

Abstract
Febrile seizures are associated with increased brain temperature and are often resistant to treatments with antiepileptic drugs, such as carbamazepine and phenytoin, which are sodium channel blockers. Although they are clearly correlated with the hyperthermic condition, the precise cellular mechanisms of febrile seizures remain unclear. We performed patch-clamp recordings from pyramidal cells in acute rat brain slices at temperatures up to 40°C and found that, at ≥37°C, L-type calcium channels are active at unexpectedly hyperpolarized potentials and drive intrinsic firing, which is also supported by a temperature-dependent, gadolinium-sensitive sodium conductance. Pharmacological data, RT-PCR, and the current persistence in Cav1.3 knock-out mice suggested a critical contribution of Cav1.2 subunits to the temperature-dependent intrinsic firing, which was blocked by nimodipine. Because intrinsic firing may play a critical role in febrile seizures, we tested the effect of nimodipine in an in vivo model of febrile seizures and found that this drug dramatically reduces both the incidence and duration of febrile seizures in rat pups, suggesting new possibilities of intervention for this important pathological condition.
Introduction
Febrile seizures (FSs) affect up to 5% of children under the age of 5 years and are the most common seizures in children of this age group (Stafstrom, 2002). They are defined by their association with hyperthermia and can be induced in rodents by increasing their body temperature (Tb) (Baram et al., 1997; Chen et al., 1999). Despite the large amount of work aimed at identifying the underlying mechanisms, the ethiopathogenesis of FSs remains unclear (Schuchmann et al., 2006). It has been shown that hyperthermia may have large effects on the excitability of hippocampal slices and leads to spreading depression (Wu and Fisher, 2000). More recently, patch-clamp recordings conducted in hippocampal slices showed that pyramidal neurons depolarize and fire spontaneously when exposed to hyperthermic temperature (Kim and Connors, 2012), whereas another study (Hedrick and Waters, 2011) showed that the firing pattern of layer 5 pyramidal neurons changes from bursting at 24–26°C to regularly firing at 36–37°C. Although these authors suggested that the phenomenon is mediated by changes in intrinsic membrane excitability, these studies did not address the detailed mechanisms of the observed effects, which remain unexplored. Intrinsic neuronal excitability is a complex integrated function that depends on multiple mechanisms, with each having some degree of temperature dependence. These mechanisms include the gating of voltage-gated ion channels (Hodgkin and Huxley, 1952), ion pump function (Carpenter and Alving, 1968; Gorman and Marmor, 1970), and the activation of channels that are bona fide temperature gated (Brauchi et al., 2006). Therefore, the effects of temperature on a highly integrated system, such as intrinsic neuronal firing, are hard to predict, yet whether or not a neuron is intrinsically active and the type of activity it displays (regular firing or burst firing; pace maker or resonant) have major impacts on the system behavior (Llinás, 1988). Seizure generation is believed to depend on network properties (Wong et al., 1986), but intrinsic excitability is also deemed to play a critical role (Traub et al., 1991). Interestingly, FSs may also be associated with loss-of-function mutations of the Nav1.1 channel, and, in this case, treatment with conventional drugs, such as phenytoin, that block sodium channels are contraindicated (Catterall et al., 2010).
Here we investigate the electrophysiological response of cortical pyramidal cells to hyperthermia and find that they display temperature-dependent intrinsic firing that primarily depends on a nimodipine-sensitive calcium current. These data suggest that this current may drive FSs. Accordingly, in vivo recordings show that nimodipine block of calcium channels strongly reduces the incidence and duration of FSs in rat pups.
Materials and Methods
Hippocampal brain slices.
All experiments followed protocols approved by the Northwestern University Center for Comparative Medicine. Ten- to 18-d-old male Long–Evans rats were anesthetized with isoflurane (200 μl to 1 L container) and killed by decapitation. This age range was chosen because it overlaps that corresponding to the maximum incidence of FSs in humans (3 months to 5 years of age; Verity et al., 1985). The brain was removed from the skull in ice-cold artificial cerebrospinal fluid (ACSF) containing the following (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1.8 CaCl2, 1 MgCl2, and 25 glucose, pH 7.4 (equilibrated with 95% O2 and 5% CO2). Transverse, 300-μm-thick, hippocampal slices were cut using a vibroslicer (Dosaka or Leica VT1200) and stored for 15 min at 35°C and subsequently at 22–24°C in a solution containing the following (in mm): 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 75 sucrose, and 25 glucose (saturated with 95% O2 and 5% CO2).
Electrophysiological recordings.
For whole-cell recordings, pipettes were pulled from Hilgenberg glass (1406180) or WPI glass (PG10-165) using a horizontal puller (P97; Sutter Instruments) and filled with internal solution consisting of the following (in mm): K-gluconate 140, NaCl 8, MgCl2 2, EGTA 10, Na2ATP 2, NaGTP 0.2, and HEPES 10, pH 7.3 with KOH. Tip resistances in working solutions ranged from 3 to 5 MΩ, yielding series resistances of 10–30 MΩ.
Voltage-clamp recordings were used to study the properties of the temperature-sensitive current. Reliable voltage-clamp conditions in slice recordings are difficult to obtain: however, the clamp quality could be improved by using a CsCl-based intracellular solution containing QX-314 (5 mm) and by substituting BaCl2 for CaCl2 in the extracellular solution. Under these conditions, the clamp quality was much improved, as assessed in separate experiments by verifying that the fast component of the tail calcium currents showed a linear current–voltage relationship and could be fitted with monoexponential functions and allowed comparison of data obtained at low and high temperature. An additional indication of the reliability of the observed temperature-induced effects on channel gating came from the consistency of the results observed with square pulses and slow potential ramps. Nevertheless, although the qualitative result showing a temperature-dependent shift toward hyperpolarized potentials of the activation curve is very strong, the precise values of the half points and slopes of the activation and inactivation curves must be taken with caution.
For extracellular recordings, pipettes were pulled from Dagan SE16 glass and had tip resistance of 2–3 MΩ. The intrapipette solution for these recordings was as follows (in mm): 138 NaCl, 4 KCl, 1.2 CaCl2, 1 MgCl2, 10 HEPES, and 25 glucose, pH 7.4 with NaOH.
Slices were visualized with an Axioskop 2FS upright microscope with a water-immersion 40× objective. The bath temperature was controlled using a Warner Instruments TC-324B temperature control unit; the temperature probe was positioned halfway between the bath perfusion inlet and the suction, very close to the position of the recording pipette, to ensure the most reliable readout of the temperature. To attain a fast temperature increase, the solution flow was kept at ∼3 ml/min, and the bath level was kept low (total volume, ∼1–2 ml). In these conditions, the rate of temperature change was ∼3–5°C/min. Extracellular recording obtained at high temperature was characterized by higher seal resistances, which explains the larger amplitudes of the action currents recorded in these conditions compared with lower temperature (Fig. 1). Pyramidal cells were visually identified on the basis of location, shape, and, for whole-cell recordings, their responses to hyperpolarizing and depolarizing current injections. Whole-cell signals were recorded using either Multiclamp 700A or Axopatch 200B amplifiers (Molecular Devices), filtered at 10 kHz, and sampled at 20 kHz. Currents elicited by slow voltage ramps were filtered at 2 kHz and sampled at 10 kHz. Traces presented in the figures represent single sweeps or the average of up to five sweeps.
Figure 1.

