

Abstract
The Neuropeptide S receptor, a Gs/Gq-coupled GPCR expressed in brain regions involved in mediating drug reward, has recently emerged as a candidate therapeutic target in addictive disorders. Here, we describe the in vitro and in vivo pharmacology of a novel, selective and brain penetrant NPSR antagonist with nanomolar affinity for the NPSR, NCGC00185684. In vitro, NCGC00185684 shows biased antagonist properties, and preferentially blocks ERK-phosphorylation over intracellular cAMP or calcium responses to NPS. In vivo, systemic NCGC00185684 blocks alcohol-induced ERK-phosphorylation in the rat central amygdala, a region involved in regulation of alcohol intake. NCGC00185684 also decreases operant alcohol self-administration, and lowers motivation for alcohol reward as measured using progressive ratio responding. These effects are behaviorally specific, in that they are observed at doses that do not influence locomotor activity or reinstatement responding following extinction. Together, these data provide an initial validation of the NPSR as a therapeutic target in alcoholism.
Introduction
Neuropeptide S (NPS) is the endogenous ligand for the Neuropeptide S receptor [NPSR; GPR154, or GPRA; (Xu et al., 2004; Reinscheid and Xu, 2005; Reinscheid, 2007)]. NPSR is coupled to both Gs and Gq G-proteins and its activation by NPS results in increased intracellular levels of cAMP and mobilization of intracellular Ca2+, respectively. In addition, activation of the NPSR leads to phosphorylation of extracellular regulated kinase (ERK) through a direct interaction with β-arrestin or activation of G-protein signaling pathways (Reinscheid et al., 2005). The existence of multiple downstream signaling pathways may suggest cell-type specific biological consequences of NPSR activation.
The NPSR is localized in anatomical regions involved in reward and emotionality (Xu et al., 2007; Leonard and Ring, 2011) and may mediate interactions of NPS with other neurotransmitters, such as corticotropin-releasing factor (CRF) in the paraventricular hypothalamus (Smith et al., 2006). Interactions between the NPS systems and these neurotransmitters could potentially mediate behavioral effects of NPS (Xu et al., 2007), as suggested by the observation that NPS fails to produce increased locomotor activity in CRF receptor-1 knock-out mice (Pañeda et al., 2009).
NPS has an unusual behavioral activity profile. Upon central administration, it produces anxiolytic-like effects, an effect accompanied by increased rather than suppressed arousal and locomotion (Xu et al., 2004) and enhanced rather than impaired memory formation (Han et al., 2009). Exogenous NPS promotes cue-induced reinstatement of alcohol and cocaine seeking (Cannella et al., 2009; Kallupi et al., 2010), and site-specific NPS injection into the lateral hypothalamus reinstates extinguished responding to alcohol (Cannella et al., 2009). The role of NPS in drug seeking and self-administration remains, however, unclear. For instance, intracerebroventricular administration of NPS to Indiana alcohol-preferring (P)-rats resulted in significantly suppressed alcohol consumption (Badia-Elder et al., 2008).
Limited availability of selective and brain penetrant NPSR antagonists hinders studies elucidating the role of endogenous NPS in addiction-related behaviors. Peptidergic NPSR antagonists have been explored, but their inability to cross the blood–brain barrier limits their utility (Camarda et al., 2009; Guerrini et al., 2009; Cifani et al., 2011). The small molecule compound SHA68 (Trapella et al., 2011) is a specific NPSR antagonist that reduces anxiolytic-like effects of exogenous NPS administration (Ruzza et al., 2010), but its utility is limited by its pharmacokinetics. We recently disclosed NCGC00185684 (NCGC84) as an NPSR antagonist with high affinity and selectivity for the NPSR (Patnaik et al., 2010). Systemically administered NCGC84 crossed the blood–brain barrier, and remained above its IC50 concentrations in the brain for over 48 h.
Here, we describe the discovery and in vitro pharmacology of NCGC84. We present in vitro pharmacology results indicating that NCGC84 is a biased NPSR antagonist, preferentially blocking NPS-induced ERK phosphorylation over intracellular Ca2+ or cAMP responses. We then examine the ability of NCGC84 to prevent alcohol-induced ERK phosphorylation in vivo in brain regions involved in regulation of alcohol-intake and reward, as well as relapse to alcohol-seeking. Finally, we use NCGC84 to examine the role of endogenous NPS in alcohol self-administration and relapse.
Materials and Methods
In vitro studies
Materials.
Ro 20-1724 (Cat # B8279) and probenecid were purchased from Sigma-Aldrich. The NPS peptide was synthesized by BiomerTech. Y10-NPS labeled with 125I was obtained from NEN PerkinElmer. All cell culture reagents were obtained from LifeTechnologies.
Cell culture.
Chinese hamster ovary (CHO-K1) cells stably expressing the NPS receptor were generated for this series of studies by transfection with a human NPS receptor clone kindly provided by Dr. Reinscheid, UC Irvine, Irvine, CA, and standard selection with zeocin and hygromycin. They were maintained in F12 Kaighn's media (ATCC) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 250 μg/ml hygromycin (Life Technologies) at 37°C with 5% CO2 in a humidified atmosphere.
Radioligand binding assay.
Radioligand binding assay was conducted as described previously (Xu et al., 2004). In brief, for the displacement binding assay, increasing concentrations of unlabeled human NPS or compounds were used to compete with 0.15 nm [125I]Y10-NPS for the binding to NPS receptor in a whole-cell binding assay. Nonspecific binding was determined in the presence of 1 μm unlabeled NPS. CHO-NPSR cells were seeded in 24-well plates at 40,000 cells/well and cultured at 37°C with 5% CO2 until reaching 95% confluence. Cells were then washed once with 1 ml of PBS and incubated with radioligand in DMEM medium containing 0.1% bovine serine albumin in the presence or absence of compounds at room temperature for 90 min. Cells were washed twice with ice-cold PBS to remove unbound radioligands and were then lysed in 0.5 ml/well 1 N NaOH. The radioactivity of bound radioligand in cell lysate was transferred to a test tube and counted in a gamma counter.
cAMP assay.
Intracellular cAMP levels were measured using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay with an HTRF cAMP kit (Cisbio) according to the manufacturer's instructions. In brief, CHO cells expressing NPSR were seeded at 20 μl/well with 10,000 cells in white, tissue-culture-treated 384-well plates (Greiner Bio-One). After overnight incubation at 37°C with 5% CO2, compound (NPS or antagonist) in assay buffer was added, followed by stimulation solution. The assay plates were then incubated for 30 min at 37°C with 5% CO2, followed by the addition of 10 μl/well detection reagent mixture of a d2-dye-conjugated cAMP (FRET acceptor) and cryptate- (Eu+) conjugated anti-cAMP antibody (FRET donor). After 30 min incubation at room temperature, the assay plates were measured in an EnVision plate reader (PerkinElmer) with TR-FRET detection mode (excitation = 320 nm; emission-1 = 615 nm and emission-2 = 665 nm with a delaying time of 60 s). The results were expressed as a ratio of the acceptor fluorescence intensity (665 nm) divided by the donor fluorescence intensity (615 nm). Because unlabeled cAMP in the cell lysate competes with the labeled cAMP, decrease in this signal reflects increase in cAMP produced in response to NPS.
Intracellular calcium assay.
