 13a). This means the reaction rate is proportional to [H2O2]. The pseudo-first-order rate constant was proportional to the concentration of 1 (Figure 13b). This suggests that 1 efficiently catalyzes the two-electron reduction of H2O2 by Fc*. The pseudo-first-order rate constant at a fixed concentration of 1 was constant with changes in concentrations of CF3COOH (Figure S10 in SI). Thus, the rate of formation of Fc*+ in the catalytic two-electron reduction of H2O2 by Fc* with 1 is given by eq 12. From this, the kcat value of
| (12) |
the catalytic two-electron reduction of H2O2 by Fc* with 1 was determined to be 4.4 × 104 M−1 s−1.
Figure 13.

(a) Rise time profiles of absorbance at 780 nm in the catalytic reduction of H2O2 (0.10 mM) by Fc* (2.0 mM) with [(PV-tmpa)CuII]2+ (1) (0 – 0.10 mM) in the presence of CF3COOH (25 mM) in deareated acetone at 298 K. (b) Plot of kobs vs [[(PV-tmpa)CuII]2+].
This value is much larger than the kcat value determined for the catalytic four-electron reduction of O2 by Fc* with [(PV-tmpa) CuII]2+ (1) (vide supra). This ensures the one-step four-electron reduction of O2 by Fc* with 1 in which once electron transfer from Fc* to 1 occurs, all the subsequent reactions are much faster, leading all the way to the four-electron reduction of O2 to water. The finding that there is no dependence of kcat on the concentration of Fc* or CF3COOH is electron transfer from Fc* to 1 (vide supra), the much faster reduction process for H2O2 by Fc* with catalyst 1 and CF3COOH must occur via the Cu(II) rather than the Cu(I) complex. Thus, we conclude that coordination of H2O2 to the Cu(II) center of 1 to produce the hydroperoxo complex ([(PV-tmpa)CuII(OOH)]+) is rate-determining. When this eventually forms, it undergoes rapid PCET reduction in the presence of Fc* and CF3COOH (eqs 9 and 10).
For the case of [(tmpa)CuII]2+ (2) as catalyst for H2O2 reduction to water, the kinetics are quite different than those for [(PV-tmpa)CuII]2+ (1) as shown in Figure 14. The rate of formation of Fc*+ obeyed zeroth-order kinetics with respect to the concentration of H2O2, (Figures 14a and 14b). The zeroth-order rate constant (kobs(0)) is proportional to concentrations of Fc*+ (Figure 14c) and CF3COOH (Figure 15d). The kcat value of the catalytic two-electron reduction of H2O2 by Fc* with 2 was determined to be 1.3 × 104 M−2 s−1 as obtained from the slope of the Figure 14c plot and the concentrations used for [(PV-tmpa) CuII]2+(0.040 mM) and CF3COOH (25 mM). Thus, the catalytic two-electron reduction of H2O2 by Fc* is described by eq 13. Thus, in this case, the coordination of H2O2 to the Cu(II)
| (13) |
center of [(tmpa)CuII]2+ (2) is not the rate-determining step in contrast to the case of [(PV-tmpa)CuII]2+ (1) (vide supra). In the latter case, the ligation step is inhibited by the presence of the amido oxygen, as was also observed for triflouroacetate binding (vide supra). This is not true for 2; H2O2 coordination and deprotonation is fast, and the rate-determining step may be the PCET reduction of the hydroperoxo complex of 2 (i.e., [(PV-tmpa) CuII(OOH)]+) by Fc* where the rate is proportional to the concentrations of Fc* and CF3COOH (eq 13). Thus, as observed for many cases, small changes in the ligand structure and/or environment in proximity to the metal center can have profound effects on the chemistry, here, by effecting a change in mechanism for hydrogen peroxide reduction to water for catalyst 1 vs 2.
Figure 14.