The apparent increase in extracellularly recorded spike amplitude at high temperature is attributable to increased seal resistance. A, Left, Example traces of loose-seal recordings of a hippocampal pyramidal cell at mild hypothermia (top), normothermia (middle), and moderate hyperthermia (bottom). Right, The spikes indicated by arrowheads are shown on an expanded time base. Seal resistance is monitored by delivering a 200 ms voltage pulse at 33.3 Hz frequency. B, Spike amplitude and seal resistance are plotted versus bath temperature. C, Plot summarizes spike amplitude (top) and seal resistance (bottom) from three hippocampal pyramidal neurons at 33°C and 38°C.
Drug stock solutions.
Picrotoxin (50 mm) was prepared in dimethylsulfoxide (DMSO) and stored at 4°C. Nimodipine (10 mm) was dissolved in DMSO and stored at 4°C (in the dark). SB366791 (10 mm) was dissolved in DMSO and stored at −30°C. Tetrodotoxin (TTX; 1 mm) was dissolved in water and stored at 4°C. Agatoxin (100 μm) and conotoxin (100 μm) were dissolved in water and stored at −30°C. Working solutions were prepared freshly on the day of the experiments. All chemicals were from Sigma, except TTX, agatoxin, and conotoxin (Alomone Labs) and SB366791 (R&D Systems).
Input resistance was measured in voltage clamp and calculated by dividing the amplitude of the voltage step (−5 mV, 30 ms, from −70 mV) by the amplitude of the current response at steady state. Membrane potential values reported were not corrected for liquid junction potentials; the junction potential difference between different extracellular solutions at different temperatures was negligible (≤1 mV).
Activation curves were calculated from peak currents measured at −80 mV; nimodipine-sensitive currents were obtained by offline subtraction of traces obtained in nimodipine from those in control. Activation and inactivation curves were fitted with functions based on the Boltzmann function:
where V is the membrane potential, V1/2 is the potential at which the value of the function is 0.5, and k is the slope factor.
Quantitative RT-PCR.
Hippocampal CA1 area was dissected out of 500 μm slices from two 11-d-old rats. Left and right hemispheres were kept separate; thus, a total of four independent samples were used. RNA was extracted from tissue using a Qiagen RNeasy Plus mini kit and reverse transcribed into cDNA using the Roche First-Strand cDNA Synthesis kit. The following primers were designed for quantitative RT-PCR: Cav1.1 (left, caatttgccgaaatggagat; right tgagggaagtcctccaaaaa), Cav1.2 (left, tggctcacagaagtgcaaga; right, agcatttctgccgtgaaaag), Cav1.3 (left, gggctacttggactggatca; right, ctggtgggcatgctagtgt), Cav1.4 (left, tgtcctcaaccttgtgcttg; right, tagccccgaaggtcctct), and GAPDH (left, ctgcaccaccaactgcttag; right, tgatggcatggactgtgg). RT-PCR was performed using a Roche LC480 cycler with Roche SYBR Green master mix, primers (0.4 μm), and CA1 cDNA. All PCR reactions were run using a 6 min incubation at 95°C, followed by 45 cycles of 10 s at 95°C, 15 s at 60°C, and 10 s at 72°C. All samples were run in duplicate; each detectable Cav signal was normalized to GAPDH, and the four CA1 samples were averaged. Melting temperature analysis demonstrated a single peak for each gene product. Samples were also run on 1.8% agarose gels to verify single gene products.
No signal was detected for Cav1.1 and Cav1.4. Standard curves were obtained using dilutions of hippocampal cDNA to calculate reaction efficiency for Cav1.2 and Cav1.3. All data were efficiency corrected using Roche LC480 software. Primers had the following efficiencies: Cav1.2 = 1.98, slope = −3.368; Cav1.3 = 2.005, slope = −3.309. Negative controls performed using samples without RT did not yield any signal for Cav1.2 and Cav1.3.
In vivo FS model and recordings.
All procedures were performed using methods approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. For our in vivo experiments, we slightly modified a well-established experimental model of FSs in immature rats (Baram et al., 1997) by combining lipopolysaccharide (LPS), a Toll-like receptor 4 ligand, with hyperthermia to induce seizures. LPS was added to potentiate the effect of hyperthermia and to decrease the maximal temperature needed to cause seizures. We believe that our model better simulates fever, a regulated increase in Tb from an immune challenge, as opposed to hyperthermia, an excessive heat load that mobilizes heat dissipation mechanisms. We used this modified rodent model of FS to investigate the therapeutic efficacy of nimodipine to suppress fever-induced seizures in immature rats. On postnatal day 14 (P14), the Long–Evans rat pups (Charles River Laboratories) were given an intraperitoneal injection of LPS (100 μg/kg) 2.5 h before induction of hyperthermic seizures and were kept in a euthermic (30.0°C) incubator until seizure induction. At 30 min before seizure induction, rats were injected with either vehicle (DMSO) or nimodipine (2.5 mg/kg dissolved in DMSO). The dose of nimodipine was chosen to minimize negative side effects observed at high doses, possibly attributable in part to inhibition of non-neuronal L-type currents in the cardiovascular system. At 30 mg/kg (Wurpel and Iyer, 1994), rats showed signs of sedation and decreased ambulation followed by frequent shivering and slow, weak, rhythmic hindlimb and forelimb clonus, as well as rhythmic chewing/chomping and head jerks. The duration of these behaviors decreased as the nimodipine concentration was decreased from 30 mg/kg (n = 7), to 15 mg/kg (n = 2), to 5 mg/kg (n = 3). At 2.5 mg/kg (n = 16), nimodipine-treated animals were indistinguishable from vehicle-injected littermates with respect to side effects.
To induce hyperthermic seizures, a pair of pups (one nimodipine-treated and one vehicle-treated) was placed in a Plexiglas chamber. Core Tb were elevated at a rate of 0.5°C every 2 min by controlling the distance of a heat lamp positioned above the chamber. Tb was continuously monitored using a rectal temperature probe (RET-4; Physitemp) connected to a digital thermocouple thermometer (WD-35427-20; Oakton Instruments). Tb was raised until a seizure occurred or a temperature >41.5°C was reached (Baram et al., 1997; Oakley et al., 2009). Behavioral seizures induced by hyperthermia consisted of sudden arrest of movement, followed by facial automatisms (chewing), clonic jerks, and finally generalized (tonic and/or clonic) seizures. Two observers blinded to the treatment independently recorded the seizures. If an animal's temperature reached >41.5°C, the animal was removed from the hyperthermia chamber and placed on a cool metal surface for at least 2 min and until Tb was below 41°C. After 30 min of hyperthermia, rats were placed on a cool surface until Tb returned to normal range for age (34–35°C) and then were returned to their dams.
A subset of P11–P12 animals (n = 9) were implanted with mouse EEG head mounts (catalog #8201; Pinnacle Technology). Rats were aligned in a stereotaxic apparatus and anesthetized with isoflurane/O2. A skin incision was made, and the head mount was placed on the exposed surface of the skull. Two pairs of screw electrodes (catalog #8209; Pinnacle Technology) were drilled through the skull to rest on the cerebral cortex, positioned bilaterally anterior to bregma and bilaterally anterior to the lambdoid suture. A two-part epoxy (SEC1233; Resinlab) was used to ensure electrical conductivity between the screw electrodes and head mount. Dental acrylic was applied over the screws and head mount to secure and insulate the apparatus. The skin incision was sutured closed, and the pup was placed on a heating pad until consciousness returned, at which point it was returned to its dam.
Ictal discharges were defined as electroclinical entities manifested by high-voltage (>200 μV) rhythmic discharges that evolve over time accompanied by generalized convulsion (tonic and/or clonic). Interictal discharges were high-voltage spikes and slow-wave or sharp-wave discharges that occur either in runs or in isolation with no time-dependent evolution and no discernible changes in animal's behavior. EEG analysis and quantification were performed by an electroencephalographer (S.K.) who was blinded to the identification of animals.