Intracellular calcium was measured using the nonwash calcium assay Fluo8 kit (AAT Bioquest) according to the manufacturer's instructions. In this assay, the fluorescent calcium dye Fluo-4 AM enters cells by passive diffusion and is deesterified by endogenous esterases in the cytosol. It becomes fluorescent upon binding of calcium, resulting in fluorescent signals proportional to the cytosol free calcium concentration. CHO cells expressing NPSR were seeded as above and incubated overnight at 37°C with 5% CO2. Next day, growth media was aspirated and calcium dye added. Following incubation for 30 min at 37°C with 5% CO2 and 30 min at room temperature, compound in assay buffer was added and assay plates incubated at room temperature for 10 min. The plates were then placed into a fluorescence kinetic plate reader (μCell, Hamamatsu). The basal fluorescence intensity was recorded 10 times at 1 Hz for 10 s, and NPS prepared in assay buffer at EC80 was then added inside the instrument followed by additional reading at 1 Hz for 3 min. The results were normalized to the average basal fluorescence intensity in ratio and the peak response (Max.) was used for the result calculation (Liu et al., 2008).
ERK phosphorylation assay.
Intracellular pERK1/2 levels were measured using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay by a HTRF Cellul'erk kit (Cisbio) according to the manufacturer's instructions. Following overnight incubation of CHO-cells expressing the NPSR at 37°C with 5% CO2, the growth medium was replaced with 100 μl/well Opti-MEM medium (ATCC) and incubated at 37°C with 5% CO2 for an additional 4 h. The compounds prepared in 100% DMSO were then added to assay plate and incubated for 10 min at 37°C with 5% CO2. NPS solution (2 nm NPS peptide prepared in the Opti-MEM medium with 0.2% BSA and 0.005% Tweeen-20) was then added. After a 20 min incubation at 37°C with 5% CO2, the medium was aspirated and the plates placed on ice for 5 min followed by the addition of cell lysis solution. The plates were then incubated at room temperature while gently rocking (for mixing) for 15 min. An aliquot of 16 μl/well of cell lysate was transferred to a 384-well Greiner white half-well plate and 2 μl/well of the d2-dye-conjugated anti-ERK1/2 antibody added, incubated in the dark for 2 h, followed by an addition of 2 μl/well of the europium cryptate-conjugated anti-ERK1/2 antibody. After overnight incubation at room temperature in the dark, the plates were measured using the EnVision plate reader similarly to the cAMP assay.
Western blot analysis of ERK1/2 phosphorylation.
CHO cells expressing NPSR were seeded 5 ml/well with 200,000 cells in 6-well tissue culture-treated plates (Corning). After overnight incubation at 37°C with 5% CO2, the medium was replaced with 1 ml/well of antagonist solution (in F12K media with 1% FBS). After 10 min incubation at 37°C with 5% CO2, the NPS solution (15 nm NPS in F12K medium with 1% FBS) was added at 0.5 ml/well followed by 20 min incubation at 37°C with 5% CO2. The cells were then washed once with 2 ml/well of ice-cold PBS. All subsequent steps were performed at 4°C. The cells were detached in 1.5 ml/well ice-cold PBS using a cell scraper. The resulting cells were centrifuged at 5000 RPM for 5 min and the cell pellet was washed once with 1.5 ml/well ice-cold PBS. The cell pellet was resuspended in 150 μl/well of RIPA cell lysis buffer (Life Technologies) supplemented with a protease inhibitor cocktail (Roche Diagnostics), and incubated on ice for 30 min. The supernatant fraction was collected by centrifugation at 11,000 RPM for 2 min and transferred to a clean tube. Protein concentration in the cell lysate was determined using the Bradford protein assay (Bio-Rad Laboratories) and normalized before loading. Samples were heated at 65°C for 10 min before being applied to the SDS-PAGE under reducing conditions. Proteins were transferred to PVDF membranes (Bio-Rad Laboratories) using the Trans Blot SD (Bio-Rad Laboratories). Rabbit monoclonal antibodies against pERK (Cell Signaling Technology, product-number 4370, 1:2000) or ERK (Cell Signaling Technology, product-number 4695, 1:1000) were used for the detection of phosphorylated ERK or nonphosphorylated ERK protein. A peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, product-number sc-2030, 1:2000) was used as secondary antibody.
In vivo studies
Subjects for in vivo studies.
Male Wistar rats (300–350 g at start of experiment; Charles-River) were used. Rats were housed on a reversed light cycle with lights on at 8:30 P.M. and off at 8:30 A.M. with behavioral testing taking place during the dark cycle. Food and water were available ad libitum. All procedures used were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the NIAAA Animal Care and Use Committee.
Drug preparation for in vivo administration.
NCGC84 was dissolved in 10% Solutol, 10% N,N-dimethylacetamide, and 80% 10 mm PBS, pH 7.4, by sonication for 2 min. Rats were injected intraperitoneally 2 h before each test at a volume of 1 ml/kg.
pERK and pCREB immunohistochemistry.
Fifteen minutes after ETOH injection, the animals were deeply anesthetized with pentobarbital sodium (60 mg/kg, i.p.) before being transcardially perfused with 300 ml of ice-cold saline (0.9% NaCl) and 400 ml of 4% paraformaldehyde (PFA). Following perfusion, brains were removed, postfixed in 4% PFA for 2 h, dehydrated in a 30% sucrose solution for 48 h, snap frozen in powdered dry ice, and stored at −80°C until further use. Cryosections were obtained [40 μm; for levels according to Paxinos and Watson (2007), see Fig. 6A], kept in ice-cold PBS, and immediately processed for immunohistochemistry according to a protocol for free-floating sections.
Figure 6.

Alcohol-induced ERK phosphorylation in the CeA and, to a lesser extent, NAc-Sh, but not in the NAc-C nor the BNST, is blocked by pretreatment with NCGC84. Pretreatment with NCGC84 inhibited ERK phosphorylation in the central nucleus of the amygdala (A–C) and the NAc-Sh (D) induced by a 1.0 g/kg, i.p. dose of alcohol. No such effect was observed within the nucleus accumbens core or the bed nucleus of the stria terminalis (E–F).
The pERK and pCREB staining protocols were adapted from the studies by Ibba et al. (2009) and Canal et al. (2008), respectively. In brief, sections were incubated in 1% H2O2 for 30 min (pERK) or 0.5% H2O2 for 20 min (pCREB), followed by blocking in 3% bovine serum albumin (BSA; pERK) or 10% normal goat serum with 2% BSA and 0.1% Triton X for 1 h. Next, sections were incubated in the primary antibody [anti-pERK: 1:300 dilution; cat. #4370, anti-pCREB(48H2): 1:8000 dilution; cat #9197; Cell Signaling Technology] overnight at 4°C on a shaker. Negative controls were performed using PBS-BSA 0.3% without primary antibody. Following incubation, sections were rinsed with 1× PBS (3 times, 15 min) and incubated for 1 h with biotinylated secondary antibody (pERK: 1:800; pCREB: 1:400). Sections were rinsed again, incubated in an avidin–biotin peroxidase complex prepared following manufacturer's instructions (Vectastain ABC kit (Vector Labs), and a 3–3′-diaminobenzidine solution (Sigma-Aldrich) was added until brown stain developed. Sections were then rinsed with 1× PBS (2 times, 15 min), mounted onto gelatin-coated slides, and processed the next day through alcohol-xylene for light microscopy examination.
pERK-positive neurons were identified in the regions of interest [central amygdala (CeA) between −2.12 and −2.56 mm bregma, bed nucleus of the stria terminalis (BNST) between −0.30 to −0.80 mm bregma, and nucleus accumbens shell and core (NAc-Sh and NAc-C) between 1.70 and 1.20 mm bregma; all bilaterally] at the lowest magnification (10×). To assess specificity, pCREB-positive neurons were identified, in adjacent sections, in the two regions where a blockade of alcohol induced pERK response by NCGC84 was detected, i.e., CeA and NAc-Sh. Quantitative analysis was performed using a Leica DM6000CS light microscope (Leica Microsystem) at 40× magnification, and images were captured by an attached digital camera (Q Imaging). Densitometry counts were obtained using BIOQUANT software (BIOQUANT Image Analysis Corporation), and data were reported as total number of immunoreactive cells divided per square millimeter of surface.