(a) Kinetic results on the catalytic reduction of H2O2 (0.15 mM) by Fc* (2.0 mM) with [(tmpa)CuII]2+ (2) (0 – 0.10 mM) in the presence of CF3COOH (25 mM) in deareated acetone at 298 K. (b) Plot of kobs(0) vs concentration of H2O2. Conditions: 2 (0.040 mM), Fc* (2.0 mM), H2O2 (0.05 – 0.20 mM), CH3COOH (25 mM) in deareated acetone (c) Plot of kobs(0) vs concentration of Fc*. Conditions: 2 (0.040 mM), Fc* (2.0 – 5.0 mM), H2O2 (0.15 mM), CH3COOH (25 mM) in deareated acetone (d) Plot of kobs(0) vs concentraiton of CF3COOH. Conditions: 2 (0.040 mM), Fc* (2.0 mM), H2O2 (0.15 mM), CH3COOH (25 – 45 mM) in deareated acetone.
Conclusion
The reaction mechanism of the four-electron reduction of O2 by Fc* with [(PV-tmpa)CuII]2+ (1) in the presence of CF3COOH in acetone at RT is summarized as shown in Scheme 2. First, electron transfer from Fc* to 1 occurs to produce Fc*+ and the corresponding CuI complex and this is the rate determining step (r.d.s.). This is followed by the fast reaction of the CuI complex with O2 to produce a superoxo-copper(II) species and which undergoes another very rapid reduction and coordination by CuI to produce the peroxo-dicopper(II) complex.29 Both the superoxo and peroxo complexes have been previously characterized.48 The peroxo complex is protonated to produce the hydroperoxo complex, which undergoes PCET reduction by Fc* and acid to give water, accompanied by regeneration of 1.
Scheme 2.

For the catalyst [(tmpa)CuII]2+ (2), which effects the same over four-electron four-proton reduction of O2 to water, Fc* reduction of 2 to its corresponding CuI complex is similarly rate determining. However, the coordination of the amido oxygen to the Cu(II) center of 1 significantly inhibits coordination of bulky CF3COO− to the Cu(II) center, whereas this trifluoroacetate anion readily binds to the Cu(II) center of 2. In contrast to the bulky CF3COO−, O2 and HO2− may easily access the coordination sphere both 1 and 2. Overall, electron transfer from Fc* to 2 is thus by comparison slowed, since the CF3COO− coordination shifts the redox potential of 2 more negative, making it harder to reduce. As a result, more efficient catalytic four-electron reduction of O2 by Fc* occurred with 1 as compared to 2 in the presence of CF3COOH.
The catalytic two-electron reduction of H2O2 by Fc* with [(PV-tmpa)CuII]2+ (1) and [(tmpa)CuII]2+ (2) in the presence of CF3COOH in acetone occurs much more rapidly than the four-electron-reduction of O2 by Fc* with 1 and 2; this is an important attribute because if H2O2 or a metal-(hydro)peroxo species is or would be an intermediate, than, the overall efficiency of the four-electron process is not compromised. The catalytic reactivity of 1 is also higher than that of 2 in this two-electron reduction of H2O2 (Figures 13a vs Fig 14a). The rate-determining step in the two-electron reduction of O2 by Fc* with 1 in the presence of CF3COOH is the coordination and deprotonation of H2O2, which is followed by fast PCET reduction of the Cu(II)-OOH complex, because the coordination of the ligand amido oxygen to the Cu(II) center of 1 hampers the coordination of HO2−. In the case of 2 in which the coordination site is open, HO2− readily coordinates to the Cu(II) center to produce the Cu(II)-OOH complex, and now the PCET reduction becomes the rate-determining step.
In summary, introduction of a pivalamido group on the tmpa ligand periphery resulted in the enhancement of the catalytic reactivity of the Cu(II) complex of PV-tmpa in both four-electron four-proton reduction of O2 and two-electron two-proton reduction dioxygen to H2O2 as compared with [(tmpa)CuII]2+ as catalyst. In the quest for efficient and selective dioxygen (a) catalytic four-electron four-proton and (b) two-electron two-proton reduction chemistry, as well as (c) efficient catalytic two-electron two-proton reduction of hydrogen peroxide, we continue to use ligand design and variations for the generation and study of new copper complex catalysts and survey of their reactivity patterns along with elucidation of their mechanisms of action.
Supplementary Material
Figure 5.