Statistical analyses.
Changes caused by treatments of individual cells were assessed using paired t tests. Gating parameters were compared using unpaired, two-tailed t tests, if not otherwise stated. Comparisons of the number of generalized tonic-clonic (GTC) FSs and duration of ictal and interictal discharges between groups were made using Fisher's exact test and Student's t tests, respectively. All data are presented as mean ± SEM. Error bars in the figures also represent SEM.
Results
Temperature-dependent spontaneous firing of pyramidal neurons
We performed patch-clamp recordings from CA1 pyramidal neurons in acute hippocampal slices at temperatures ranging from mild hypothermia (32–34°C) to moderate hyperthermia (38–40°C). A first set of recordings was performed extracellularly (loose seal, pipette containing HEPES-buffered ACSF; Fig. 2A,B) to preserve physiological ionic gradients and intracellular pathways. Mild temperature increases induced neuronal firing; 59% of pyramidal neurons recorded in control ACSF (n = 17) and 35% of the 63 cells recorded in the presence of blockers of fast synaptic transmission (2 mm kynurenic acid and 0.1 mm picrotoxin) became spontaneously firing with moderate hyperthermia. The threshold temperature for firing was 38.3 ± 1°C in control ACSF and 38.3 ± 0.3°C in the presence of synaptic blockers. Thus, intact hippocampal pyramidal cells often fire intrinsically starting at temperature ∼38°C (Fig. 2C). We wondered whether this finding is restricted to the hippocampus or represents a general feature of pyramidal neurons; to this end, extracellular recordings were performed in the entorhinal and occipital cortices in the presence of blockers of fast synaptic transmission. Figure 2D shows that temperature-dependent firing was detected in these areas as well, although in the occipital cortex the fraction of cells showing this phenotype was smaller than in the other areas.
Figure 2.

Temperature-dependent intrinsic firing of pyramidal neurons. A, B, Extracellular recordings of pyramidal neurons in acute hippocampal slices in physiologic ACSF (A) and in the presence of blockers of fast synaptic transmission (B). When the temperature (bottom traces) reached ∼38°C, cells started firing. The apparent increase in spike amplitude at high temperature is attributable to higher seal resistance (Fig. 1). C, Firing frequency in intact neurons plotted against bath temperature for cells recorded in ACSF (open symbols) and in the presence of kynurenic acid and picrotoxin (filled symbols). Bars above the plots show approximately the boundaries for hypothermia (white), normothermia (gray), and hyperthermia (black). D, Temperature-dependent firing was also detected in pyramidal cells in the entorhinal (EC; 35.7% of the cells, n = 14) and occipital (OC; 18.2% of the cells, n = 22) cortices. All the recordings in these areas were performed after blockade of fast synaptic transmission. E, Whole-cell recording from a pyramidal neuron in a hippocampal slice in ACSF while increasing the temperature (bottom trace) to 40°C. F, Similar recording in the presence of blockers of fast synaptic transmission.
Whole-cell recordings were performed from CA1 pyramidal neurons, an area that shows a robust effect of hyperthermia, to investigate the underlying mechanisms. For these experiments, the bath contained blockers of fast synaptic transmission. In keeping with the extracellular measurements, these recordings showed temperature-dependent depolarizations large enough to elicit intrinsic firing both in control conditions and after blockade of fast synaptic transmission (Fig. 2E,F). Similar to the extracellular recordings (Fig. 3A), the hyperthermia-induced firing was reversible during cooling and did not lead to any apparent permanent cell damage, because cells responded with normal firing patterns to injection of depolarizing current even after being held at 38–40°C for ≥20 min (Fig. 3B,C). Also, the fraction of cells that depolarized (39%; 16 of 41 cells) was similar to that in extracellular recordings performed in the presence of blockers of fast synaptic transmission. However, several neurons showed a hyperpolarizing response or did not respond [44% (18 of 41 cells) and 17% (7 of 41 cells), respectively]. The response variability may in part depend on the composition of the intracellular solution; for instance, intracellular gluconate, which was used for these recordings, may induce a partial block of high-threshold calcium currents (Velumian et al., 1997). In keeping with this hypothesis, when cells were patched using a K-phosphate internal solution, five of six cells fired in response to hyperthermia and only one of six hyperpolarized (Fig. 4). Although this observation is based on a relatively small sample, when considered together with the fact that temperature led to a decrease in input resistance in all the cells in which it was monitored (it decreased from 262.8 ± 18.7 MΩ at 33°C to 185.4 ± 25.6 MΩ at 39°C, independently of the effect on resting potential; n = 6), it suggests that multiple temperature-sensitive channels having different ionic permeability contribute to the temperature response of hippocampal neurons; however, the temperature-dependent depolarization is a robust phenomenon (Kim and Connors, 2012) that can be maintained after dialysis of the intracellular medium.
Figure 3.

Temperature up to 40°C does not lead to acute neuronal damage. A, Tridimensional plot showing that temperature-induced firing is reversible during cooling of the bath solution. Firing frequency was extracellularly recorded from a pyramidal neuron and plotted against temperature and recording time. Note the reversibility of the temperature-induced firing. B, Whole-cell recordings of the voltage responses of a pyramidal neuron to hyperpolarizing and depolarizing current injections before (−100 and +60 pA) and during (−120 and +60 pA) a brief exposure to hyperthermic temperature; the cell showed very similar firing patterns before and during hyperthermia. C, Pyramidal neurons exhibit normal electrophysiological responses to current injections (−340 and +160 pA) after >20 min in hyperthermic conditions. The resting membrane potential of this cell after 20 min at 39°C was −59 mV. Intracellular recordings shown here were performed using K-gluconate internal solution.
Figure 4.

Temperature-dependent depolarization is independent of the intracellular solution. A, When CA1 pyramidal neurons were recorded using K-phosphate-based intracellular solution, they showed temperature responses indistinguishable from cells recorded using K-gluconate solution. B, Qualitative summary of temperature responses observed in cells recorded using the K-phosphate solution (6 cells).
