

Abstract
Loss of function of galactosylceramidase lysosomal activity causes demyelination and vulnerability of various neuronal populations in Krabbe disease. Psychosine, a lipid-raft-associated sphingolipid that accumulates in this disease, is thought to trigger these abnormalities. Myelin-free in vitro analyses showed that psychosine inhibited fast axonal transport through the activation of axonal PP1 and GSK3β in the axon. Abnormal levels of activated GSK3β and abnormally phosphorylated kinesin light chains were found in nerve samples from a mouse model of Krabbe disease. Administration of GSK3β inhibitors significantly ameliorated transport defects in vitro and in vivo in peripheral axons of the mutant mouse. This study identifies psychosine as a pathogenic sphingolipid able to block fast axonal transport and is the first to provide a molecular mechanism underlying dying-back degeneration in this genetic leukodystrophy.
Introduction
Krabbe disease is an autosomal-recessive leukodystrophy caused by the loss of function of galactosylceramidase (GALC), a lysosomal enzyme that catabolizes galactosylated sphingolipids. Psychosine (galactosyl-sphingosine) is a lipid-raft-associated neurotoxin (White et al., 2009) known to accumulate and to trigger pathogenesis in Krabbe patients (Igisu and Suzuki, 1984; Suzuki, 1998). The diffuse damage to myelin sheaths, a hallmark of the disease, is thought to result from dysfunction of both oligodendrocytes and Schwann cells consequent to the accumulation of psychosine (Tanaka et al., 1989; Jatana et al., 2002). Krabbe patients also show progressive and severe neurological deficiencies in both the central and peripheral nervous systems. Symptoms include muscle rigidity and atrophy, ataxic movement, hearing and vision defects, and rapid loss of cognitive and motor skills (Duffner et al., 2009; Escolar et al., 2009). The pathogenic mechanisms mediating these deficiencies are unknown, but the disease presents as a dying-back degeneration of mutant axons (Castelvetri et al., 2011) and involves dysfunction of selective neuronal populations (Krabbe, 1916).
Axonal dystrophy in conjunction with increased phosphatase (PP1 and PP2A) and caspase-3 activity lead to defects in cytoskeletal components in the twitcher mouse, a well characterized animal model of Krabbe disease (Castelvetri et al., 2011; Smith et al., 2011; Cantuti-Castelvetri et al., 2012). The selectivity (i.e., the disparity in which neuronal and axonal populations are affected), the dying-back pattern affecting axons, and the characteristic appearance of axonal swellings point to defects in fast axonal transport (FAT). The physiological consequences of deficient FAT have been established for several neurological disorders (Pigino et al., 2003; Morfini et al., 2004, 2006, 2009a,b; Ori-McKenney et al., 2010). However, the pathogenic role of defective FAT in a leukodystrophy such as Krabbe disease had not been addressed previously.
This study demonstrates that psychosine inhibits FAT and requires the activation of axonal phosphotransferases PP1 and GSK3β and the abnormal phosphorylation of the molecular motor protein kinesin light chains (KLCs). In vitro and in vivo neuroprotection experiments demonstrated the pathogenic relevance of this pathway in neurodegeneration in Krabbe disease.
Materials and Methods
Animals.
Breeder twitcher heterozygous mice (C57BL/6J, twi/+) were originally purchased from The Jackson Laboratory and maintained under standard housing conditions. Protocols were according to animal care and use committee protocols of our institution. The twitcher mutation was analyzed as described previously (Sakai et al., 1996). Male and female twitcher mice were used indistinctly.
Cell cultures.
Cortical neurons were prepared as described previously (Kaech and Banker, 2006). Briefly, cortices were isolated from E16 C57BL embryos. Tissue was minced, treated with 0.25% trypsin, and passed through a fire-polished pipette. Cells were then plated in DMEM (Mediatech) supplemented with 10% FBS for 2 h and further incubated in neurobasal medium supplemented with B27. NSC34 cells were grown in 5% FBS DMEM, l-glutamine (Invitrogen), and penicillin/streptomycin (Invitrogen). NSC34 cells were serum deprived for 12 h before treatments. Glass coverslips were precleaned with nitric acid, water, and ethanol and then autoclaved before use. Lipids were purchased from Sigma. Enantiomeric psychosine was obtained from M.S. and resuspended in ethanol. Psychosine was added to in vitro cell systems at 1–5 μm, which falls within the range of concentrations measured in the central and peripheral nervous systems of the twitcher mouse (1–10 nmol/g tissue or 1–10 μm; Kobayashi et al., 1987; White et al., 2009). Okadaic acid (Calbiochem) was added together with psychosine, whereas TDZD8 or L803 inhibitors (Calbiochem) (Leopold et al., 1992; Chen et al., 2004; Watzlawik et al., 2010) were added 30 min before the addition of psychosine. Psychosine treatment lasted for 3 h. The final concentration of ethanol was maintained at 0.01% v/v.
Mitochondrial motility measurements.
Cells were treated as described above and incubated at 37°C for 20 min with 75 nm Mitotracker G diluted in growth medium. After replenishing with fresh medium, live video recording (1 frame every 10 s for 10 min) was performed using an Axiocam Hrm camera attached to an Axiovert 200M microscope (both Carl Zeiss). To measure the speed and number of the moving/stationary mitochondria, kymographs were generated and analyzed using ImageJ. The vehicle of psychosine (0.01% ethanol in DMEM) was used as control. Each condition was tested three times on separate dates.
Transmission electron microscopy.
Tissue was fixed in 2% paraformaldehyde, 0.5% glutaraldehyde overnight at 4°C. Samples were postfixed for 1 h in 2% OsO4 in 0.1% sodium cacodylate, dehydrated in graded steps of acetone, and embedded in Araldite. Ultrathin sections (60 nm) were cut with a DiATOME diamond knife on a Leica Ultracut UCT microtome, collected on Formvar-coated one-hole grids, and counterstained with uranile/lead. Samples were observed in a JEOL 1220 electron microscope.
Western blotting.