Alcohol self-administration.
Self-administration training and testing was performed in operant chambers (MedAssociates) as previously described (Cippitelli et al., 2008). Briefly, rats were trained 5 d/week to lever press for a 0.1 ml volume of solution, delivered into a drinking-cup on a Fixed Ratio 1 (FR1) schedule over a 30 min session. A saccharin-fading procedure was used to reach self-administration of 10% alcohol in water. After 5 d of 10% alcohol on the FR1 schedule, rats were switched to an FR3 schedule and maintained on this schedule until stable self-administration rates were reached (∼15 d). For treatment, a counter-balanced, Latin Square repeated measures design was used, with 2 d of self-administration in between each test day.
Progressive ratio.
Rats (n = 8 per group) were trained to self-administer 10% alcohol as described above. When stable responding was reached, rats were tested for progressive ratio (PR)-responding. The progression of lever presses required to receive an alcohol reinforcer was 1, 2, 3, 4, 6, 8, 10, 12, and 16, after which the ratio increased in steps of 4 (Ciccocioppo et al., 2004). The breakpoint was defined as the last ratio completed before 30 min passed without the completion of the next ratio.
Cue- and stress-induced reinstatement.
Reinstatement experiments were performed as previously described (Cippitelli et al., 2008). Briefly, for cue-induced reinstatement, rats (n = 16) were trained to self-administer 10% alcohol as described above, in the presence of an orange scent as a contextual cue. After stable responding was reached, rats went through an extinction period during which alcohol and cues were absent. Once the extinction criterion (<15 lever-presses per 30 min session) was reached, animals were randomly assigned to treatment with vehicle or 1 mg/kg NCGC84. Rats within each treatment group were tested on 2 d, once while exposed to extinction conditions, and once while exposed to reinstatement conditions (orange scent, levers available, but no alcohol delivered), using a mixed design with drug treatment as a between subjects, and reinstatement condition as a within subjects, counter-balanced factor. Under these conditions, the presentation of alcohol-associated cues reliably leads to resumption of lever pressing on the previously alcohol-associated lever, or cue-induced reinstatement.
In the stress-induced reinstatement experiment (n = 16 per group), rats were placed in the self-administration chambers 1 h 45 min after intraperitoneal injection with NCGC84 (1 mg/kg) or vehicle, and subjected to 15 min of intermittent foot-shock, followed by a 30 min session under reinstatement conditions (no cues, levers available, but no alcohol delivered). Under these conditions, exposure to the 15 min foot-shock stress reliably leads to resumption of lever pressing on the previously alcohol-associated lever, or stress-induced reinstatement. Rats were tested on 4 d with 2 extinction days between each test day using a counterbalanced Latin Square repeated measures design.
Loss of righting reflex.
A 3.5 g/kg volume of 20% alcohol was administered to animals (n = 20). Rats were then placed in a supine position upon loss of consciousness, and the time was measured until the animals regained their righting reflex (defined as the ability to turn over into the upright position after being manually flipped back to the supine position 3 times within 1 min). In addition to the latency until regaining the righting reflex, blood alcohol concentration (BAC) upon waking was determined.
Locomotor activity.
Locomotor activity was tested in sound-attenuated enclosed chambers (MedAssociates) equipped with an infra-red beam detection system. Rats (n = 12) were injected with saline 1 h 30 min before being habituated to the boxes for 1 h on Day 1. Testing was on Day 2.
Saccharin self-administration.
Rats were trained to self-administer 0.0125% saccharin on a FR1 reinforcement schedule in the same chambers as those used for alcohol self-administration. Once stable response rates were reached (∼7 d), rats were switched to an FR3 schedule with a cue light signaling saccharin delivery. Testing was performed after stable response rates were reestablished (∼15 d). Rats (n = 7 per group; one animal removed from subsequent statistical analysis due to lack of responding on either lever during testing) were tested in a counter-balanced Latin Square repeated measures design with a 2 h pretreatment of either vehicle or NCGC00185684 (1 mg/kg) with two self-administration sessions in between test days.
Conditioned place aversion.
This procedure was based on the study by Hou et al. (2009), #1166. The two-chamber conditioned place aversion (CPA) apparatus [17.5” length (L) × 16” width (W) × 11” depth (D)] consisted of two distinct chambers distinguished by visual and tactile cues that were separated by a removable guillotine door (Med Associates). One chamber (17.5”L × 8” W) had horizontal black and white striped walls and a rod floor. The other chamber (17.5”L × 8” W) had vertical black and white striped walls with a grid floor. For the pretest, each rat was removed from its home cage and placed into the center of the CPA apparatus, near the guillotine door opening. The animals were allowed to explore both chambers for 15 min and the time spent in each chamber was recorded. For conditioning trials, each rat received a pretreatment and treatment injection on each of the 6 conditioning days and was placed in either chamber for 30 min. On drug-paired conditioning days, rats were pretreated with an intraperitoneal injection of either NCGC84 (1 mg/kg, n = 8) or 0.15 m NaCl vehicle (LiCl Group, n = 8) 1 h 45 min before placing in the drug-paired chamber. Immediately before the trial, each rat was given an injection of either 0.15 m NaCl (NPS antagonist group) or LiCl (LiCl group, 64 mg/kg) and immediately placed in the drug-paired chamber. On vehicle-paired conditioning days, rats were treated identically to the drug-paired conditioning days but were given vehicle instead of either NCGC84 or LiCl. On the test day, each rat was removed from its home cage and placed into the center of the CPA apparatus, near the guillotine door opening. The animals were allowed to explore both chambers for 15 min and the time spent on the drug-paired and vehicle-paired side was recorded. Locomotor activity was also recorded. CPA scores were calculated as the time in the drug-paired compartment during the testing phase minus that during the pretest exploration period. A negative score indicates conditioned place aversion, while a positive score is indicative of place preference.
Statistical analyses
In vitro studies.
The compound library screen data were analyzed using software developed internally at NIH Chemical Genomics Center. Concentration-response curves were analyzed and EC50 values (mean ± SD) calculated using Prism software (GraphPad). Results in figures are expressed as mean of triplicates ± SD unless they are specified.
In vivo studies.
Densitometry data were evaluated using Prism5 (GraphPad Prism Software) by a two-way measures ANOVA with ETOH and NPS treatment as factors.