L-type calcium channels and NALCN-like channels mediate intrinsic firing
The observation that CA1 pyramidal neurons depolarize in response to moderate hyperthermia even in the absence of fast synaptic transmission suggests that temperature acts on intrinsic neuronal properties. To promote firing, the depolarizing channels must show substantial activity at resting membrane potential; this suggests that they may be voltage independent. Indeed, a temperature-dependent response mediated by transient receptor potential TRPV4 channels was reported previously in cultured hippocampal neurons (Shibasaki et al., 2007); additionally, expression of temperature-gated TRPV1 channels (Caterina et al., 1997) has been suggested in the hippocampus using immunohistochemical methods (Tóth et al., 2005), and TRPV1 channels have been implicated in long-term plasticity of synapses on hippocampal interneurons (Gibson et al., 2008). However, contrary to our expectations, bath application of the unspecific TRP channel blocker ruthenium red failed to prevent intrinsic firing (whole-cell recordings, n = 6; data not shown). In fact, in the presence of ruthenium red, several cells became intrinsically firing even at low temperature, probably because of blockade of background leakage conductance mediated by TASK3 channels, which are effectively blocked by ruthenium red (Berg et al., 2004; Kang et al., 2004) and abundantly expressed in hippocampal pyramidal neurons (Taverna et al. 2005). Therefore, to further investigate whether a TRPV1-mediated current may be involved in the temperature-dependent depolarization of these cells, we performed voltage-clamp recordings to examine the effect of the TRPV1-specific blocker SB366791 [4′-Chloro-3-methoxycinnamanilide] (Gunthorpe et al., 2004) on the background current (recorded at −65 mV) at high temperature (39°C). However, these experiments failed to identify any temperature-activated TRPV1-mediated background current in pyramidal neurons (Fig. 5), suggesting that other channels mediate the temperature-dependent depolarizing response. Voltage-clamp recordings were then used to characterize the temperature-sensitive depolarizing background conductance. For these recordings, we used a CsCl-based intracellular solution containing 5 mm QX-314 (to block voltage-gated sodium and potassium channels), and the neurons were voltage clamped with slow (0.12 V/s) ramps from −100 to 20 mV; the ramps were applied at a frequency of 0.6 Hz while the temperature of the bath solution was increased from ∼25 to 40°C at a rate of ∼3–5°C/min (Fig. 6A). In these conditions, the temperature increase produced a substantial background depolarizing current associated with a large and reversible leftward shift in the activation range of a voltage-sensitive inward current. The potential at which the current peaked was left shifted by 1.04 mV/°C, from −18.7 ± 1.9 mV at 29°C to −30.1 ± 2 mV at 40°C (in 2 mm [Ca2+]o; Fig. 6B). Very similar data were obtained when the intracellular solution was K-gluconate, the same used for current-clamp recordings; in this case, the temperature-dependent peak shift was 0.97 mV/°C (data not shown). This QX-314-resistant, temperature-sensitive current was blocked by bath application of cadmium (50 μm; Fig. 6C,D), suggesting that it was carried by calcium ions, and its activation in response to a slow voltage ramp suggested the involvement of L-type channels. Therefore, we tested whether nimodipine (10 μm), a selective L-type Ca2+ channel blocker, had any effect on the temperature-sensitive current. For these experiments, 2 mm extracellular barium was used as charge carrier, and phosphocreatine (10 mm) was added to the pipette solution to maximize current stability. Nimodipine promptly reduced the current (by 52 ± 4.3%; Fig. 6E,F). In contrast, neither the P/Q current blocker ω-agatoxin IVa (500 nm) nor N-type blocker ω-conotoxin GVIa (20 μm) nor 50 μm Ni2+ (a concentration selective for α1H channels (Lee et al., 1999) exhibited any detectable effect on the temperature-sensitive current (data not shown). Although current mediated by the Cav1.3 subunit only accounts for a very small current in pyramidal neurons (Moosmang et al., 2005), this current activates at relatively hyperpolarized membrane potentials, and therefore, we thought it may mediate, at least in part, the background calcium current. Because the Cav1.3 current is less sensitive to dihydropyridines relative to other L-type subunits (Xu and Lipscombe, 2001), we tested lower concentrations of nimodipine to unmask a possible contribution of this current. However, contrary to our expectations, we found that the block saturated already at 3 μm, and that 500 nm nimodipine blocked ∼65% of the current sensitive to 3 μm nimodipine (Fig. 6F), suggesting that Cav1.3 subunits were unlikely to mediate a large fraction of this current.
Figure 5.
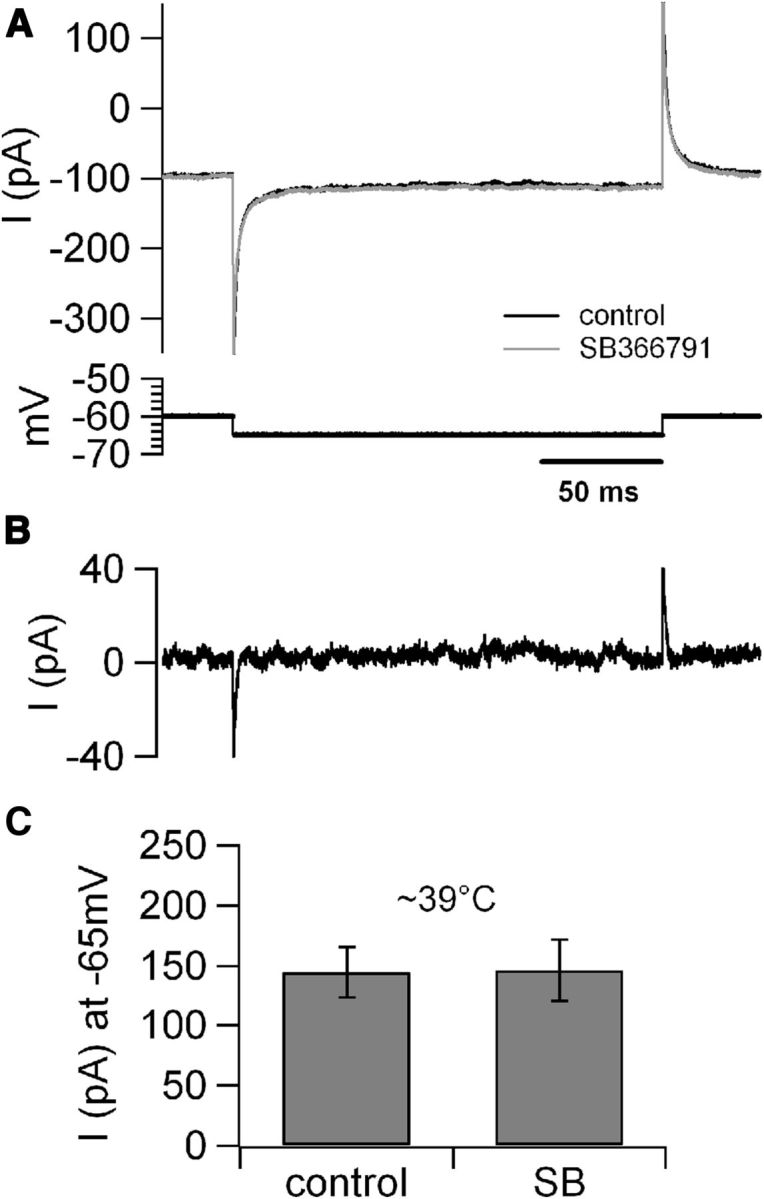
Temperature increase does not produce detectable TRPV1-mediated background current in hippocampal pyramidal neurons. A, Top traces, Voltage-clamp recordings obtained at ∼39°C from a CA1 pyramidal cell in control conditions and in the presence of bath-applied SB366791 (10 μm); bottom trace, voltage protocol. B, Offline digital subtraction of the trace in SB366791 from the control trace shows no evidence for a TRPV1-sensitive background current at ∼39°C (4 cells). C, Bar chart summary of the background current recorded at ∼39°C in control conditions and in the presence of SB366791 (SB).
Figure 6.

Temperature modulates L-type calcium current in CA1 neurons. A, Whole-cell current recording of a CA1 pyramidal cell at 26.5°C (black trace) and at 39.2°C (gray trace) in response to a slow voltage ramp from −100 to +20 mV (0.12V/s, top trace). B, The voltage at which the current peaked plotted versus recording temperature. These data were obtained from recordings performed in extracellular solution containing 2 mm Ca2+ (n = 6) or 2 mm Ba2+ (n = 11); in both conditions, temperature increase induced a shift in the hyperpolarizing direction of ∼1mV/°C. C, The temperature-sensitive current was carried by calcium ions, because it was blocked by CdCl2 (50 μm, dotted trace). D, Average peak current measured at high temperature (38.6 ± 0.2°C, 6 cells) in 2 mm external Ba2+ in control conditions and in the presence of 50 μm CdCl2. E, Current recordings obtained at 39.2°C in control conditions (gray trace) and after focal application of 10 μm nimodipine (black dotted trace). The traces in A, C, and E were obtained from the same neuron with sequential drug applications. F, Left, Different concentrations of nimodipine (3 μm, n = 4 and 10 μm, n = 5, applied focally at 35.4–39.3°C) induced similar current block. Right, In a different set of recordings, 500 nm nimodipine blocked ∼65% of the nimodipine-sensitive current (defined as the current blocked by 3 μm nimodipine in the same cells, n = 3, recorded at 36.1 ± 0.5°C). *p < 0.05.