Tissues were homogenized in lysis buffer (1 mm PMSF, 2 mm sodium orthovanadate, 1 mm NaF, 20 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 5 mm MgCl2, 300 nm okadaic acid), briefly sonicated on ice, and centrifuged at 5000 rpm for 5 min to remove debris. Protein content was quantified using the Bradford assay (Bio-Rad). Equal amounts of proteins (10–20 μg) were loaded on a 4–12% Bis-Tris gel (Invitrogen) and electrophoresed at 80 mV for 4 h before transfer of gels on a PVDF membrane (Bio-Rad) for 2 h. Membranes were blocked in 5% nonfat milk, 1% BSA, 0.05% Tween 20 in Tris-glycine buffer (blocking solution) and probed with primary antibodies at 4°C overnight. After incubation with secondary HRP-conjugated antibodies, bands were visualized using the enhanced luminescence kit (Thermo Scientific) and quantified using ImageJ. Antibodies used detected the following: actin (rabbit; Sigma), heat shock protein 60 (HSP60, mouse; Santa Cruz Biotechnology), SNAP 25 (goat; Abcam), phosphorylated ser-9 GSK3β (rabbit; Cell Signaling Technology), total GSK3β (rabbit; Cell Signaling Technology), kinesin heavy chain (KHC; mouse; H2), KLC (mouse; L2), and dephosphorylated KLC (mouse; 63-90). The latter three antibodies were donated by S.T.B. For the quantification of the 63-90 antibody (dephosphorylated KLC), bands were normalized to KHC using ImageJ. Although total KLC is a more appropriate loading control, normalization toward total KHC or KLC gave the same results. For the quantification of total protein levels, actin was used as a loading control.
Vesicular axonal transport in sciatic nerve.
To analyze for the accumulation of transported cargoes, spinal cord and distal portions of sciatic nerves and proximal stumps from ligated sciatic nerves (Hirokawa et al., 1991) were processed for immunoblotting.
Vesicle motility assays.
Axoplasm was extruded from giant axons of the squid Loligo pealei (Marine Biological Laboratory) as described previously (Brady et al., 1982). Sphingolipids were diluted into X/2 buffer (175 mm potassium aspartate, 65 mm taurine, 35 mm betaine, 25 mm glycine, 10 mm HEPES, 6.5 mm MgCl2, 5 mm EGTA, 1.5 mm CaCl2 and 0.5 mm glucose, pH 7.2) supplemented with 2–5 mm ATP and 20 μl of this mixture was added to perfusion chambers. Preparations were analyzed on a Zeiss Axiom microscope with a 100×, 1.3 numerical aperture objective, and DIC optics. Hamamatsu Argus 20 and model 2400 CCD cameras were used for image processing and analysis. Organelle velocities were measured with a Photonics Microscopy C2117 video manipulator (Hamamatsu) by matching calibrated cursor movements to the speed of vesicles moving in the axoplasm. Individual organelles are below the resolution limit and their individual properties (frequency of pauses, number of moving organelles) cannot be analyzed. However, the average velocity measurements were correlated with the number of vesicles moving in a given treatment: low rates reflect reduced numbers of vesicles and slower mean velocities (Brady et al., 1993; Ratner et al., 1998). This method reliably reproduces the rates of transport of individual organelles moving on axoplasms.
In vivo administration of GSK3β inhibitor.
After genotyping, twitcher pups were injected intraperitoneally with a myristoylated form of L803 (a GSK3β inhibitor) or vehicle (3 mice/group) beginning on postnatal day 3 and daily thereafter until postnatal day 20. Each injection delivered 400 nm (Wang et al., 2008) of the inhibitor in a 25 μl bolus of vehicle (saline). Motor conduction velocities were recorded in sciatic nerves before tissue collection as described previously (Dolcetta et al., 2005) and axonal transport analysis was done as described above.
Statistical analysis.
Results are expressed as mean ± SE in three to six independent experiments. Data were analyzed by two-tailed unpaired t test (confidence interval 95%) or by ANOVA test followed by the Newman–Keuls multiple-comparison test to analyze for all possible comparisons, where applicable. p < 0.05 was considered statistically significant.
Results
Psychosine inhibits FAT through PP1 and GSK3β
Recent descriptions of axonal degeneration in twitcher axons suggest that selected populations of neurons undergo a dying-back degeneration (Castelvetri et al., 2011; Smith et al., 2011; Cantuti-Castelvetri et al., 2012). Axonal swellings are pathognomonic of structural and physiological deficiencies in diseased axons, which may include focal accumulation of unsorted cargoes due to defects in FAT (Coleman and Ribchester, 2004; De Vos et al., 2008). We investigated whether psychosine is a pathogenic compound able to interfere with FAT using ex vivo axoplasm preparations from L. pealei, a model that has been used successfully for quantitative real-time analyses of the effects of bioactive molecules on both anterograde and retrograde components of FAT (Brady et al., 1982). Extruded axoplasms were perfused with 1 or 5 μm psychosine and anterograde and retrograde rates were recorded during 50 min. Psychosine induced a significant (p = 0.001) inhibitory response in both FAT components (Fig. 1a,b), whereas axoplasms perfused with d-sphingosine, the sphingoid base of psychosine (Fig. 1c), or with an enantiomeric form of psychosine (Fig. 1d) had no effect on FAT.
Figure 1.

Psychosine is a pathogenic lipid that inhibits fast axonal transport through PP1 and GSK3β activities. Measurements of FAT rate (μm/s) for membrane-bound organelles were performed on squid axoplasms perfused with psychosine or control lipids. Black arrowheads and line and gray arrowheads and line represent anterograde and retrograde FAT components, respectively. Perfusion of 1 μm (a) and 5 μm (b) psychosine into squid axoplasms led to a strikingly reduced rate of anterograde and retrograde transport. The effects of 1 and 5 μm psychosine were comparable, suggesting that much lower concentrations may also affect FAT. Perfusion with 5 μm d-sphingosine (c) or 1 μm enantiomeric psychosine (d) had no effect on transport (n = 3–6 axoplasms). Coperfusion of 5 μm psychosine with 200 nm okadaic acid (a general phosphatase inhibitor) (e) or 50 nm inhibitor 2 (I2) (a selective PP1 inhibitor) (f) led to protection of transport rates. Coperfusion of 1 μm psychosine with 100 nm ING-35 (a selective GSK3β inhibitor) (g) led to an almost complete protection of FAT, whereas coperfusion of 1 μm psychosine with 250 nm tetrabromocinnamic acid, a selective casein kinase II inhibitor (h), did not protect against the effects of psychosine. Anterograde (i) and retrograde (j) transport rates at the end of the treatments indicated a significant recovery (p = 0.001, ANOVA) in axoplasms coperfused with the inhibitors I2 and ING 35. Immunoblotting with antibodies against phosphorylated (ser-9) GSK3β (pGSK3β) and total GSK3β (tGSK3β) revealed significant differences (p = 0.04, unpaired t test) in activated (dephosphorylated) GSK3β levels in 30-day sciatic nerves and 10-day cultured primary cortical neurons, but not in extracts from 30-day spinal cords (n = 6 per condition per group) (k).