Behavioral data were evaluated using Statistica 8 (StatSoft). One-way repeated-measures ANOVA was used to analyze the self-administration data with treatment as factor. Cue-induced reinstatement data were analyzed using a two-way ANOVA (with session as the within-subjects factor and treatment as the between-subjects factor), while the stress-induced reinstatement data were analyzed with a two-way, repeated-measures ANOVA (with session as the within-subjects factor and treatment as the between-subjects factor). Locomotor activity was analyzed using a one-way ANOVA. Unpaired t tests were used for the progressive ratio and loss of righting reflex data. The Newman–Keul test was used for all post hoc analyses.
Results
Identification of NPSR antagonists by high throughput screening
A cell-based NPS cAMP assay in a 1536-well plate format was used in the primary screen of compound collections to identify the NPSR antagonists (Patnaik et al., 2010). The active compounds identified from the primary screen were further confirmed in the NPS-stimulated intracellular calcium release assay and the [125I]Y10-NPS binding assay. The first screen of 77,000 available compounds resulted in the identification of the initial lead compound, MLS000558527 (McCoy et al., 2010). Additional screening of 220,874 compounds in our increased library collection led to the identification of the second NPSR antagonist, MLS001018695, with IC50 values of 731.9, 76.9, and 108.1 nm in cAMP, calcium assay, and radioligand binding assays, respectively.
NCGC00185684 is an optimized, competitive and selective NPSR antagonist
A subsequent structural optimization of MLS001018695 yielded a series of potent imidazopyridine NPSR antagonists (Patnaik et al., 2010). NCGC00185684 (NCGC84) is one of these optimized compounds. Its activities were first determined in the cAMP, intracellular Ca2+, and radioligand displacement assays. The IC50 values of this compound in the NPS-induced cAMP response and calcium response were 22.1 nm and 36.5 nm, respectively, representing 33- and 2-fold improvements over MLS001018695. The IC50 value of NCGC84 for displacement of 125I-NPS binding to NPSR was 5.0 nm, a 22-fold improvement compared with the original lead compound (Fig. 1; Table 1).
Figure 1.

Structure of NPSR antagonist NCGC84 and its inhibitory activities in Ca2+, cAMP, and [125I]Y10-NPS displacement assay. A, NCGC84 was chemically optimized from MLS001018695, an original HTS hit identified from a screen using a cAMP quantitation assay. B–D, Concentration response curves of NCGC00185686 in the intracellular Ca2+ assay (B); cAMP assay (C), and radioligand displacement assay (D). The IC50 values of NCGC84 in the Ca2+, cAMP, and binding assays are 22.1 ± 1.9, 36.5 ± 6.4, and 5.0 ± 0.05 nm, respectively.
Table 1.
Comparison of pharmacological properties of NCGC84 with SHA68
| Compound ID | NCGC00185684 | NCGC00184846 |
|---|---|---|
| Code name | NCGC84 | SHA68 |
| Structure | 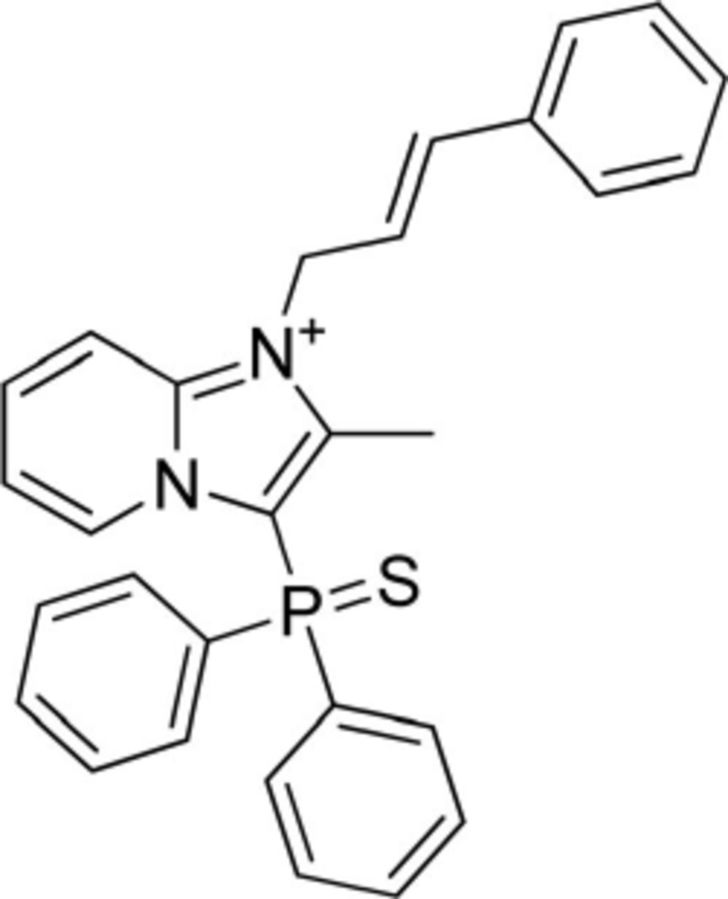 |
 |
| IC50 - cAMP | 22.1 ± 1.9 | 208.3 ± 36.3 |
| IC50 - Ca2+ | 36.5 ± 6.4 | 569.6 ± 130.3 |
| IC50 - ERK-phospho | 9.3 ± 11.5 | 15.8 ± 2.7 |
| IC50 - binding (95% CI) | 5.0 (3.88–6.42) | 9.5 (53.2–73.4) |
| pA2 (95% CI) | 8.98 (9.03–8.94) | 8.16 (8.25–8.08) |
| Schild slope | 1.03 ± 0.01 | 1.00 ± 0.03 |
To characterize the mechanism of inhibition on NPSR, a Schild analysis of NCGC84 was performed in the NPS cAMP assay in comparison with the known NPSR antagonist SHA68. In the presence of increased concentrations of NCGC84, the concentration–response curves of NPS shifted in parallel to the right, similarly to that of SHA68 (Fig. 2A–C). The pA2 value for NCGC84 was 8.98 (95% CI: 9.03–8.94). The slope of the Schild regression plot was close to 1.0 (1.03 ± 0.01). Together, these results show that NCGC84 is a specific, competitive antagonist at the NPSR.
Figure 2.

Schild regression analysis of NCGC84 in the cAMP assay, compared with the known NPSR antagonist SHA68. A, With increases in the concentration of NCGC84, the NPS concentration–response curves were shifted to the right in parallel. pA2 is 8.98 (CI: 9.03–8.94). B, SHA68 also shifted the concentration response curves of NPS to the right. C, Schild plot in which the slopes for NCGC84 as well as SHA68 are ∼1.0, indicative of a competitive antagonist.
Because of NPSR sequence similarity to the vasopressin receptor, we examined the selectivity of NCGC84 on the vasopressin 1b (V1b) receptor and on the endogenous purinergic receptor in HEK293 cells (Schachter et al., 1997), by measuring inhibition of the intracellular Ca2+ response to the respective agonist added at EC80. NCGC84 did not inhibit the vasopressin V1b or the endogenous purinergic receptor at concentrations up to 10 μm, demonstrating a complete selectivity of NPSR antagonist NCGC84 for the NPSR over the vasopressin V1b and purinergic receptors (Fig. 3).
Figure 3.

NCGC84 concentrations up to 10 μm did not affect intracellular Ca2+-level in cells expressing vasopressin V1b or purinergic receptors following application of EC80 of their respective ligand. The NPSR has the closest protein sequence homology with the vasopressin V1b receptor (∼28%).