Optimized structure of [(PV-tmpa)CuII]2+ obtained by DFT calculations with the UCAM-B3LYP density-functional and the 6-311G(d) basis set. The SUMO obrital is shown in the right panel.
ACKNOWLEDGMENT
This work was supported by Grants-in-Aid (No. 20108010 to S.F. and 23750014 to K.O.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and by KOSEF/MEST through the WCU Project (R31-2008-000-10010-0). K.D.K. also acknowledges support from the USA National Institutes of Health.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Kinetic analyses (Figures S1 – S10) and full author list for ref 55 (S11). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]; (b) Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pereira MM, Santana M, Teixeira M. Biochim. Biophys. Acta. 2001;1505:185. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]; (b) Winter M, Brodd R. J. Chem. Rev. 2004;104:4245. doi: 10.1021/cr020730k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ferguson-Miller S, Babcock GT. Chem. Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]; (b) Hosler JP, Ferguson-Miller S, Mills DA. Annu. Rev. Biochem. 2006;75:165. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kaila VRI, Verkhovsky MI, Wikström M. Chem. Rev. 2010;110:7062. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 4.(a) Christoph von Ballmoos C, Lachmann P, Gennis RB, Ädelroth P, Brzezinski P. Biochemistry. 2012;51:4507. doi: 10.1021/bi300132t. [DOI] [PubMed] [Google Scholar]; (b) Belevich I, Verkhovsky MI. Antioxid. Redox Signaling. 2008;10:1. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- 5.(a) Collman JP, Devaraj NK, Decréau RA, Yang Y, Yan Y-L, Ebina W, Eberspacher TA, Chidsey CED. Science. 2007;315:1565. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Collman JP, Decréau RA, Lin H, Hosseini A, Yang Y, Dey A, Eberspacher TA. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7320. doi: 10.1073/pnas.0902285106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Collman JP, Ghosh S, Dey A, Decréau RA, Yang Y. J. Am. Chem. Soc. 2009;131:5034. doi: 10.1021/ja9001579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Rosenthal J, Nocera DG. Acc. Chem. Res. 2007;40:543. doi: 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]; (b) Chang CJ, Loh Z-H, Shi C, Anson FC, Nocera DG. J. Am. Chem. Soc. 2004;126:10013. doi: 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]; (c) Dogutan DK, Stoian SA, McGuire R, Schwalbe M, Teets TS, Nocera DG. J. Am. Chem. Soc. 2011;133:131. doi: 10.1021/ja108904s. [DOI] [PubMed] [Google Scholar]; (d) Teets TS, Cook TR, McCarthy BD, Nocera DG. J. Am. Chem. Soc. 2011;133:8114. doi: 10.1021/ja201972v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kadish KM, Shen J, Frémond L, Chen P, El Ojaimi M, Chkounda M, Gros CP, Barbe J-M, Ohkubo K, Fukuzumi S, Guilard R. Inorg. Chem. 2008;47:6726. doi: 10.1021/ic800458s. [DOI] [PubMed] [Google Scholar]; (b) Kadish KM, Frémond L, Shen J, Chen P, Ohkubo K, Fukuzumi S, El Ojaimi M, Gros CP, Barbe J-M, Guilard R. Inorg. Chem. 2009;48:2571. doi: 10.1021/ic802092n. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kadish KM, Frémond L, Ou Z, Shao J, Shi C, Anson FC, Burdet F, Gros CP, Barbe JM, Guilard R. J. Am. Chem. Soc. 2005;127:5625. doi: 10.1021/ja0501060. [DOI] [PubMed] [Google Scholar]; (b) Ou Z, Lü A, Meng D, Huang S, Fang Y, Lu G, Kadish KM. Inorg. Chem. 2012;51:8890. doi: 10.1021/ic300886s. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hatay I, Su B, Li F, Méndez MA, Khoury T, Gros CP, Barbe J-M, Ersoz M, Samec Z, Girault HH. J. Am. Chem. Soc. 2009;131:13453. doi: 10.1021/ja904569p. [DOI] [PubMed] [Google Scholar]; (b) Partovi-Nia R, Su B, Li F, Gros CP, Barbe J-M, Samec Z, Girault HH. Chem.–Eur. J. 2009;15:2335. doi: 10.1002/chem.200801807. [DOI] [PubMed] [Google Scholar]; (c) Hatay I, Su B, Méndez MA, Corminboeuf C, Khoury T, Gros CP, Bourdillon M, Meyer M, Barbe J-M, Ersoz M, Zális S, Samec Z, Girault HH. J. Am. Chem. Soc. 2010;132:13733. doi: 10.1021/ja103460p. [DOI] [PubMed] [Google Scholar]; (d) Su B, Hatay I, Trojánek A, Samec Z, Khoury T, Gros CP, Barbe J-M, Daina A, Carrupt P-A, Girault HH. J. Am. Chem. Soc. 2010;132:2655. doi: 10.1021/ja908488s. [DOI] [PubMed] [Google Scholar]
- 10.(a) Chen R, Li H, Chu D, Wang G. J. Phys. Chem. C. 2009;113:20689. [Google Scholar]; (b) Shi Z, Zhang J. J. Phys. Chem. C. 2007;111:7084. [Google Scholar]
- 11.Bakac A. Inorg. Chem. 2010;49:3584. doi: 10.1021/ic9015405. [DOI] [PubMed] [Google Scholar]
- 12.Vielstich W, Lamm A, Gasteiger HA. Handbook of fuel cells: fundamentals, technology, and applications. Chichester, UK; Hoboken, NJ: Wiley; 2003. [Google Scholar]
- 13.(a) Zagal JH, Griveau S, Silva JF, Nyokong T, Bedioui F. Coord. Chem. Rev. 2010;254:2755. [Google Scholar]; (b) Li ZP, Liu BH. J. Appl. Electrochem. 2010;40:475. [Google Scholar]; (c) Zagal JH, Gulppi M, Isaacs M, Cárdenas-Jirón G, Aguirre MJ. Electrochim. Acta. 1998;44:1349. [Google Scholar]
- 14.Li W, Yu A, Higgins DC, Llanos BG, Chen Z. J. Am. Chem. Soc. 2010;132:17056. doi: 10.1021/ja106217u. [DOI] [PubMed] [Google Scholar]
- 15.(a) Gewirth AA, Thorum MS. Inorg. Chem. 2010;49:3557. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]; (b) Stambouli AB, Traversa E. Renew. Sust. Energy Rev. 2002;6:295. [Google Scholar]; (c) Markovic NM, Schmidt TJ, Stamenkovic V, Ross PN. Fuel Cells. 2001;1:105. [Google Scholar]; (d) Steele BCH, Heinzel A. Nature. 2001;414:345. doi: 10.1038/35104620. [DOI] [PubMed] [Google Scholar]
- 16.(a) Solomon EI, Ginsbach JW, Heppner DE, Kieber-Emmons MT, Kjaergaard CH, Smeets PJ, Tian L, Woertink JS. Faraday Discuss. 2011;148:11. doi: 10.1039/c005500j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 17.Farver O, Pecht I. In: Multi-Copper Oxidases. Messerschmidt A, editor. Singapore: World Scientific; 1997. [Google Scholar]
- 18.(a) Djoko KY, Chong LX, Wedd AG, Xiao Z. J. Am. Chem. Soc. 2010;132:2005. doi: 10.1021/ja9091903. [DOI] [PubMed] [Google Scholar]; (b) Kosman DJ. J. Biol. Inorg. Chem. 2010;15:15. doi: 10.1007/s00775-009-0590-9. [DOI] [PubMed] [Google Scholar]
- 19.Tan Y, Deng W, Li Y, Huang Z, Meng Y, Xie Q, Ma M, Yao S. J. Phys. Chem. B. 2010;114:5016. doi: 10.1021/jp100922t. [DOI] [PubMed] [Google Scholar]
- 20.Farver O, Tepper AWJW, Wherland S, Canters GW, Pecht I. J. Am. Chem. Soc. 2009;131:18226. doi: 10.1021/ja908793d. [DOI] [PubMed] [Google Scholar]
- 21.Riva S. Trends Biotechnol. 2006;24:219. doi: 10.1016/j.tibtech.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Ward AL, Elbaz L, Kerr JB, Arnold J. Inorg. Chem. 2012;51:4694. doi: 10.1021/ic2026957. [DOI] [PubMed] [Google Scholar]