Various signaling pathways have been reported to regulate FAT (Morfini et al., 2002; 2006; Pigino et al., 2009). A signaling pathway involving the activities of PP1 and GSK3β regulates FAT by phosphorylation of KLCs (subunits of the motor kinesin) (Morfini et al., 2004) and promotes release from its transported cargoes (Morfini et al., 2002). To determine whether this pathway was involved in psychosine-mediated inhibition of FAT, motility assays were performed by coperfusing psychosine with okadaic acid and inhibitor 2, which are selective inhibitors of PP1. Interestingly, both inhibitors significantly (p = 0.001) protected anterograde and retrograde components of FAT (Fig. 1e,f,i,j). Similarly, ING-35, a specific inhibitor of GSK3β, almost completely abolished (p = 0.001) the effect of psychosine (Fig. 1g,i,j). To evaluate the role of alternative pathways, inhibitors of other kinases, such as the casein kinase 2 inhibitor tetrabromocinnamic acid (Martin and Pagano, 1994), were coperfused with psychosine, but these inhibitors did not exhibit a significant protective effect (Fig. 1h). These experiments indicate that psychosine impairs FAT mainly through deregulation of PP1 and GSK3β activities.
Nerve tissue from twitcher mutants contain higher levels of activated GSK3β
Activation of GSK3β is regulated primarily by phosphorylation-dephosphorylation of ser-9, a process mediated by a variety of kinases including Akt (Woodgett, 1994) and phosphatases including PP1 (Morfini et al., 2002). We previously demonstrated the presence of increased PP1 activity levels in twitcher mutants (Cantuti-Castelvetri et al., 2012), which is consistent with the prediction of higher levels of GSK3β activity in vivo. Immunoblot analyses for phosphorylated (ser-9) and total GSK3β revealed a decrease in the level of the phosphorylated (inactive) kinase in peripheral (sciatic) nerves, but not in the spinal cord of twitcher mice (Fig. 1k). Differences in GSK3β activity in these two regions of the nervous system are likely due to the ubiquitous nature of this kinase in the nervous system, including both neuronal and non-neuronal cells. This ubiquity may mask changes in neuronal activity that occurred in the spinal cord. To analyze GSK3β phosphorylation levels in neurons, we acutely isolated neurons from twitcher cortices. Indeed, mutant neurons contained significantly lower levels of phosphorylated (ser-9) GSK3β than wild-type neurons (Fig. 1k), which is consistent with our observations in nerve tissue and ex vivo axoplasm analyses. These data suggest that the deficiency of GALC and the subsequent accumulation of psychosine are correlated with higher levels of activated GSK3β.
KLC is less abundant and more phosphorylated in mutant axons
Activation of the axonal PP1-GSK3β pathway by psychosine predicts an increase in phosphorylated KLC and the premature release of their cargoes. To test this hypothesis, we first used immunoblotting to assess levels of KHCs and KLCs in postmortem nerve tissues isolated from affected mutants. We found no significant differences in the abundance of either kinesin in twitcher spinal cord extracts at 7 or 30 d of age (Fig. 2a,c,e), whereas both kinesins were significantly decreased (p = 0.03) in 30-day twitcher sciatic nerve extracts (Fig. 2b,d,f). This strong decrease in kinesins suggests a reduced trafficking of motors in twitcher mice during the onset of neurological manifestations (∼postnatal day 25), which is consistent with a distal axonopathy.
Figure 2.
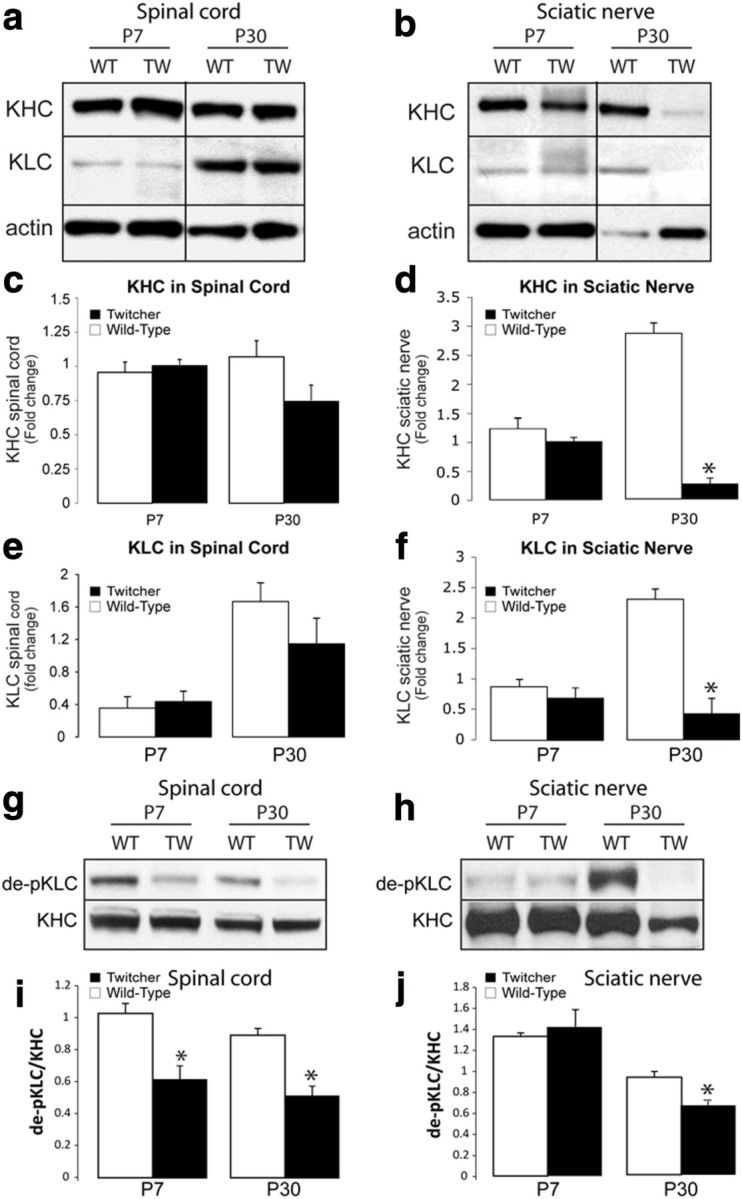
Alteration in molecular motor kinesins in twitcher sciatic nerves. Immunoblotting analyses for total KHC and KLC to test for changes in relative amounts of these motors 7- and 30-day (P7 and P30) spinal cords (a) and sciatic nerves (b) (n = 4/condition/group; actin used as loading control) revealed no significant differences in either KHC (c) and KLC (e) in spinal cord extracts from the mutants at either time point, although the P30 time point shows a trend of reduced amount of these motors (p > 0.05; t test unpaired). Levels of KHC (d) and KLC (f) differed significantly (p = 0.03, unpaired t test) in P30 twitcher sciatic nerves but were less affected in P7 mutant sciatic nerves. Immunoblotting analyses using monoclonal antibody 63-90, which detects changes in phosphorylation of KLC by GSK3β in 7- and 30-day-old (P7 and P30) spinal cords (g) and sciatic nerves (h) (n = 4/condition/group), revealed significant (p = 0.002–0.015, unpaired t test) differences in the level of dephosphorylated KLC (de-pKLC) in all twitcher samples except in P7 mutant sciatic nerves (i,j).