In keeping with the hypothesis that the nimodipine-sensitive current initiates the temperature-induced depolarization, the size of this current was substantial even at hyperpolarized membrane potentials (−108 ± 28 pA at −65 mV; Fig. 7A,B). However, L-type currents are typically high threshold. To further investigate the nature of the channels mediating the temperature response, the activation of the nimodipine-sensitive component was studied at low and high temperature. Current–voltage responses were recorded (using 2 mm Ba2+ as charge carrier and in the presence of intracellular QX-314) in control and in the presence of 3 μm nimodipine either at low or high temperature. Intriguingly, the half-activation potential of the nimodipine-sensitive current was barely changed when the temperature was raised from 26°C to 35°C, whereas an additional increase of only 3°C caused an ∼9 mV leftward shift in the activation midpoint (it was −34 ± 3.5 mV at 35°C and −43.5 ± 1.1 mV at ≥38°C for four and five cells, respectively; Fig. 7C). Although it is important to keep in mind that these values were obtained in whole-cell recordings in brain slices and therefore in suboptimal clamp conditions, this observation may suggest a nonlinear dependence of gating on temperature. More studies will be required to verify this observation. Next, we investigated the temperature effects on the inactivation curve. However, for these experiments, pharmacological subtraction of the nimodipine-sensitive current could not be performed because of the strong voltage sensitivity of nimodipine binding, which resulted in dramatically different binding during the long (5 s) prepulses used for these measurements. Therefore, to study inactivation, Ba2+ currents were recorded using a QX-314-containing CsCl internal solution in control conditions and in the presence of extracellular cadmium (50 μm) and then isolated by offline subtraction. These recordings showed that the inactivation properties of native neuronal calcium currents of pyramidal neurons are strongly temperature sensitive: increasing the temperature by ∼12°C shifted the midpoint of the inactivation curve to more depolarized potentials by ∼10 mV (−47.8 ± 1 mV at >38°C, n = 6, vs −57.9 ± 1.8 mV at 27°C, n = 7; Fig. 7D); the slope of the curve was also strongly affected (11.8 ± 1.0 mV at >38°C vs 18.3 ± 0.2 mV at <26°C, respectively; Fig. 7D).
Figure 7.

High temperature affects both activation and inactivation of native neuronal L-type current. A, Current recordings obtained at −65 mV at 28.5°C (black trace), 39°C (light gray), and 39°C in the presence of 10 μm nimodipine (focally applied, dark gray). The traces represent the CdCl2-sensitive components obtained by offline digital subtraction. B, Average CdCl2-sensitive current recorded at −65 mV in control temperature (27 ± 0.2°C) and at >37°C in control conditions and in the presence of nimodipine (3–10 μm, n = 8). C, Temperature increase induced a leftward shift in the current–voltage curve. The temperature dependence of this effect was most pronounced around the normothermia–hyperthermia boundary. Fitting Boltzmann curves to the data points yielded the following midpoints (indicated by the dotted lines) and slopes: at ≤26°C, V1/2 = −37.9 ± 0.9 mV, k = 8.2 ± 0.8 mV (n = 6); at 35°C, V1/2 = −34 ± 3.5 mV, k = 10.3 ± 1.2 mV (n = 4); and at ≥38°C, V1/2 = −43.5 ± 1.1 mV, k = 5.46 ± 1.3 mV (n = 5). Thus, an ∼9°C increase did not produce any significant shift in half-point or slope, whereas an additional 3°C increase caused a 9.5 mV shift in half-point and an ∼40% change in slope. D, Temperature also affected the inactivation curve. At <28°C, V1/2 was −57.9 ± 1.8 mV, and the slope was 18.3 ± 2.0 mV (n = 6). At temperatures >38°C, V1/2 was −47.8 ± 1.0 mV, and the slope was 11.8 ± 1.0 mV (n = 7). These differences resulted in a large change in channel availability at membrane potential around resting: at −66 mV, the availability increased from 68% at low temperature to 83% at >38°C, as evidenced by the dotted lines.
These data revealed that the gating of a nimodipine-sensitive calcium current of hippocampal pyramidal neurons is strongly regulated by temperature and thus may mediate temperature-dependent firing in these cells. However, the molecular identity of the channels mediating the current remained unclear. Quantitative RT-PCR from isolated CA1 tissue showed abundant expression of Cav1.3 and Cav1.2 subunits (Fig. 8A,B), whereas Cav1.1 and Cav1.4 were not detected (data not shown). Although Cav1.3 expression was more abundant, the high nimodipine sensitivity of the current (Fig. 6F) and the minimal residual L-current in transgenic animals lacking the Cav1.2 subunit (Moosmang et al., 2005) suggest a more prominent role for Cav1.2. To verify this hypothesis, we studied the temperature response in hippocampal pyramidal cells from Cav1.3 knock-out (KO) mice. These measurements showed that the temperature-sensitive current in these cells was almost indistinguishable from that in cells from our control animals (Fig. 8C,D). Similarly, current-clamp recordings (n = 8) in slices from mice lacking the Cav1.3 subunit found that the response to temperature increase of pyramidal cells was similar to that of cells from wild-type rats (Fig. 8E,F). Thus, both voltage- and current-clamp data strongly support the idea of a critical role for Cav1.2 subunits.
Figure 8.

The temperature-sensitive current is primarily mediated by Cav1.2 subunits. A, Agarose gel depicting a single product for Cav1.2 and Cav1.3 after real-time PCR amplification of CA1 tissue from 11-d-old rats. B, Amount of Cav1.2 and Cav1.3 mRNA normalized to GAPDH. C, Recordings of the current response of a CA1 pyramidal cell in a slice from a Cav 1.3 KO mouse in response to a slow voltage ramp (using 2 mm Ba2+ as charge carrier) obtained at low (28.2°C, black trace) and high (38.6°C, gray trace) temperature. All five of the cells that were tested in these mice showed the temperature-sensitive current. D, The voltage at which the current peaked plotted versus temperature for cells obtained from control animals (open symbols; n = 11) and Cav 1.3 KO animals (filled symbols; n = 5). The voltage shift in Cav1.3 KO mice (−0.87 mV/°C) was similar to that in control animals (−0.90 mV/°C), suggesting that Cav1.3 subunits are not critical for the temperature sensitivity of the current. E, Voltage trace showing the temperature response of a CA1 hippocampal pyramidal cell from a Cav1.3 KO mouse measured using whole-cell current clamp; bottom trace, simultaneous bath temperature recording. F, Summary of the fraction of cells that depolarized in response to temperature increase in wild-type rats (control) and in Cav1.3 KO mice. Internal solution was K-gluconate (see Materials and Methods) in both cases. Data from 41 and 8 cells, respectively.