- 23.(a) Zhang J, Anson FC. J. Electroanal. Chem. 1992;341:323. [Google Scholar]; (b) Zhang J, Anson FC. J. Electroanal. Chem. 1993;348:81. [Google Scholar]
- 24.(a) Zhang J, Anson FC. Electrochim. Acta. 1993;38:2423. [Google Scholar]; (b) Lei Y, Anson FC. Inorg. Chem. 1994;33:5003. [Google Scholar]
- 25.Weng YC, Fan F-RF, Bard AJ. J. Am. Chem. Soc. 2005;127:17576. doi: 10.1021/ja054812c. [DOI] [PubMed] [Google Scholar]
- 26.Thorum MS, Yadav J, Gewirth AA. Angew. Chem. Int. Ed. 2009;48:165. doi: 10.1002/anie.200803554. [DOI] [PubMed] [Google Scholar]
- 27.(a) McCrory CCL, Ottenwaelder X, Stack TDP, Chidsey CED. J. Phys. Chem. A. 2007;111:12641. doi: 10.1021/jp076106z. [DOI] [PubMed] [Google Scholar]; (b) McCrory CCL, Devadoss A, Ottenwaelder X, Lowe RD, Stack TDP, Chidsey CED. J. Am. Chem. Soc. 2011;133:3696. doi: 10.1021/ja106338h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Pichon C, Mialane P, Dolbecq A, Marrot J, Riviere E, Keita B, Nadjo L, Secheresse F. Inorg. Chem. 2007;46:5292. doi: 10.1021/ic070313w. [DOI] [PubMed] [Google Scholar]; (b) Dias VLN, Fernandes EN, da Silva LMS, Marques EP, Zhang J, Marques ALB. J. Power Sources. 2005;142:10. [Google Scholar]; (c) Losada J, del Peso I, Beyer L. Inorg. Chim. Acta. 2001;321:107. [Google Scholar]
- 29.(a) Thorseth MA, Tornow CE, Tse ECM, Gewirth AA. Coord. Chem. Rev. 2013;257:130. [Google Scholar]; (b) Thorseth MA, Letko CS, Rauchfuss TB, Gewirth AA. Inorg. Chem. 2011;50:6158. doi: 10.1021/ic200386d. [DOI] [PubMed] [Google Scholar]
- 30.(a) Fukuzumi S, Okamoto K, Gros CP, Guilard R. J. Am. Chem. Soc. 2004;126:10441. doi: 10.1021/ja048403c. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S, Okamoto K, Tokuda Y, Gros CP, Guilard R. J. Am. Chem. Soc. 2004;126:17059. doi: 10.1021/ja046422g. [DOI] [PubMed] [Google Scholar]
- 31.(a) Fukuzumi S, Mochizuki S, Tanaka T. Inorg. Chem. 1989;28:2459. [Google Scholar]; (b) Fukuzumi S, Mochizuki S, Tanaka T. Inorg. Chem. 1990;29:653. [Google Scholar]; (c) Fukuzumi S, Mochizuki S, Tanaka T. J. Chem. Soc. Chem. Commun. 1989;391 [Google Scholar]; (d) Fukuzumi S. Chem. Lett. 2008;37:808. [Google Scholar]
- 32.(a) Olaya AJ, Schaming D, Brevet P-F, Nagatani H, Zimmermann T, Vanicek J, Xu H-J, Gros CP, Barbe J-M, Girault HH. J. Am. Chem. Soc. 2012;134:498. doi: 10.1021/ja2087322. [DOI] [PubMed] [Google Scholar]; (b) Peljo P, Murtomäki L, Kallio T, Xu H-J, Meyer M, Gros CP, Barbe J-M, Girault HH, Laasonen K, Kontturi K. J. Am. Chem. Soc. 2012;134:5974. doi: 10.1021/ja3004914. [DOI] [PubMed] [Google Scholar]
- 33.Halime Z, Kotani H, Li Y, Fukuzumi S, Karlin KD. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13990. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuzumi S, Tahsini L, Lee Y-M, Ohkubo K, Nam W, Karlin KD. J. Am. Chem. Soc. 2012;134:7025. doi: 10.1021/ja211656g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Karlin KD, Zuberbuhler ADIn. In: Bioinorganic Catalysis. 2nd ed. Reedijk J, Bouwman E, editors. New York: Marcel Dekker; 1999. pp. 469–534. [Google Scholar]; (b) Hatcher LQ, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 36.Kitajima N, Moro-oka Y. Chem. Rev. 1994;94:737. [Google Scholar]