To determine whether phosphorylation of KLCs was changed in vivo, we examined tissue extracts by immunoblotting with the 63-90 antibody (Stenoien and Brady, 1997), which recognizes changes of phosphorylation in this motor protein (Morfini et al., 2004). Analyses showed a strong decrease in the immunoreactivity of monoclonal antibody 63-90 (binding of the antibody is inversely proportional to phosphorylation) in spinal cord (Fig. 2g,i; p = 0.002) and sciatic nerves (Fig. 2h,j; p = 0.01), particularly from 30-day-old twitcher mice. This finding is consistent with the increase in activated GSK3β (Fig. 1) and PP1 activity (Cantuti-Castelvetri et al., 2012). Significant (p = 0.015) phosphorylation of KLCs was also observed in cord extracts from 7-day-old twitcher mice, indicating an early deregulation of motors in this mutant.
In vivo and in vitro FAT is reduced in twitcher neurons
To determine whether twitcher mutants develop axonal transport defects in vivo, we measured transport efficiency by biochemical analysis of accumulated cargoes in wild-type and twitcher mice after sciatic nerve ligation. This simple paradigm has been used successfully for many years to measure changes in transport (Hirokawa et al., 1991), providing one of the first demonstrations that FAT existed. Proximal (relative to the spinal cord) stumps of the nerve (∼1 mm) were used for immunoblotting analyses. Measurement of the relative accumulation of molecular motor KHC, mitochondrial molecular chaperone HSP60 (Walls et al., 2012), and t-SNARE protein SNAP-25 (Walch-Solimena et al., 1995) indicated a significant (p = 0.001) decrease in their abundance in 7- and 30-day-old mutant proximal stumps (Fig. 3a,b). Transmission electron microscopy showed clear accumulations of vesicular bodies in all of the wild-type control nerves analyzed (Fig. 3g, inset), whereas mutant nerves showed a mix of axons without any visible accumulation of vesicular bodies (Fig. 3d) and with accumulated material (Fig. 3e). Vesicular structures were observed on the cytoplasmic face of mutant axoplasms (Fig. 3c, arrows), which were absent in wild-type axoplasms (Fig. 3f). The presence of these vesicular accumulations suggests alterations in the sorting of transported membranous organelles (Edgar et al., 2004), which is consistent with impaired FAT.
Figure 3.

Defective axonal transport in the twitcher sciatic nerve. Immunoblotting analyses of the proximal end of ligated nerves in 7-day (P7) (a) and 30-day (P30) (b) twitcher mice (n = 6 independent ligations) for KHC, HSP60 (a mitochondrial marker), and SNAP 25 revealed significantly reduced (p = 0.003–0.048, unpaired t test) accumulation of each marker at both time points. Transmission electron microscopic images of nonligated and ligated twitcher (c–e) and wild-type (f,g) nerves (ligation site on the top right corner) revealed abundant accumulation of vesicular material toward the site of ligation in wild-type axons (inset in g), whereas markedly fewer axons with accumulated material (d,e, inset in e) were seen in the ligated mutant nerve. Bar indicates 5 μm. Arrows in c point to vesicular material underneath the mutant axolemma.
Two experiments were performed to determine whether deficits in FAT are due to a cell-autonomous mechanism. First, wild-type cortical neuronal cultures were exposed to psychosine and the anterograde and retrograde movement of fluorescently labeled mitochondria was measured. Kymographs (n = 30 per group) determined that psychosine reduced both the number (Fig. 4a,b) and speed (Fig. 4c,d) of anterograde and retrograde moving mitochondria. Second, similar mitochondrial mobility analyses were performed on cultures of mutant neurons. Kymograph analyses showed that twitcher neurons displayed comparable deficits in anterograde and retrograde moving mitochondria (Fig. 4e,f) and reduced speeds (Fig. 4g,h), indicating that mutant neurons are autonomously affected.
Figure 4.

Defective transport of mitochondria in psychosine-elevated conditions. Anterograde and retrograde movement of fluorescently labeled mitochondria was measured using kymograph analyses. Wild-type neurons exposed to 1 μm psychosine are shown to have significantly (p = 0.01, unpaired t test) fewer mitochondria moving either anterogradely (a) or retrogradely (b) compared with the control (vehicle treatment, 0.01% ethanol in DMEM). Kymographs permitted the measurement of significant (p = 0.02, unpaired t test) reductions in anterograde (c) and retrograde (d) average moving speeds. Significant (p = 0.01, unpaired t test) reductions in moving mitochondria (e,f) and movement speed (g,h) were measured in cultures containing twitcher neurons.