Although these data support the notion that the temperature-activated, voltage-dependent depolarizing current of hippocampal pyramidal neurons is mediated by Cav1.2 subunits, the size of the background current at −65 mV appears larger than what may be expected from the activation curve of these channels, even considering a window-current effect. Therefore, we studied in more detail the ionic selectivity of the background current. Because a fraction of this current persisted in the presence of nimodipine and cadmium (Fig. 6C,E), we hypothesized that it was not calcium mediated and explored the possibility that it may be carried by sodium ions, possibly through NALCN (sodium leak channel nonselective protein)-like channels (Lu et al., 2007). Two sets of experiments were performed to address this point. In a first set of measurements, the size of the temperature-sensitive background (active at −65 mV) current was studied in control conditions and after substitution of extracellular sodium by choline (while keeping the calcium concentration constant). Figure 9A shows that choline substitution did not produce detectable effects at 30°C, although it inhibited a sizable current at 39°C. Interestingly, the size of the choline-blocked current at high temperature was similar when calcium channels were blocked by bath application of 0.1 mm cadmium (Fig. 9B), suggesting that this current is not carried or regulated by calcium ions. In a second set of recordings, we tested the effect of gadolinium (0.1 mm, a blocker of NALCN currents) on the size of the temperature-sensitive background inward current. Because gadolinium is unspecific and also blocks calcium currents (Lansman, 1990), it was always tested in the presence of 0.1 mm CdCl2 to avoid contamination from blockade of calcium currents. Intriguingly, gadolinium did not have any detectable effect in control temperature (∼30°C; Fig. 9C,D) but inhibited a sizable current at high temperature (at ∼39°C, the Gd-sensitive current at −65 mV was −32.3 ± 5.3 pA, four cells; Fig. 9C,D), supporting the hypothesis that a calcium-independent cationic current becomes active at rest in elevated temperature conditions and may contribute to neuronal firing.
Figure 9.

Sodium current contributes to the temperature-sensitive background conductance. A, Comparison of conductance at −65 mV in control ACSF and after substitution of 100 mm external Na+ with choline. In choline–ACSF, the background conductance was reduced by 13% (from 0.67 ± 0.26 to 0.59 ± 0.28 nS) at 30 ± 0.3°C and by 49% (from 0.81 ± 0.38 to 0.41 ± 0.29 nS) at 38.8 ± 0.5°C. B, Average conductance recorded in control ACSF + 100 μm CdCl2 and in choline–ACSF + 100 μm CdCl2. Conductance was reduced by 21% (from 0.41 ± 0.13 to 0.33 ± 0.12 nS) at 30.3 ± 0.2°C and by 41% (from 0.65 ± 0.24 to 0.38 ± 0.16 nS in choline) at 39.4 ± 0.3°C. C, Gadolinium-sensitive current (obtained by offline digital subtraction) recorded at 28.9°C (black trace) and 38.7°C (gray trace). D, Average gadolinium-sensitive current measured in CA1 pyramidal cells at −65 mV. No significant current (0.51 ± 0.3 pA, n = 3) was observed at 29.9 ± 0.5°C, whereas at 38.7 ± 0.2°C (n = 4), the current had a mean amplitude of −32.3 ± 5.3 pA.
Nimodipine prevents temperature-dependent firing in vitro and FSs in vivo
Our data show that hippocampal pyramidal cells express at least two different temperature-activated depolarizing currents: (1) a nimodipine-sensitive calcium current; and (2) a calcium- and voltage-independent sodium current. What is their relative contribution to intrinsic firing? If the temperature-dependent firing depends on the nimodipine-sensitive current, it should be blocked by this drug. Current-clamp recordings confirmed this hypothesis; in control conditions (K-gluconate intracellular solution, extracellular solution containing 2 mm CaCl2, 2 mm kynurenic acid, and 50 μm picrotoxin), 39% of the tested neurons (n = 41) fired in response to an ∼10°C temperature increase (Fig. 10A). In contrast, in the presence of even low concentration of nimodipine (500 nm), intrinsic firing was dramatically decreased because it was detected in only 15% of the tested neurons (n = 13), a number similar to that observed when the nimodipine concentration was raised to 10 μm (depolarization was detected in only 1 of 13 cells tested; Fig. 10B,C). These data support the critical role of nimodipine-sensitive currents in temperature-dependent hyperexcitability of cortical neurons, which may be relevant to FSs. Thus, we took advantage of a rodent model to test whether nimodipine could prevent the development of FSs in vivo. These experiments showed that nimodipine (2.5 mg/kg) reduced both the incidence and duration of GTC FSs in rat pups (Fig. 10D,E); A total of 81% (13 of 16) of vehicle-treated rats progressed to GTC seizures, whereas only 18% (3 of 16) of nimodipine-treated rats experienced GTC seizures (p < 0.05, Fisher's exact test). Electrographic monitoring further strengthened these results, showing that nimodipine reduced the duration of ictal discharges from 59 ± 21 s in control to 5 ± 5 s in nimodipine-treated rats (four and five animals, respectively; p < 0.05, Student's t test) during the period of hyperthermia, whereas the duration of abnormal interictal activity was unaffected (449 ± 151 s in control and 422 ± 118 s in nimodipine-treated rats).
Figure 10.

Nimodipine prevents temperature-induced firing in vitro and FSs in vivo. A, Voltage response (top trace) of a CA1 pyramidal cell to a temperature increase (bottom trace) in ACSF containing 2 mm Ca2+. B, Similar recording obtained in the presence of 500 nm nimodipine in the bath. C, Bar chart summarizing the percentage of CA1 pyramidal cells that depolarized and/or fired after an increase to hyperthermic temperatures in control conditions and in 500 nm and 10 μm nimodipine. The fraction of cells that depolarized dropped from 39% in control to 15% in 500 nm and to 8% in the presence of 10 μm nimodipine (n = 41, 13, and 13 cells, respectively). D, Bar chart summarizing the effect of nimodipine (nimo; 2.5 mg/kg) on FSs in vivo. Increasing the Tb to 41.5°C led 81% of control animals to develop GTC FSs (n = 16), whereas only 18% of nimodipine-treated animals (n = 16) exhibited GTC seizures (p < 0.05). E, EEG recording of a GTC FS in a control animal (top). Only interictal epileptiform discharges were captured in one of four control and in four of five nimodipine-treated animals (bottom). F, Bar chart summarizing the duration of ictal electrographic discharges in control animals (n = 4) and nimodipine-treated animals (n = 5). Nimodipine significantly decreased the average duration of ictal discharges (p < 0.05).
Discussion
We performed patch-clamp recordings from acute rat brain slices to investigate the electrophysiological properties of cortical pyramidal neurons in normothermic and hyperthermic conditions. We found that several hippocampal and extrahippocampal pyramidal neurons are intrinsically firing at temperatures ∼38°C (normothermic in the rat) and that febrile temperature (39–40°C) dramatically increases the fraction of intrinsically active neurons. We also found that intrinsic firing of pyramidal neurons depends on a temperature-dependent calcium current, mediated in large part by Cav1.2 subunits and is supported by a temperature-activated sodium conductance. Finally, we showed that nimodipine prevents FSs in rat pups. The importance of these results is twofold: (1) they offer new insights into the etiology and treatment of FSs; and (2) they suggest that the idea of a strict dichotomy in cortical cell phenotype, in which pyramidal neurons rest in the absence of excitatory synaptic input whereas few types of “pace-making” neurons spontaneously generate action potentials, needs to be reconsidered.