- 37.(a) Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (b) Solomon EI, Szilagyi RK, De-Beer George S, Basumallick L. Chem. Rev. 2004;104:419. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]; (c) Chen P, Solomon EI. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13105. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Itoh S, Fukuzumi S. Acc. Chem. Res. 2007;40:592. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]; (b) Itoh S, Fukuzumi S. Bull. Chem. Soc. Jpn. 2002;75:2081. [Google Scholar]
- 39.(a) Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (b) Tolman WB. Acc. Chem. Res. 1997;30:227. [Google Scholar]; (c) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (d) Que L, Jr, Tolman WB. Angew. Chem. Int. Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 40.(a) Klinman JP. Chem. Rev. 1996;96:2541. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; (b) Klinman JP. J. Biol. Chem. 2006;281:3013. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 41.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 42.Rodgers CJ, Blanford CF, Giddens SR, Skamnioti P, Armstrong FA, Gurr SJ. Trends Biotechnol. 2010;28:63. doi: 10.1016/j.tibtech.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Rolff M, Schottenheim J, Decker H, Tuczek F. Chem. Soc. Rev. 2011;40:4077. doi: 10.1039/c0cs00202j. [DOI] [PubMed] [Google Scholar]
- 44.(a) Kim E, Chufán EE, Kamaraj K, Karlin KD. Chem. Rev. 2004;104:1077. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]; (b) Chufán EE, Puiu SC, Karlin KD. Acc. Chem. Res. 2007;40:563. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]; (c) Karlin KD, Kaderli S, Zuberbühler AD. Acc. Chem. Res. 1997;30:139. [Google Scholar]
- 45.Tahsini L, Kotani H, Lee Y-M, Cho J, Nam W, Karlin KD, Fukuzumi S. Chem. Eur. J. 2012;18:1084. doi: 10.1002/chem.201103215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuzumi S, Kotani H, Lucas HR, Doi K, Suenobu T, Peterson RL, Karlin KD. J. Am. Chem. Soc. 2010;132:6874. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew. Chem. Int. Ed. 1998;37:798. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. Yamaguchi S, Wada A, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Chem. Lett. 2004;33:1556. doi: 10.1021/ic0496572. (c) Note, for [(PV-tmpa) CuII(OOH)]+, Masuda47d obtained an A⊥ value of 95 G for an EPR spectrum of this species in MeCN solvent; we observed a very similar EPR spectrum when this hydroperoxo complex is generated and recorded in the MeCN under the same experimental conditions. Yamaguchi S, Masuda H. Sci. Technol. Adv. Mater. 2005;6:34.
- 48.(a) Yamaguchi S, Wada A, Funahashi Y, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Eur. J. Inorg. Chem. 2003;24:4378. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]; (b) Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J. Am. Chem. Soc. 2011;133:1702. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.For the catalytic reduction of O2 by chemical reductants with cobalt complexes, see: refs 7 and 30. Fukuzumi S, Mandal S, Mase K, Ohkubo K, Park H, Benet-Buchholz J, Nam W, Llobet A. J. Am. Chem. Soc. 2012;134:9906. doi: 10.1021/ja303674n. Mase K, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2013;135:2800–2808. doi: 10.1021/ja312199h.