Neuroprotection of axonal transport by interference of GSK3β activity
We previously reported that PP1 activity is elevated in nerve tissues of the twitcher mouse and that this is a contributing factor to cytoskeletal abnormalities and the dying-back pathology detected in this Krabbe disease model (Cantuti-Castelvetri et al., 2012). Further, psychosine was sufficient to stimulate higher activities of PP1 in the motor-neuron-like cell line NSC34 (Cantuti-Castelvetri et al., 2012) and to inhibit FAT through PP1 and GSK3β (Fig. 1). Our ex vivo axoplasm experiments using PP1 and GSK3β inhibitors provided further evidence that this effect is specific, suggesting that FAT might be protected from psychosine effects with appropriate neuroprotective compounds. To test this hypothesis further and to evaluate potential clinical applications, we conducted in vitro and in vivo neuroprotective experiments using inhibitors of PP1 and GSK3β.
First, activation of PP1 was induced on NSC34 cells as described previously (Cantuti-Castelvetri et al., 2012) and the level of downstream activation of GSK3β through PP1 was assessed by immunoblotting in the presence and absence of okadaic acid. Okadaic acid was sufficient to abolish psychosine-triggered activation of GSK3β (Fig. 5a,b), indicating the need for PP1 in activating GSK3β dephosphorylation. To examine the functional relevance of interfering with PP1 and GSK3β after psychosine treatment, we tested for changes in the antibody 63-90 immunoreactivity of KLC in extracts of treated NSC34 cells. As expected, psychosine alone induced a significant decrease in the immunoreactivity of kinesin to antibody 63-90 (Fig. 5a). Cotreatment of neurons either with okadaic acid to inhibit PP1 or with TDZD8 to inhibit GSK3β completely abolished the effect of psychosine on kinesin phosphorylation (Fig. 5a,c).
Figure 5.

In vitro neuroprotection using PP1 and GSK3β inhibitors. Immunoblotting of NSC34 cells for phosphorylated (ser-9) GSK3β (pGSK3β) and for the 63-90 epitope (de-phosphorylated KLC [de-pKLC]) after treatment of cells with 10 nm okadaic acid (a general inhibitor of phosphatases) and TDZD8 (a selective inhibitor of GSK3β; a) indicated a potent normalization effect by okadaic acid on levels of activated GSK3β (p = 0.05 for 1 μm psychosine and p = 0.001 for 5 μm psychosine, unpaired t test; b). Pretreatment of NSC34 cells with okadaic acid or TDZD8 led to a marked reduction of phosphorylation of KLC (measured as an increase of dephosphorylated KLC) by inhibition of PP1 and GSK3β (a,c), whereas pretreatment of NSC34 cells with myristoylated L803 (a selective GSK3β inhibitor) significantly (p = 0.01, unpaired t test) improved the proportion of mitochondria moving either anterogradely (d) or retrogradely (e) as well as their anterograde (f) and retrograde (g) movement speeds. Data are shown as mean ± SE of three to five independent experiments.
To test the functional impact of interfering with GSK3β on mitochondrial motility, NSC34 cells were coincubated with psychosine and a cell-permeable inhibitor of GSK3β myristoylated L803. Kymograph analyses of fluorescently labeled mitochondria showed that L803 was sufficient to restore mitochondrial transport in psychosine-treated cells (Fig. 5d–g). Control experiments with cells treated only with L803 showed no significant change in mitochondrial motility.
To evaluate the in vivo effects of interfering with GSK3β, twitcher mice were injected intraperitoneally with myristoylated L803 in saline from day 2 of age until day 20. Mutant mice were not conditioned with any other treatment nor did they receive traditional bone marrow transplantation to correct for the biochemical defect in psychosine catabolism. Extracts from nerves were used to measure for changes in 63-90 immunoreactivity. Untreated mutants showed the expected decrease in 63-90 immunoreactivity (Fig. 6a), whereas mutant mice treated with the inhibitor revealed significant increases in 63-90 immunoreactivity (Fig. 6a; p = 0.01).
Figure 6.

In vivo neuroprotection using GSK3β inhibitors. (a) Twitcher mice treated with the myristoylated L803 showed a significant normalization of dephosphorylated KLC levels, indicating a recovery in KLC function (a; p = 0.01, unpaired t test) as well as in levels of KHC and HSP60 in proximal sciatic nerve stumps (b; p = 0.01, unpaired t test). Data are expressed as percentage of protein marker accumulated in ligated nerves from treated mutant versus that in nontreated mutants. c, Motor conduction velocities (MCV, in meters per second) were recorded on the sciatic nerves of twitcher (Twi) mice treated with L803 and in untreated mutants and wild-type mice of matching age (n = 4 mice per group). Although MCV in treated mutants showed a mild improvement, this was not significant.
To test for possible protection against axonal transport defects in twitcher mice treated with the GSK3β inhibitor, we measured the percent recovery of the transport of kinesin and HSP60 in ligated sciatic nerves. Administration of the L803 inhibitor led to a significant (p = 0.01) recovery in the amount of transported kinesin and HSP60 (Fig. 6b). Finally, we examined the extent to which L803 treatment ameliorated some of the clinical symptoms of mutant mice. Because this treatment was not combined with any kind of metabolic correction (e.g., bone marrow transplantation or gene therapy; Galbiati et al., 2009), L803-treated twitcher had no recovery in psychosine or GALC levels (data not shown) and their life span was not expected to be affected. Measurement of motor conduction velocity in sciatic nerves showed a mild but not significant improvement in treated mutants (Fig. 6c). However, these results are encouraging because they show that this axonal defect may be treated positively in vivo using pharmacological strategies and underline the potential benefit of combinatorial treatments. Further experiments will determine the synergy of these neuroprotective therapies in combination with bone marrow transplantation and gene therapy.
Discussion
Since its description in 1916 (Krabbe, 1916), neuronal vulnerability has been an unclear aspect of Krabbe disease, but recent work is shedding light on the underlying pathogenic mechanism(s) (Castelvetri et al., 2011; Smith et al., 2011; Cantuti-Castelvetri et al., 2012). The present study provides further evidence that Krabbe disease develops defects in vesicular transport and identifies psychosine as the first pathogenic sphingolipid inhibiting FAT through deregulated activities of PP1-GSK3β.