NCGC84 preferentially inhibits NPS-induced ERK phosphorylation in vitro
NPS-stimulated ERK phosphorylation was determined in CHO cells expressing NPSR using a two antibody-based homogenous TR-FRET assay. NPS-induced intracellular Ca2+ response and cAMP level in the same cell line were measured in parallel as comparisons. NPS induced ERK phosphorylation in a concentration-dependent manner with an EC50 value of 0.27 nm. The activity of NPS on ERK phosphorylation was three- to fourfold more potent than these observed in the intracellular calcium and cAMP assays (Fig. 4). NCGC84 inhibited NPS-induced ERK phosphorylation in a concentration-dependent manner, and the resulting IC50 value, 9.3 nm, is 2.3- and 3.9-fold more potent than the cAMP and intracellular calcium response, respectively (Fig. 5A). The inhibition of NPS-induced ERK phosphorylation by NCGC84 was confirmed by Western blot analysis (Fig. 5B).
Figure 4.

NPS activity in the ERK phosphorylation, cAMP, and intracellular calcium assays. A, NPS-induced ERK phosphorylation in CHO cells expressing NPSR with an EC50 value of 0.27 ± 0.048 nm. B, NPS-stimulated intracellular calcium response with an EC50 of 0.91 ± 0.044 nm. C, NPS-increased cellular cAMP level with an EC50 of 1.12 ± 0.030 nm.
Figure 5.

Inhibition of NPS-induced ERK phosphorylation in CHO cells expressing NPSR. A, The NPS-induced ERK phosphorylation was inhibited by NCGC84 with an IC50 value of 9.3 ± 11.5 nm. B, ERK phosphorylation (pERK) induced by NPS was significantly inhibited by NCGC84 in Western blot analysis, while levels of nonphosphorylated ERK protein (ERK) remained constant.
A summary of the obtained IC50 values compared with those for the published small molecule antagonist SHA68 can be seen in Table 1. The data clearly indicate that NCGC84 exhibits the most potent inhibition on NPS-induced ERK phosphorylation that is 2.3- and 3.9-fold more potent than these on cAMP and intracellular Ca2+ responses, respectively. Compared with previously reported NPSR antagonists, the imidazopyridinium NCGC84 has a unique structure and possesses drug-like properties with good brain penetrance (Patnaik et al., 2010). It therefore offers an attractive research tool to probe the role of endogenous NPS for brain function.
Systemic NCGC84 inhibits alcohol-induced central ERK phosphorylation in vivo
Next, we therefore evaluated the ability of systemic NCGC84 to block ERK-phosphorylation induced by systemic alcohol administration in vivo. Acute administration of alcohol has been reported to induce phosphorylation of multiple protein-kinases, including ERK, in brain regions involved in regulation of alcohol intake, such as CeA and NAc-Sh (Ibba et al., 2009; Björk et al., 2010). In agreement with those reports, intraperitoneal administration of alcohol (1 g/kg) produced a robust ERK phosphorylation in CeA, a response that was abolished by pretreatment with NCGC84 at a dose (1 mg/kg, i.p.) selected based on preliminary experiments that showed a lack of nonspecific motor effect at this dose [Fig. 6A–C; two-way ANOVA: main effect of alcohol: (F(1,27) = 5.1, p = 0.03); main effect of NPS antagonist: (F(1,27) = 7.7, p = 0.01); interaction: (F(1,27) = 6.3, p = 0.02)]. In the NAc-Sh, the main effect of alcohol did not reach significance (F(1,27) = 2.0, p = 0.17), but the pattern was similar, and both the main effect of NCGC84 (F(1,27) = 4.766, p = 0.04), and the group × treatment interaction (F(1,27) = 4.4, p = 0.04) were significant (Fig. 6D). NCGC84 did not affect unstimulated ERK phosphorylation in CeA or NAc-Sh. In addition, NCGC84 did not affect the status of ERK phosphorylation in the NAc-C or BNST after vehicle or alcohol administration (Fig. 6E,F). Inhibition of ERK phosphorylation by NCGC84 was selective. In comparison, CREB phosphorylation, a readout of PKA activity, was unaffected in CeA (main drug treatment effect: F(1,27) = 0.56; p = 0.46; main alcohol effect: F(1,27) = 0.30; p = 0.59; drug treatment × alcohol interaction: F(1,27) = 0.01; p = 0.92). A limitation of the CeA pCREB data was that a lack of alcohol response in this structure could have rendered this outcome insensitive to effects of NCGC84. However, in the NAc-Sh, there was a main effect of alcohol (F(1,25) = 4.6; p = 0.04), yet neither the main effect of drug treatment (F(1,25) = 1.6; p = 0.22) nor the drug treatment × alcohol interaction (F(1,25) = 0.17; p = 0.68) were significant. In fact, the pCREB levels in NAc-Sh after alcohol + NCGC84 (1818.1 ± 106.6; N = 7) were numerically slightly higher than those following alcohol + vehicle (1624.6 ± 53.0; N = 8).
Systemic NCGC84 decreases operant alcohol self-administration
Because CeA is critically involved in regulation of alcohol consumption (Hyytiä and Koob, 1995; Möller et al., 1997), the ability of NCGC84 to prevent alcohol-induced ERK phosphorylation in this structure suggested that the NPS antagonist may also modulate the motivation to obtain and consume alcohol. Accordingly, pretreatment with NCGC84 significantly reduced operant alcohol self-administration (Fig. 7A; F(2,45) = 7.9; p = 0.0002). Post hoc tests showed a significant reduction following the 1 mg/kg, i.p. dose, but not the 0.3 mg/kg dose (Newman–Keuls, p < 0.005). Inactive lever responses were negligible, and unaffected by NCGC84 (F(2,45) = 1.37; p = 0.27; data not shown).
Figure 7.

NCGC84 decreases alcohol self-administration and motivation to obtain alcohol. A, I,P. administration of 1 mg/kg NCGC84 significantly suppressed alcohol self-administration (Newman–Keuls, **p < 0.005). Values (mean ± SEM) represent total rewards received in a 30 min session at an FR3 schedule of reinforcement. Inactive rewards received were negligible and unaffected by NCGC84 (data not shown). B, Additionally, NCGC84 (1 mg/kg) significantly decreased the progressive ratio breakpoint for alcohol self-administration (t = 2.42; p = 0.03).
As a measure of motivation to obtain alcohol, we also determined the break-point, i.e., the highest ratio completed on a progressive ratio self-administration schedule (Hodos, 1961). NCGC84 at the dose that suppressed alcohol self-administration (1 mg/kg, i.p.) also significantly decreased this measure of motivation (Fig. 7B; t = 2.42; p = 0.03).