We found that nimodipine-sensitive calcium channels that are active at hyperpolarized membrane potentials are critical for the temperature-dependent firing. This notion may appear in contrast with the classical definition of L-type currents as “high threshold” but fits well with a previous study reporting that background activation of a nimodipine-sensitive current regulates (at 32°C) basal intracellular calcium in CA1 pyramidal neurons (Magee et al., 1996). Additionally, a temperature-dependent increase in native L-type conductance is not surprising, because shifts in the activation and inactivation properties were reported for native cardiac calcium currents (Tsien et al., 1987; Nobile et al., 1990), as well as recombinant Cav1.2 and Cav1.4 channels (Peloquin et al., 2008). Similar to our data, this paper reported temperature-induced changes in the channel gating, including a left shift in the activation and, to a lesser extent, inactivation curves. The native temperature-sensitive calcium currents in pyramidal neurons appear primarily mediated by Cav1.2 channels. This conclusion is supported by two sets of data: (1) already at 500 nm, nimodipine blocks ∼40% of the current, similar to a Cav1.2 current (Xu and Lipscombe, 2001); and (2) calcium currents in CA1 pyramidal cells from mice lacking the Cav1.3 subunit had a temperature response very similar to controls. Accordingly, our RT-PCR data show that abundant expression of Cav1.2 subunit in hippocampal CA1 area. Cav1.3 expression is higher, but the most likely explanation for this abundant expression is the high Cav1.3 expression in CA1 interneurons (Hell et al., 1993). Such an interpretation is further supported by the finding that, in transgenic mice lacking the Cav1.2 subunits, only a very small nimodipine-sensitive current is detectable in pyramidal cells [∼4% of the peak calcium current (Moosmang et al., 2005)], in keeping with the idea that the L-type current in pyramidal neurons is almost entirely mediated by Cav1.2 subunits.
Although the L-type current is the main temperature-sensitive conductance, we also found evidence for a second temperature-dependent depolarizing current. This current, which is carried by sodium ions, is mediated by NALCN-like channels, because it is primarily voltage insensitive and blocked by gadolinium. Acid-sensing ion channels share some of these properties, but they are extremely unlikely to mediate this current because they are inhibited—rather than activated—by temperature increases (Blanchard and Kellenberger, 2011). The temperature-sensitive sodium current did not appear to be dramatically calcium dependent, because it was present when intracellular calcium was strongly buffered (10 mm EGTA) and extracellular calcium influx was prevented by adding cadmium to the bath solution. Because nimodipine could prevent temperature-dependent firing, we suggest that, although the leakage sodium current may be crucial for keeping the cells within reach of the spike threshold, it is the nimodipine-sensitive conductance that contributes to the final push to reach across the threshold for spike generation. Similarly, although our experiments did not completely rule out a contribution of TRPV channels to the temperature sensitivity of these neurons (particularly at depolarized potentials), the efficacy of nimodipine in blocking the temperature-induced depolarization and firing supports the central role of L-type currents in this phenomenon.
Our data show that, in the presence of blockers of fast synaptic transmission, ∼35% of the hippocampal pyramidal cells recorded extracellularly fired in response to a temperature increase (to ∼40°C), similar to the ∼40% of cells recorded in whole-cell configuration that depolarized in response to a similar stimulus. Multiple explanations may account for cells that did not fire in extracellular configuration: (1) they could be cells that depolarized but did not reach threshold; (2) cells that did not depolarize; or (3) cells that were incapable of firing, possibly because they were in depolarization block. However, in whole-cell configuration, the temperature-dependent depolarizing current was found in all the cells that were recorded. This discrepancy can be reconciled considering that several temperature-sensitive channels coexist in pyramidal neurons and their relative functional impact may change in different experimental conditions, so that in some cells the effect of the depolarizing conductance is counterbalanced by hyperpolarizing currents. Background potassium (KCNK) channels play an important role in resting potential regulation of hippocampal pyramidal neurons (Taverna et al., 2005), and several of these channels display some degree of temperature sensitivity (Kang et al., 2005); thus, they may cause hyperpolarizing responses like the ones we observed in some current-clamp recordings. Because KCNK channels are inhibited by diverse metabolic pathways (Chemin et al., 2003), they may be mostly silent in intact cells and available to activation in the whole-cell configuration; alternatively, KCNK channels that are stretch-activated (Bang et al., 2000; Maingret et al., 2000) may be activated by the osmotic effects of intracellular dialysis in the whole-cell configuration. The presence of such hyperpolarizing currents in the whole-cell configuration is supported by the fact that nimodipine block of calcium current uncovered a small hyperpolarizing effect of temperature in whole-cell recordings (in 10 μm nimodipine, the membrane potential was −65.9 ± 1.1 mV at 27.6 ± 0.6°C and −68 ± 1.1 mV at 38.8 ± 0.4°C, n = 9). Finally, it is also likely that the size of the calcium current runs down in some whole-cell recordings as a consequence of intracellular dialysis (Marty and Neher, 1985; Belles et al., 1988; Rosenberg et al., 1988).
Another intriguing aspect of our data is that, contrary to the prevalent view of cortical glutamatergic neurons (McCormick and Shepherd, 2004), we found a significant fraction of pyramidal neurons to fire intrinsically at physiological temperature. Taddese and Bean (2002) showed that the gating properties of voltage-gated sodium channels are such that every neuron is potentially capable of intrinsic firing and that the presence of other background conductances prevents spontaneous firing in most cells. Here we suggest that nimodipine-sensitive calcium currents show a nonlinear temperature response that appears right at the border between normothermia and hyperthermia so that, at temperatures ≥38°C, nimodipine-sensitive calcium currents help tip the balance in favor of the sodium channels in a large fraction of pyramidal neurons, overcoming the effects of other conductances working in the opposite direction.
Finally, our data may be critical for understanding the etiology of FSs and for new, disease-specific, therapeutic approaches. Animal models show that hyperthermia promptly leads to FSs, although the mechanisms are not fully understood (Schuchmann et al., 2006). Currently, the treatment of FSs is limited and inadequate, often resulting in seizures lasting well over 40 min (Hesdorffer et al., 2011) and leading to detrimental long-term consequences on brain development (Dubé et al., 2009; Hesdorffer et al., 2011). Children with FSs are at increased risk for later epilepsy (Nelson and Ellenberg, 1976; Annegers et al., 1987), and febrile status epilepticus may be a predisposing factor to the subsequent development of chronic temporal lobe epilepsy (Hesdorffer and Hauser, 2002; Dubé et al., 2009). We show that, at febrile temperature, L-type calcium channels endow pyramidal neurons with a large depolarizing current that is active at the resting membrane potential and that may support intrinsic firing, which has been suggested to play a role in epileptic discharge (Traub et al., 1991). Additionally, the increased levels of intracellular calcium may induce potentiation of excitatory synaptic transmission, further promoting synchronous firing. Thus, this temperature-sensitive calcium current may represent a pharmacological target for the treatment of FSs. In keeping with this hypothesis, our in vivo data show that nimodipine dramatically reduced the incidence and duration of FSs in rats. Although dihydropyridines have well-known effects on the cardiovascular system, the behavioral effect observed with the low (2.5 mg/kg) concentration is most likely caused by direct effects on the brain, as is suggested by the EEG recordings. The clinical relevance of this result is further increased when we consider that conditions such as Dravet's syndrome and genetic epilepsy with febrile seizures plus are caused by loss-of-function mutations of the Nav1.1 sodium channel, which is predominantly expressed by inhibitory interneurons (Catterall et al., 2010). Thus, the use of traditional anti-epileptic drugs that act as sodium channel blockers (such as phenytoin, lamotrigine, and carbamazepine) is contraindicated in these patients. However, nimodipine acts via the inhibition of calcium channels and therefore could prove effective in these cases.
Footnotes
This work was supported by the Epilepsy Foundation (MM), NIH NS064091 (MM) & NS073768 (SK). We thank Dr. J. D. Surmeier for helpful discussions and Dr. G. Maccaferri for critical reading of this manuscript. Drs. J. D. Surmeier (Northwestern University, Chicago, IL) and Jörg Striessnig (University of Innsbruck, Innsbruck, Austria) provided the Cav1.3 KO mice.
The authors declare no competing financial interests.