FAT ensures neuronal communication, regulating the timely delivery of cargoes to neuronal domains. The pathogenic relevance of impaired transport is best exemplified by the discovery of specific mutations in molecular motors in hereditary spastic paraplegia HSP10 and some forms of sensory and motor neuron disease (Zhao et al., 2001; Reid et al., 2002; Puls et al., 2003). These deficiencies decrease cargo transport, initiating an axonal dying-back pathology that kills vulnerable neurons. Therefore, dysfunctional FAT might be seen as a common pathogenic mechanism activated in several unrelated neurological diseases. In the case of Krabbe disease, a deficit of FAT likely adds to the severity of demyelination, leading to a complex neurological phenotype. It is possible to speculate how deficient kinesin-based delivery affects neurons. For example, impaired mitochondrial transport could lead to inefficient energy distribution along the axon, reducing its capacity to maintain physiological ion concentrations. Higher influx of calcium through the sodium-calcium exchanger (Craner et al., 2004) and subsequent activation of calcium-dependent proteases may contribute to the axonal swellings (Beirowski et al., 2005; Castelvetri et al., 2011). Similarly, defective transport of ion channels will affect the functionality of nodes of Ranvier, impairing nerve conduction. Kinesin-based transport is also fundamental for the delivery of myelin components (Carson et al., 1997; Lyons et al., 2009). Therefore, defects in motor-based-transport such as those described in this study may also occur in other cell types such as oligodendrocytes and Schwann cells.
Because myelination is a known regulator of axonal transport (de Waegh and Brady, 1990; de Waegh et al., 1992), our results do not exclude the possibility that abnormal myelination might contribute to FAT defects. However, demyelination effects on FAT are likely to be secondary to direct effects of psychosine, given that twitchers show normal myelination during early postnatal life (Nagara et al., 1982) and that our in vitro studies using twitcher neurons confirmed a cell autonomous defect. However, after disease onset and with ongoing demyelination, FAT defects are likely exacerbated by demyelination. Psychosine inhibits specifically both anterograde and retrograde components of FAT. The GSK3β pathway has been primarily associated with regulation of anterograde transport (Morfini et al., 2004). However, our studies using GSK3β inhibitors revealed an additional regulatory role on retrograde transport. Further studies are needed to characterize the molecular partners mediating this effect.
In vivo, transport defects were correlated with the progressive accumulation of psychosine (Igisu and Suzuki, 1984; Castelvetri et al., 2011). It seems highly unlikely that psychosine inhibits FAT by eliciting nonspecific effects such as detergent action (i.e., disruption of organelle membranes) or by disrupting microtubules because: (1) membranous organelles and microtubules in extruded axoplasms exposed to psychosine were structurally unaffected for the duration of our experiments (data not shown); (2) electron microscopy showed normal structure of membranous organelles and microtubules within twitcher axons; and (3) coperfusion of psychosine with PP1 and GSK3β inhibitors protected transport significantly. Furthermore, inhibition of FAT by psychosine does not require either myelin or myelinating glia, because motility axoplasm assays and in vitro experiments on NSC34 cells and primary cortical neurons were conducted in the absence of mutant myelin and glia. The finding of psychosine in acutely isolated neurons further suggests an in situ source of this neurotoxin (Castelvetri et al., 2011).
If psychosine is sufficient to block FAT, it must somehow be spatially related to the transport machinery to exert this inhibitory role. Psychosine may accumulate in the axon from different sources, including neuronal synthesis and transport via membrane-bound cargoes (M.B. Santos and E.R.B., manuscript in preparation), and lipid transfer from myelin sheaths/surrounding glia (Vance et al., 1994, 2000; Krijnse-Locker et al., 1995). To date, it remains unclear whether one or more of these sources contribute to psychosine accumulation in axons in vivo.
Krabbe disease is not associated with mutations in molecular motors or cytoskeletal proteins, so FAT inhibition must be caused by one or more pathogenic components intrinsic of the disease. Psychosine, which activates pathogenic responses in glia (Khan et al., 2005; Giri et al., 2006), also increases axonal PP1 and PP2A activities (Cantuti-Castelvetri et al., 2012). Here we found that psychosine exacerbates axonal GSK3β activity through PP1, a process shown to increase phosphorylation of KLCs, releasing kinesin from cargoes (Morfini et al., 2002). One possible mechanism by which psychosine elicits these changes is through membrane alterations. Because psychosine is a lipid-raft-associated sphingolipid (White et al., 2009; 2011) and because axonal PP1 and GSK3β colocalize in transported vesicles (Morfini et al., 2004) and associate with rafts (Sui et al., 2006; Henkhaus et al., 2008), psychosine may modify PP1 and/or GSK3β activities by regulating their interaction with membranes. For example, caveolin-1 is a raft-specific protein that can inhibit PP1 (Henkhaus et al., 2008). Psychosine may affect PP1-caveolin-1 in rafts by releasing PP1. Because the PP1/GSK3β pathway was also found to deregulate FAT in other pathological conditions (Kanaan et al., 2011), these findings suggest that the PP1/GSK3β pathway is a common mechanism in unrelated neurological conditions.
Although abnormal GSK3β activity may affect transport of many membrane-bound organelles through deregulation of kinesin-based motors, other organelles such as mitochondria may be targeted through other still unclear mechanisms. For example, recent studies suggest that mitochondria may not require KLCs (Cai and Sheng, 2009) for transport but rather a complex of various docking proteins (Stowers et al., 2002; Guo et al., 2005; Glater et al., 2006), whereas other studies show that both KLC and KHC and GSK3β are associated with mitochondria (Leopold et al., 1992; Chen et al., 2008). In our experiments, an inhibitor of GSK3β acted as a neuroprotective agent, improving axonal mobilization of mitochondria and confirming a fundamental role of this pathway in regulating mitochondrial transport. Furthermore, PP1/GSK3β inhibitors also ameliorated psychosine-inhibitory effects on the retrograde component of FAT, suggesting that the PP1/GSK3β and/or some other functionally interacting pathways may have regulatory roles in both transport components. These pathways could decrease FAT by modifying other components of the transport machinery, such as the microtubular cytoskeleton and its regulatory proteins. For example, mutations or changes in the phosphorylation of the microtubule-binding protein Tau are known to significantly inhibit axonal transport (Stoothoff et al., 2009; Kanaan et al., 2011; Bull et al., 2012; Shahpasand et al., 2012). Because Tau is also a target of PP1 and GSK3β, it is possible that changes in Tau phosphorylation might indirectly affect the rates of transport in both directions (Bull et al., 2012). Although the recovery of transport using PP1/GSK3β inhibitors was significant, it was not complete, suggesting the involvement of other regulatory components in psychosine pathogenesis.