Control experiments suggested that effects of NCGC84 on alcohol self-administration were unlikely to be caused by nonspecific effects or common confounds. At the 1 mg/kg that potently suppressed alcohol self-administration, saccharin self-administration was not significantly affected (mean ± SEM; vehicle: 89.3 ± 17.4; NCGC84: 56.8 ± 18.8; F(1,5) = 2.6, p = 0.16). Furthermore, alcohol sensitivity, measured as the time to regain the righting reflex after a high (3.5 g/kg) bolus dose of alcohol was unaffected by the 1 mg/kg, i.p. dose of NCGC84, as was the blood alcohol concentration at the time of regaining the reflex (t = 1.51 and t = 0.93; p = 0.16 and p = 0.37, respectively; n = 5–9 per group; data not shown). The 1 mg/kg dose that potently suppressed alcohol self-administration also left locomotor activity unaffected (saline vs NCGC84, mean ± SEM, n = 6: 3030 ± 218 vs 2975 ± 363, n.s.). Finally, NCGC84 did not have aversive properties, as shown by the CPA experiment. In fact, the preference for score for the NCGC84 group was positive (61.1 ± 38.2), while that for the aversive control, LiCl, was negative as expected (−165 ± 81.2; F(1,15) = 5.8, p = 0.03).
NCGC84 does not affect cue- or stress-induced relapse to alcohol seeking
Exogenous NPS has been shown to promote relapse-like behavior, assessed as reinstatement of alcohol seeking by exposure to alcohol-associated cues following extinction (Cannella et al., 2009). To assess whether this reflects the function of endogenous NPS, we examined the ability of NCGC84 to block relapse-like behavior. Alcohol seeking was robustly reinstated both by alcohol-associated cues, and also by foot-shock stress-exposure, another established relapse-promoting stimulus (active lever-responding: F(1,14) = 21.20; p = 0.0004 and F(1,15) = 34.13; p < 0.0001, respectively; Fig. 8A,C). However, NCGC84 (1 mg/kg) did not affect cue- (F(1,14) = 0.002; p = 0.97) nor stress- (F(1,15) = 1.24; p = 0.28) induced reinstatement.
Figure 8.

NCGC84 does not affect stress- or cue-induced reinstatement of alcohol-seeking. A, C, Alcohol seeking was robustly reinstated both by alcohol-associated cue and stress exposure. However, NCGC84 (1 mg/kg) did not have a significant effect neither on cue- nor stress-induced reinstatement. As is typically found, inactive lever responses in the cue-induced reinstatement experiment were also somewhat increased during reinstatement but this increase was marginal (B). Similarly, in the stress-induced reinstatement test, inactive lever responses were also slightly but significantly increased during reinstatement (D).
As is typically found, inactive lever responses in the cue-induced reinstatement experiment were also somewhat increased during reinstatement (F(1,14) = 10.37; p = 0.006), but this increase was marginal compared with that observed on the alcohol-associated lever, and was also unaffected by NCGC84-administration (F(1,14) = 2.6; p = 0.13; Fig. 8B). Similarly, in the stress-induced reinstatement test, inactive lever responses were also slightly but significantly increased during reinstatement (F(1,15) = 12.89; p = 0.003) with no significant effect of NCGC84 on this measure (F(1,15) = 1.43; p = 0.25; Fig. 8D).
Discussion
We report the characterization of a novel, small-molecule NPSR antagonist, NCGC84. In vitro, NCGC84 bound selectively to the NPSR with nanomolar affinity, and behaved as a competitive antagonist that blocked intracellular ERK phosphorylation, cAMP, and calcium responses to NPS. The activity profile of NCGC84 was biased toward ERK inhibition, an NPS-response predominant at low, physiological NPS concentrations. In vivo, systemic NCGC84 decreased alcohol self-administration and motivation to obtain alcohol at an NCGC84-dose not influencing locomotor activity, cue- or stress-induced reinstatement of alcohol seeking, or other control behaviors. The effects on self-administration are thus behaviorally specific.
Similar to other GPCRs, NPSR is coupled to multiple signaling pathways. We found EC50 values of NPS for intracellular calcium and cAMP responses of 0.91 and 1.12 nm, respectively, similar to previous findings (Xu et al., 2004). The Kd for NPS binding to the NPSR was 0.33 nm, as determined by an [125I]Y10-NPS binding assay in NPSR-expressing CHO cells. Thus, the binding affinity of NPS to NPSR is three- to fourfold higher than its functional activities in intracellular calcium and cAMP assays. In addition to intracellular calcium and cAMP, NPSR is coupled to ERK phosphorylation. The EC50 value for NPS stimulated ERK phosphorylation has been reported at 0.32 nm in HEK cells expressing NPSR Ile107, and 1.23 nm in HEK cells expressing NPSR Asn107 (Reinscheid et al., 2005). Using a TR-FRET ERK phosphorylation assay, we found that the EC50 value for NPS-stimulated ERK phosphorylation in CHO cells expressing NPSR (Asn107) was 0.27 nm, and thus close to the observed NPS binding affinity. This suggests that ERK-MAPK signaling may be a preferred pathway for NPS, or a predominant signaling pathway at low physiological NPS concentration.
ERK phosphorylation in response to GPCR activation has been implicated in the regulation of GPCR internalization/desensitization, and in the modulation of other GPCR-signaling pathways (Calebiro et al., 2010; Feinstein et al., 2011). For instance, induction of the ERK phosphorylation cascade by activation of the oxytocin receptor induces long-term potentiation and CREB signaling in mice (Tomizawa et al., 2003). Oxytocin also reduces anxiety-like behavior in rats via the ERK phosphorylation pathway, an effect abolished by the MEK inhibitor U0126 (Blume et al., 2008). In vitro, NCGC84 preferentially inhibited NPSR-mediated ERK phosphorylation compared with intracellular calcium and cAMP responses to NPS, and its affinity in the radioligand binding assay was similar to its EC50 for ERK phosphorylation. Together with the results for preferential activation of ERK phosphorylation by NPS, these data suggest that the ERK phosphorylation pathway may play a major role in transduction of the NPS signal.
ERK signaling plays a key role in aspects of cocaine addiction (Lu et al., 2006). Central ERK activation has also been found during withdrawal from alcohol (Sanna et al., 2002), but its role in alcohol reinforcement and relapse to alcohol-seeking is less well understood. Acute alcohol administration induces ERK phosphorylation in CeA (Ibba et al., 2009), a finding replicated in our study. We have previously shown that ibotenic acid lesion of CeA led to decreased alcohol consumption (Möller et al., 1997). We presently found that systemic pretreatment with NCGC84 (1 mg/kg) inhibited ERK phosphorylation in CeA after a 1.0 g/kg, i.p. dose of alcohol. This NCGC84-dose also suppressed lever-pressing for a 10% alcohol-solution and reduced motivation to obtain alcohol on a progressive ratio schedule. At a minimum, the blocked alcohol-induced ERK phosphorylation following NCGC84 pretreatment serves as a biomarker of central target engagement upon systemic administration of the 1 mg/kg NCGC84 dose, in agreement with our initial data indicating acceptable brain availability of this compound (Patnaik et al., 2010). Together with the behavioral findings, the data additionally suggest the possibility that endogenous NPS participates in regulation of alcohol intake through ERK signaling in CeA. Unfortunately, the limited solubility of NCGC84 renders it less useful for site-directed microinjection experiments.
Neither cue- nor stress-induced reinstatement of alcohol-seeking was significantly affected by NCGC84. The lack of effect on cue-induced reinstatement was unexpected, since we have previously shown exogenous NPS to promote cue-induced relapse to alcohol (Cannella et al., 2009) and cocaine seeking (Kallupi et al., 2010). The latter study also supported a role for endogenous NPS in cue-induced cocaine relapse, because reinstatement responding was attenuated by the NPSR antagonist SHA68 given alone. In contrast, NPSR antagonist data to examine a role of endogenous NPS in reinstatement of alcohol seeking have until now been lacking. Our current results do not provide support for such a role. A possible interpretation is that reinstatement behavior associated with alcohol and cocaine relapse is controlled by overlapping but distinct neurocircuitry. This possibility is illustrated by prior findings that activation of nociceptin receptors blocks relapse behavior for alcohol but not for cocaine (Martin-Fardon et al., 2000). Another possibility is that NPS triggers relapse-like behavior through activation of cAMP or calcium signaling, responses that may not be effectively blocked by NCGC84 at doses sufficient to block ERK responses.