References
- Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med. 1987;316:493–498. doi: 10.1056/NEJM198702263160901. [DOI] [PubMed] [Google Scholar]
- Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem. 2000;275:17412–17419. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- Baram TZ, Gerth A, Schultz L. Febrile seizures: an appropriate-aged model suitable for long-term studies. Brain Res Dev Brain Res. 1997;98:265–270. doi: 10.1016/S0165-3806(96)00190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belles B, Malécot CO, Hescheler J, Trautwein W. “Run-down” of the Ca current during long whole-cell recordings in guinea pig heart cells: role of phosphorylation and intracellular calcium. Pflugers Arch. 1988;411:353–360. doi: 10.1007/BF00587713. [DOI] [PubMed] [Google Scholar]
- Berg AP, Talley EM, Manger JP, Bayliss DA. Motoneurons express heteromeric TWIK-related acid-sensitive K+ (TASK) channels containing TASK-1 (KCNK3) and TASK-3 (KCNK9) subunits. J Neurosci. 2004;24:6693–6702. doi: 10.1523/JNEUROSCI.1408-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard MG, Kellenberger S. Effect of a temperature increase in the non-noxious range on proton-evoked ASIC and TRPV1 activity. Pflugers Arch. 2011;461:123–139. doi: 10.1007/s00424-010-0884-3. [DOI] [PubMed] [Google Scholar]
- Brauchi S, Orta G, Salazar M, Rosenmann E, Latorre R. A hot-sensing cold receptor: C-terminal domain determines thermosensation in transient receptor potential channels. J Neurosci. 2006;26:4835–4840. doi: 10.1523/JNEUROSCI.5080-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DO, Alving BO. A contribution of an electrogenic Na+ pump to membrane potential in Aplysia neurons. J Gen Physiol. 1968;52:1–21. doi: 10.1085/jgp.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Girard C, Duprat F, Lesage F, Romey G, Lazdunski M. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J. 2003;22:5403–5411. doi: 10.1093/emboj/cdg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Baram TZ, Soltesz I. Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat Med. 1999;5:888–894. doi: 10.1038/11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev. 2009;31:366–371. doi: 10.1016/j.braindev.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AL, Marmor MF. Temperature dependence of the sodium-potassium permeability ratio of a molluscan neurone. J Physiol. 1970;210:919–931. doi: 10.1113/jphysiol.1970.sp009249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis JB. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004;46:133–149. doi: 10.1016/S0028-3908(03)00305-8. [Erratum (2004) 46:905] [DOI] [PubMed] [Google Scholar]
- Hedrick T, Waters J. Spiking patterns of neocortical L5 pyramidal neurons in vitro change with temperature. Front Cell Neurosci. 2011;5:1. doi: 10.3389/fncel.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA. Febrile seizures and the risk of epilepsy. In: Baram TZ, Shinnar S, editors. Febrile seizures. San Diego: Academic; 2002. pp. 63–76. [Google Scholar]
- Hesdorffer DC, Benn EK, Bagiella E, Nordli D, Pellock J, Hinton V, Shinnar S, FEBSTAT Study Team Distribution of febrile seizure duration and associations with development. Ann Neurol. 2011;70:93–100. doi: 10.1002/ana.22368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Han J, Talley EM, Bayliss DA, Kim D. Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J Physiol. 2004;554:64–77. doi: 10.1113/jphysiol.2003.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Choe C, Kim D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol. 2005;564:103–116. doi: 10.1113/jphysiol.2004.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Connors BW. High temperatures alter physiological properties of pyramidal cells and inhibitory interneurons in hippocampus. Front Cell Neurosci. 2012;6:27. doi: 10.3389/fncel.2012.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB. Blockade of current through single calcium channels by trivalent lanthanide cations. Effect of ionic radius on the rates of ion entry and exit. J Gen Physiol. 1990;95:679–696. doi: 10.1085/jgp.95.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129:371–383. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D. Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:3460–3470. doi: 10.1152/jn.1996.76.5.3460. [DOI] [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E. TREK-1 is a heat-activated background K+ channel. EMBO J. 2000;19:2483–2491. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A, Neher E. Potassium channels in cultured bovine adrenal chromaffin cells. J Physiol. 1985;367:117–141. doi: 10.1113/jphysiol.1985.sp015817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick D, Shepherd GM, editors. The synaptic organization of the brain. Ed 5. Oxford: Oxford UP; 2004. [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med. 1976;295:1029–1033. doi: 10.1056/NEJM197611042951901. [DOI] [PubMed] [Google Scholar]
- Nobile M, Carbone E, Lux HD, Zucker H. Temperature sensitivity of Ca currents in chick sensory neurones. Pflugers Arch. 1990;415:658–663. doi: 10.1007/BF02584002. [DOI] [PubMed] [Google Scholar]
- Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci U S A. 2009;106:3994–3999. doi: 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peloquin JB, Doering CJ, Rehak R, McRory JE. Temperature dependence of Cav1.4 calcium channel gating. Neuroscience. 2008;151:1066–1083. doi: 10.1016/j.neuroscience.2007.11.053. [DOI] [PubMed] [Google Scholar]
- Rosenberg RL, Hess P, Tsien RW. Cardiac calcium channels in planar lipid bilayers. L-type channels and calcium-permeable channels open at negative membrane potentials. J Gen Physiol. 1988;92:27–54. doi: 10.1085/jgp.92.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchmann S, Schmitz D, Rivera C, Vanhatalo S, Salmen B, Mackie K, Sipil ä ST, Voipio J, Kaila K. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat Med. 2006;12:817–823. doi: 10.1038/nm1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki K, Suzuki M, Mizuno A, Tominaga M. Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. J Neurosci. 2007;27:1566–1575. doi: 10.1523/JNEUROSCI.4284-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE. The incidence and prevalence of febrile seizures. San Diego: Academic; 2002. [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/S0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Taverna S, Tkatch T, Metz AE, Martina M. Differential expression of TASK channels between horizontal interneurons and pyramidal cells of rat hippocampus. J Neurosci. 2005;25:9162–9170. doi: 10.1523/JNEUROSCI.2454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth A, Boczán J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135:162–168. doi: 10.1016/j.molbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66:635–650. doi: 10.1152/jn.1991.66.2.635. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Fox AP, Hess P, McCleskey EW, Nilius B, Nowycky MC, Rosenberg RL. Multiple types of calcium channel in excitable cells. Soc Gen Physiol Ser. 1987;41:167–187. [PubMed] [Google Scholar]
- Velumian AA, Zhang L, Pennefather P, Carlen PL. Reversible inhibition of IK, IAHP, Ih and ICa currents by internally applied gluconate in rat hippocampal pyramidal neurones. Pflugers Arch. 1997;433:343–350. doi: 10.1007/s004240050286. [DOI] [PubMed] [Google Scholar]
- Verity CM, Butler NR, Golding J. Febrile convulsions in a national cohort followed up from birth. I–Prevalence and recurrence in the first five years of life. Br Med J (Clin Res Ed) 1985;290:1307–1310. doi: 10.1136/bmj.290.6478.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RK, Traub RD, Miles R. Cellular basis of neuronal synchrony in epilepsy. Adv Neurol. 1986;44:583–592. [PubMed] [Google Scholar]
- Wu J, Fisher RS. Hyperthermic spreading depressions in the immature rat hippocampal slice. J Neurophysiol. 2000;84:1355–1360. doi: 10.1152/jn.2000.84.3.1355. [DOI] [PubMed] [Google Scholar]
- Wurpel JN, Iyer SN. Calcium channel blockers verapamil and nimodipine inhibit kindling in adult and immature rats. Epilepsia. 1994;35:443–449. doi: 10.1111/j.1528-1157.1994.tb02458.x. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]