How neuronal vulnerability is regulated is still unclear. Previous experiments showed that PP1 is not homogenously activated in twitcher axons (Cantuti-Castelvetri et al., 2012), suggesting that the PP1-GSK3β pathway may be differentially affected in subsets of axons. Selective damage of neurons is observed in other neurological diseases such as Parkinson's and Alzheimer's diseases. The mechanism regulating neuronal vulnerability in these diseases and in Krabbe disease may depend on the occurrence of various pathogenic factors concomitantly in the same cell. In Krabbe disease, this may mean that there is differential accumulation of psychosine in subsets of neurons or that other pathogenic/protective pathways may exist in specific neurons.
Krabbe disease has remained incurable to date. The only traditional treatment providing some improvement (albeit not a cure) is hematogenous replacement (Yeager et al., 1984; Escolar et al., 2005). Our study indicates that pharmacological intervention to protect FAT may be beneficial. We and others (McGraw et al., 2005; Hawkins-Salsbury et al., 2011) have hypothesized that simultaneous engagement of various pathogenic mechanisms limits hematogenous replacement efficacy. Because the pharmacological treatments used in this study were not combined with any metabolic correction, treated mutants were not expected to avoid developing neurological symptoms of the disease. However, the normalization of phosphorylated KLC, the amelioration of FAT, and the mild (albeit not significant) improvement in nerve conduction in twitcher mice treated with GSK3β inhibitors underline the potential benefit of such neuroprotective interventions. The potential synergy of combining these neuroprotective treatments with hematogenous replacement is currently being evaluated in our laboratory.
In conclusion, our data demonstrate the occurrence of FAT defects in cellular and animal models of Krabbe disease and identify the sphingolipid psychosine as a FAT inhibitor. Elevated psychosine inhibits FAT by activation of the PP1-GSK3β pathway. Preliminary data on postmortem human brain samples indicate that similar pathogenic activation of GSK3β occurs in the nervous system of Krabbe patients, underlining the relevance of this axonal defect in children with this disease. Neuroprotection experiments have the potential of treating Krabbe disease neurodegeneration by reversing FAT defects, opening new and promising areas of clinical research relevant for Krabbe disease and other leukodystrophies.
Footnotes
This work was supported by a Chancellor Award (to L.C.C.), the National Institutes of Health (Grant #RNS065808A), the Morton Cure Paralysis Foundation, the Legacy of Angels Foundation, and the Board of Trustees at the University of Illinois (to E.R.B.). We dedicate this work to the memory of our colleague and friend Agnieszka Kaminska.
References
- Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;6:6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Brady ST, Richards BW, Leopold PL. Assay of vesicle motility in squid axoplasm. Methods Cell Biol. 1993;39:191–202. doi: 10.1016/S0091-679X(08)60171-5. [DOI] [PubMed] [Google Scholar]
- Bull ND, Guidi A, Goedert M, Martin KR, Spillantini MG. Reduced axonal transport and increased excitotoxic retinal ganglion cell degeneration in mice transgenic for human mutant P301S tau. PLoS One. 2012;7:e34724. doi: 10.1371/journal.pone.0034724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Sheng ZH. Mitochondrial transport and docking in axons. Exp Neurol. 2009;218:257–267. doi: 10.1016/j.expneurol.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Zhu H, Givogri MI, Chidavaenzi RL, Lopez-Rosas A, Bongarzone ER. Psychosine induces the dephosphorylation of neurofilaments by deregulation of PP1 and PP2A phosphatases. Neurobiol Dis. 2012;46:325–335. doi: 10.1016/j.nbd.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JH, Worboys K, Ainger K, Barbarese E. Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil Cytoskeleton. 1997;38:318–328. doi: 10.1002/(SICI)1097-0169(1997)38:4<318::AID-CM2>3.0.CO%3B2-%23. [DOI] [PubMed] [Google Scholar]
- Castelvetri LC, Givogri MI, Zhu H, Smith B, Lopez-Rosas A, Qiu X, van Breemen R, Bongarzone ER. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol. 2011;122:35–48. doi: 10.1007/s00401-011-0814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen S, Owens GC, Edelman DB. Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS One. 2008;3:e2804. doi: 10.1371/journal.pone.0002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Ribchester RR. Programmed axon death, synaptic dysfunction and the ubiquitin proteasome system. Curr Drug Targets CNS Neurol Disord. 2004;3:227–238. doi: 10.2174/1568007043337436. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- de Waegh S, Brady ST. Altered slow axonal transport and regeneration in a myelin-deficient mutant mouse: the trembler as an in vivo model for Schwann cell-axon interactions. J Neurosci. 1990;10:1855–1865. doi: 10.1523/JNEUROSCI.10-06-01855.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-D. [DOI] [PubMed] [Google Scholar]
- Dolcetta D, Amadio S, Guerrini U, Givogri MI, Perani L, Galbiati F, Sironi L, Del Carro U, Roncarolo MG, Bongarzone E. Myelin deterioration in Twitcher mice: motor evoked potentials and magnetic resonance imaging as in vivo monitoring tools. J Neurosci Res. 2005;81:597–604. doi: 10.1002/jnr.20574. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Caviness VS, Jr, Erbe RW, Patterson MC, Schultz KR, Wenger DA, Whitley C. The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet Med. 2009;11:450–454. doi: 10.1097/GIM.0b013e3181a16e04. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Smith JK, Gilmore JH, Kurtzberg J, Lin W, Styner M. Diffusion tensor imaging detects abnormalities in the corticospinal tracts of neonates with infantile Krabbe disease. AJNR Am J Neuroradiol. 2009;30:1017–1021. doi: 10.3174/ajnr.A1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Givogri MI, Cantuti L, Rosas AL, Cao H, van Breemen R, Bongarzone ER. Combined hematopoietic and lentiviral gene-transfer therapies in newborn twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J Neurosci Res. 2009;87:1748–1759. doi: 10.1002/jnr.22006. [DOI] [PubMed] [Google Scholar]