- 50.Fukuzumi S, Yamada Y, Karlin KD. Electrochim. Acta. 2012;82:493. doi: 10.1016/j.electacta.2012.03.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.(a) Yamada Y, Fukunishi Y, Yamazaki S, Fukuzumi S. Chem. Commun. 2010;46:7334. doi: 10.1039/c0cc01797c. [DOI] [PubMed] [Google Scholar]; (b) Yamada Y, Yoshida S, Honda T, Fukuzumi S. Energy Environ. Sci. 2011;4:2822. [Google Scholar]; (c) Shaegh SAM, Nguyen N-T, Ehteshami SMM, Chan SH. Energy Environ. Sci. 2012;5:8225. [Google Scholar]
- 52.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 5th ed. Oxford: Butterworth-Heinemann; 2003. [Google Scholar]
- 53.(a) Mair RD, Graupner AJ. J. Anal. Chem. 1964;36:194. [Google Scholar]; (b) Fukuzumi S, Kuroda S, Tanaka T. J. Am. Chem. Soc. 1985;107:3020. [Google Scholar]
- 54.Mann CK, Barnes KK. Electrochemical Reactions in Nonaqueous Systems. New York: Marel Dekker; 1990. [Google Scholar]
- 55.Frisch MJ, et al. Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc; 2009. (Full author list is shown in SI) [Google Scholar]
- 56.(a) Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 57.(a) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270. [Google Scholar]; (b) Curtiss LA, McGrath MP, Blaudeau J-P, Davis NE, Binning RC, Jr, Radom L. J. Chem. Phys. 1995;103:6104. [Google Scholar]
- 58.(a) Yanai T, Tew DP, Handy NC. Chem. Phys. Lett. 2004;393:51. [Google Scholar]; (b) Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K. J. Chem. Phys. 2004;120:8425. doi: 10.1063/1.1688752. [DOI] [PubMed] [Google Scholar]
- 59.Takaichi J, Ohkubo K, Sugimoto H, Nakano M, Usa D, Maekawa H, Fujieda N, Nishiwaki N, Seki S, Fukuzumi S, Itoh S. Dalton Trans. 2013;42:2438. doi: 10.1039/c2dt32413j. [DOI] [PubMed] [Google Scholar]
- 60.Dennington R, II, Keith T, Millam J, Eppinnett K, Hovell WL, Gilliland R. Gaussview. Shawnee Mission, KS: Semichem, Inc; 2003. [Google Scholar]
- 61. No potential shift was observed by the addition of NaClO4 under the similar experimental conditions (see Figure S3 in SI)
- 62. Electron transfer from Fc* to 1 is an equilibrium process and thus the intercept in Figure 8 corresponds to back electron transfer from the Cu(I) complex to Fc*+. In the presence of O2, electron transfer becomes irreversible because of the very fast reaction of the Cu(I) complex with O2 in comparison to the back electron transfer.
- 63. The shoulder absorption around 400 nm is assigned to Cu(II) hydroperoxo species, which is partially formed by the reaction with residual water in acetone without CF3COOH.47
- 64.The Cu(II)-OOH intermediate was also detected by UV-vis absorption spectral measurements as a shoulder absorption around 400 nm (Figure 8), which agrees with the absorbance of Cu(II)-OOH formed with H2O2 and Me4NOH (Figure 10).
- 65.(a) Kunishita A, Scanlon JD, Ishimaru H, Honda K, Ogura T, Suzuki M, Cramer CJ, Itoh S. Inorg. Chem. 2008;47:8222. doi: 10.1021/ic800845h. [DOI] [PubMed] [Google Scholar]; (b) Kunishita A, Kubo M, Ishimaru H, Ogura T, Sugimoto H, Itoh S. Inorg. Chem. 2008;47:12032. doi: 10.1021/ic801568g. [DOI] [PubMed] [Google Scholar]
- 66.Kim S, Saracini C, Siegler MA, Drichko N, Karlin KD. Inorg. Chem. 2012;51:12603. doi: 10.1021/ic302071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.The molar extinction coefficient value for [(PV-tmpa)CuII(OOH)]+ is apparently lower than that of [(tmpa)CuII(OOH)]+, based on the EPR quantification carried out on the hydroperoxo species formed with H2O2 and Me4NOH.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.