- Giri S, Khan M, Rattan R, Singh I, Singh AK. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006;47:1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum Mol Genet. 2011;20:R54–R60. doi: 10.1093/hmg/ddr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkhaus RS, Roy UK, Cavallo-Medved D, Sloane BF, Gerner EW, Ignatenko NA. Caveolin-1-mediated expression and secretion of kallikrein 6 in colon cancer cells. Neoplasia. 2008;10:140–148. doi: 10.1593/neo.07817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Singh AK. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett. 2002;330:183–187. doi: 10.1016/S0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011;31:9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, Singh AK. Peroxisomal participation in psychosine-mediated toxicity: implications for Krabbe's disease. J Neurosci Res. 2005;80:845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Shinoda H, Goto I, Yamanaka T, Suzuki Y. Globoid cell leukodystrophy is a generalized galactosylsphingosine (psychosine) storage disease. Biochem Biophys Res Commun. 1987;144:41–46. doi: 10.1016/S0006-291X(87)80472-2. [DOI] [PubMed] [Google Scholar]
- Krabbe K. A new familial, infantile form of diffuse brain sclerosis. Brain. 1916;39:74–114. doi: 10.1093/brain/39.1-2.74. [DOI] [PubMed] [Google Scholar]
- Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol Biol Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold PL, McDowall AW, Pfister KK, Bloom GS, Brady ST. Association of kinesin with characterized membrane-bounded organelles. Cell Motil Cytoskeleton. 1992;23:19–33. doi: 10.1002/cm.970230104. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Scholze A, Talbot WS. Kif1b is essential for mRNA localization in oligodendrocytes and development of myelinated axons. Nat Genet. 2009;41:854–858. doi: 10.1038/ng.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OC, Pagano RE. Internalization and sorting of a fluorescent analogue of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanisms. J Cell Biol. 1994;125:769–781. doi: 10.1083/jcb.125.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw P, Liang L, Escolar M, Mukundan S, Kurtzberg J, Provenzale JM. Krabbe disease treated with hematopoietic stem cell transplantation: serial assessment of anisotropy measurements–initial experience. Radiology. 2005;236:221–230. doi: 10.1148/radiol.2353040716. [DOI] [PubMed] [Google Scholar]
- Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Björkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009a;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009b;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Nagara H, Kobayashi T, Suzuki K, Suzuki K. The twitcher mouse: normal pattern of early myelination in the spinal cord. Brain Res. 1982;244:289–294. doi: 10.1016/0006-8993(82)90087-7. [DOI] [PubMed] [Google Scholar]
- Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol. 2010;12:1228–1234. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST. A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J Neurosci. 1998;18:7717–7726. doi: 10.1523/JNEUROSCI.18-19-07717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J Neurochem. 1996;66:1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Galbiati F, Castelvetri LC, Givogri MI, Lopez-Rosas A, Bongarzone ER. Peripheral neuropathy in the Twitcher mouse involves the activation of axonal caspase 3. ASN Neuro. 2011;3:e00066. doi: 10.1042/AN20110019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K, Fan Z, Xie H, Bacskai B, Edd J, Irimia D, Hyman BT. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J Neurochem. 2009;111:417–427. doi: 10.1111/j.1471-4159.2009.06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/S0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Sui Z, Kovács AD, Maggirwar SB. Recruitment of active glycogen synthase kinase-3 into neuronal lipid rafts. Biochem Biophys Res Commun. 2006;345:1643–1648. doi: 10.1016/j.bbrc.2006.05.087. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–259. doi: 10.1023/A:1022436928925. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nagara H, Kobayashi T, Goto I, Suzuki K. The twitcher mouse: attenuated processes of Schwann cells in unmyelinated fibers. Brain Res. 1989;503:160–162. doi: 10.1016/0006-8993(89)91719-8. [DOI] [PubMed] [Google Scholar]
- Vance JE, Pan D, Campenot RB, Bussière M, Vance DE. Evidence that the major membrane lipids, except cholesterol, are made in axons of cultured rat sympathetic neurons. J Neurochem. 1994;62:329–337. doi: 10.1046/j.1471-4159.1994.62010329.x. [DOI] [PubMed] [Google Scholar]
- Vance JE, Campenot RB, Vance DE. The synthesis and transport of lipids for axonal growth and nerve regeneration. Biochim Biophys Acta. 2000;1486:84–96. doi: 10.1016/S1388-1981(00)00050-0. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C, Blasi J, Edelmann L, Chapman ER, von Mollard GF, Jahn R. The t-SNAREs syntaxin 1 and SNAP-25 are present on organelles that participate in synaptic vesicle recycling. J Cell Biol. 1995;128:637–645. doi: 10.1083/jcb.128.4.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls KC, Coskun P, Gallegos-Perez JL, Zadourian N, Freude K, Rasool S, Blurton-Jones M, Green KN, Laferla FM. Swedish Alzheimer's mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of APP and beta-amyloid. J Biol Chem. 2012;287:30317–30327. doi: 10.1074/jbc.M112.365890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Garcia CA, Rehani K, Cekic C, Alard P, Kinane DF, Mitchell T, Martin M. IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-beta. J Immunol. 2008;181:6797–6802. doi: 10.4049/jimmunol.181.10.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzlawik J, Warrington AE, Rodriguez M. Importance of oligodendrocyte protection, BBB breakdown and inflammation for remyelination. Expert Rev Neurother. 2010;10:441–457. doi: 10.1586/ern.10.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, Bongarzone ER. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. J Neurosci. 2009;29:6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AB, Galbiati F, Givogri MI, Lopez Rosas A, Qiu X, van Breemen R, Bongarzone ER. Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J Neurosci Res. 2011;89:352–364. doi: 10.1002/jnr.22564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol. 1994;5:269–275. [PubMed] [Google Scholar]
- Yeager AM, Brennan S, Tiffany C, Moser HW, Santos GW. Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science. 1984;225:1052–1054. doi: 10.1126/science.6382609. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/S0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]