Although NPSR antagonism did not block relapse in nondependent rats, future studies will be needed to determine whether dependence-induced neuroadaptations may engage the NPS system in these behaviors. This possibility is suggested by observations of increased NPSR expression in both BLA and CeA for at least a week after withdrawal from alcohol exposure (Ruggeri et al., 2010). This may parallel prior experience with other neuropeptide systems, such as CRF (Heilig and Koob, 2007). In nondependent rats, CRFR1 antagonists are inactive in several behavioral models. In vivo activity is, however, revealed following a history of alcohol dependence resulting in a pathological activation of the CRF system (Gehlert et al., 2007; Sommer et al., 2008). A similar sensitization to CRFR1 antagonists can result from an innate upregulation of the CRF system due to genetic factors (Hansson et al., 2006; Hansson et al., 2007). Given the demonstrated ability for NPSR activation to promote relapse, it will be of interest to determine whether a history of alcohol dependence, or genetic variation that alters the expression of NPS or its receptor, will enhance the potency of NPSR antagonists to suppress alcohol self-administration, or reveal an ability to block relapse.
Meanwhile, the negative reinstatement data provide a control for activity-suppressing effects of NCGC84, since the reinstatement models rely on a behavioral output identical to that used to assess self-administration of alcohol, i.e., lever pressing, with the only difference being the absence of alcohol delivery. Under these conditions, the 1 mg/kg NCGC84 dose that suppressed lever pressing for alcohol did not influence reinstatement response rates. Additionally, 1 mg/kg NCGC84 given systemically did not influence locomotor activity in the open field. Furthermore, NCGC84 did not affect the loss of righting reflex, a measure of sensitivity to the sedative properties of alcohol. Finally, NCGC84 did not alter blood-alcohol levels at the time of regaining the righting reflex, indicating that elimination of alcohol was not altered by the antagonist. Together, these observations suggest that suppression of alcohol self-administration and progressive ratio responding by NCGC84 is a specific, mechanism-mediated effect. Similarly, self-administration of a sweetened solution was not significantly suppressed by an NCGC84 dose that robustly suppressed alcohol self-administration. However, a numeric trend for suppression of this behavior was observed, suggesting that selectivity for alcohol self-administration over natural rewards may be relative rather than absolute.
These data suggest a utility of NCGC84 as a tool to probe the function of endogenous NPS. Further optimization would, however, clearly increase its utility, since we have found that NCGC84 has limited solubility, a steep dose–response relationship, and a relatively narrow separation between behaviorally specific and sedative effects, with measurable sedation setting in at systemic doses of ≥3× those required to produce specific suppression of alcohol self-administration (data not shown).
To date, the majority of data on NPS-induced intracellular responses, including intracellular calcium, cAMP, and ERK phosphorylation, have been obtained in cells stably expressing NPSR. It is presently unclear whether different functional in vivo effects of NPS, such as arousal, anxiolytic-like actions, or appetitive effects, are mediated through different intracellular responses to NPSR activation. If that is the case, then the emerging complexity of intracellular signaling pathways mediating NPSR responses would have the potential to open new opportunities for achieving pharmacological selectivity through the use of antagonists biased toward different signaling pathways transducing NPS responses. Our data suggest that the motivation to obtain and consume alcohol may be particularly sensitive to NPSR antagonists biased toward blockade of ERK phosphorylation. A full analysis of how the different cellular signaling pathways activated by NPSR contribute to different in vivo effects will require extensive future studies.
In conclusion, we have identified a new series of potent, selective, and brain penetrant imidazopyridine NPSR antagonists that potently inhibit NPS-stimulated cellular calcium, cAMP, and ERK phosphorylation responses. Using the lead compound, we report data suggesting a role of endogenous NPS in the motivation to obtain alcohol. These data suggest that the NPSR may merit further exploration as a candidate therapeutic target in alcoholism.
Footnotes
This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Programs of the National Institute on Alcohol Abuse and Alcoholism, NIH. We thank Dr. Reinscheid for generously providing the NPSR clone originally used to generate NPSR-CHO cell line, Sam Michael for assistance in robotic screen, and Paul Shinn for compound management.
The authors declare no competing financial interests.
References
- Badia-Elder NE, Henderson AN, Bertholomey ML, Dodge NC, Stewart RB. The effects of neuropeptide S on ethanol drinking and other related behaviors in alcohol-preferring and -nonpreferring rats. Alcoholism, clinical and experimental research. 2008;32:1380–1387. doi: 10.1111/j.1530-0277.2008.00713.x. [DOI] [PubMed] [Google Scholar]
- Björk K, Terasmaa A, Sun H, Thorsell A, Sommer WH, Heilig M. Ethanol-induced activation of AKT and DARPP-32 in the mouse striatum mediated by opioid receptors. Addiction biology. 2010;15:299–303. doi: 10.1111/j.1369-1600.2010.00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume A, Bosch OJ, Miklos S, Torner L, Wales L, Waldherr M, Neumann ID. Oxytocin reduces anxiety via ERK1/2 activation: local effect within the rat hypothalamic paraventricular nucleus. Eur J Neurosci. 2008;27:1947–1956. doi: 10.1111/j.1460-9568.2008.06184.x. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Persani L, Lohse MJ. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci. 2010;31:221–228. doi: 10.1016/j.tips.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Camarda V, Rizzi A, Ruzza C, Zucchini S, Marzola G, Marzola E, Guerrini R, Salvadori S, Reinscheid RK, Regoli D, Cal ò G. In vitro and in vivo pharmacological characterization of the neuropeptide s receptor antagonist [D-Cys(tBu)5]neuropeptide S. J Pharmacol Exp Ther. 2009;328:549–555. doi: 10.1124/jpet.108.143867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Intra-amygdala injections of CREB antisense impair inhibitory avoidance memory: role of norepinephrine and acetylcholine. Learn Mem. 2008;15:677–686. doi: 10.1101/lm.904308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannella N, Economidou D, Kallupi M, Stopponi S, Heilig M, Massi M, Ciccocioppo R. Persistent increase of alcohol-seeking evoked by neuropeptide S: an effect mediated by the hypothalamic hypocretin system. Neuropsychopharmacology. 2009;34:2125–2134. doi: 10.1038/npp.2009.37. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Fedeli A, Angeletti S, Weiss F, Heilig M, Massi M. Attenuation of ethanol self-administration and of conditioned reinstatement of alcohol-seeking behaviour by the antiopioid peptide nociceptin/orphanin FQ in alcohol-preferring rats. Psychopharmacology. 2004;172:170–178. doi: 10.1007/s00213-003-1645-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifani C, Micioni Di Bonaventura MV, Cannella N, Fedeli A, Guerrini R, Calo G, Ciccocioppo R, Ubaldi M. Effect of neuropeptide S receptor antagonists and partial agonists on palatable food consumption in the rat. Peptides. 2011;32:44–50. doi: 10.1016/j.peptides.2010.10.018. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, Defonseca FR, Piomelli D, Ciccocioppo R. Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology. 2008;198:449–460. doi: 10.1007/s00213-008-1104-0. [DOI] [PubMed] [Google Scholar]
- Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Cippitelli A, Thorsell A, L ê AD, Hipskind PA, Hamdouchi C, Lu J, Hembre EJ, Cramer J, Song M, McKinzie D, Morin M, Ciccocioppo R, Heilig M. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo [1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. doi: 10.1523/JNEUROSCI.4985-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R, Camarda V, Trapella C, Cal ò G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Synthesis and biological activity of human neuropeptide S analogues modified in position 5: identification of potent and pure neuropeptide S receptor antagonists. J Med Chem. 2009;52:524–529. doi: 10.1021/jm8012294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han RW, Yin XQ, Chang M, Peng YL, Li W, Wang R. Neuropeptide S facilitates spatial memory and mitigates spatial memory impairment induced by N-methyl-d-aspartate receptor antagonist in mice. Neurosci Lett. 2009;455:74–77. doi: 10.1016/j.neulet.2009.03.023. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L, Terasmaa A, Massi M, Heilig M, Ciccocioppo R. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci U S A. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Ciccocioppo R, Heilig M. Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addict Biol. 2007;12:30–34. doi: 10.1111/j.1369-1600.2007.00050.x. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodos W. Progressive ratio as a measure of reward strength. Science. 1961;134:943–944. doi: 10.1126/science.134.3483.943. [DOI] [PubMed] [Google Scholar]
- Hou YY, Lu B, Li M, Liu Y, Chen J, Chi ZQ, Liu JG. Involvement of actin rearrangements within the amygdala and the dorsal hippocampus in aversive memories of drug withdrawal in acute morphine-dependent rats. J Neurosci. 2009;29:12244–12254. doi: 10.1523/JNEUROSCI.1970-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyyti ä P, Koob GF. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-B. [DOI] [PubMed] [Google Scholar]
- Ibba F, Vinci S, Spiga S, Peana AT, Assaretti AR, Spina L, Longoni R, Acquas E. Ethanol-induced extracellular signal regulated kinase: role of dopamine D1 receptors. Alcohol Clin Exp Res. 2009;33:858–867. doi: 10.1111/j.1530-0277.2009.00907.x. [DOI] [PubMed] [Google Scholar]
- Kallupi M, Cannella N, Economidou D, Ubaldi M, Ruggeri B, Weiss F, Massi M, Marugan J, Heilig M, Bonnavion P, de Lecea L, Ciccocioppo R. Neuropeptide S facilitates cue-induced relapse to cocaine seeking through activation of the hypothalamic hypocretin system. Proc Natl Acad Sci U S A. 2010;107:19567–19572. doi: 10.1073/pnas.1004100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard SK, Ring RH. Immunohistochemical localization of the neuropeptide S receptor in the rat central nervous system. Neuroscience. 2011;172:153–163. doi: 10.1016/j.neuroscience.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Liu K, Titus S, Southall N, Zhu P, Inglese J, Austin CP, Zheng W. Comparison on functional assays for Gq-coupled GPCRs by measuring inositol monophospate-1 and intracellular calcium in 1536-well plate format. Current chemical genomics. 2008;1:70–78. doi: 10.2174/1875397300801010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Martin-Fardon R, Ciccocioppo R, Massi M, Weiss F. Nociceptin prevents stress-induced ethanol- but not cocaine-seeking behavior in rats. Neuroreport. 2000;11:1939–1943. doi: 10.1097/00001756-200006260-00026. [DOI] [PubMed] [Google Scholar]
- McCoy JG, Marugan JJ, Liu K, Zheng W, Southall N, Huang W, Heilig M, Austin CP. Selective modulation of Gq/Gs pathways by naphtho pyrano pyrimidines as antagonists of the neuropeptide S receptor. ACS Chem Neurosci. 2010;1:559–574. doi: 10.1021/cn100040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller C, Wiklund L, Sommer W, Thorsell A, Heilig M. Decreased experimental anxiety and voluntary ethanol consumption in rats following central but not basolateral amygdala lesions. Brain Res. 1997;760:94–101. doi: 10.1016/S0006-8993(97)00308-9. [DOI] [PubMed] [Google Scholar]
- Pañeda C, Huitron-Resendiz S, Frago LM, Chowen JA, Picetti R, de Lecea L, Roberts AJ. Neuropeptide S reinstates cocaine-seeking behavior and increases locomotor activity through corticotropin-releasing factor receptor 1 in mice. J Neurosci. 2009;29:4155–4161. doi: 10.1523/JNEUROSCI.5256-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik S, Marugan J, Liu K, Zheng W, Thorsell A, Eskay R, Southall N, Heilig M, Inglese J, Austin C. Probe reports from the NIH Molecular Libraries Program. Bethesda, MD: NIH; 2010. Identification of small molecule antagonists of the neuropeptide-S receptor. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates: hard cover edition. San Diego: Academic; 2007. [Google Scholar]
- Reinscheid RK. Phylogenetic appearance of neuropeptide S precursor proteins in tetrapods. Peptides. 2007;28:830–837. doi: 10.1016/j.peptides.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid RK, Xu YL. Neuropeptide S and its receptor: a newly deorphanized G protein-coupled receptor system. Neuroscientist. 2005;11:532–538. doi: 10.1177/1073858405276405. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O. Pharmacological characterization of human and murine neuropeptide s receptor variants. J Pharmacol Exp Ther. 2005;315:1338–1345. doi: 10.1124/jpet.105.093427. [DOI] [PubMed] [Google Scholar]
- Ruggeri B, Braconi S, Cannella N, Kallupi M, Soverchia L, Ciccocioppo R, Ubaldi M. Neuropeptide S receptor gene expression in alcohol withdrawal and protracted abstinence in postdependent rats. Alcohol Clin Exp Res. 2010;34:90–97. doi: 10.1111/j.1530-0277.2009.01070.x. [DOI] [PubMed] [Google Scholar]
- Ruzza C, Rizzi A, Trapella C, Pela' M, Camarda V, Ruggieri V, Filaferro M, Cifani C, Reinscheid RK, Vitale G, Ciccocioppo R, Salvadori S, Guerrini R, Calo' G. Further studies on the pharmacological profile of the neuropeptide S receptor antagonist SHA 68. Peptides. 2010;31:915–925. doi: 10.1016/j.peptides.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Simpson C, Lutjens R, Koob G. ERK regulation in chronic ethanol exposure and withdrawal. Brain Res. 2002;948:186–191. doi: 10.1016/S0006-8993(02)03191-8. [DOI] [PubMed] [Google Scholar]
- Schachter JB, Sromek SM, Nicholas RA, Harden TK. HEK293 human embryonic kidney cells endogenously express the P2Y1 and P2Y2 receptors. Neuropharmacology. 1997;36:1181–1187. doi: 10.1016/S0028-3908(97)00138-X. [DOI] [PubMed] [Google Scholar]
- Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV, Ghatei MA, Bloom SR. Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology. 2006;147:3510–3518. doi: 10.1210/en.2005-1280. [DOI] [PubMed] [Google Scholar]
- Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS, Heilig MA. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psych. 2008;63:139–145. doi: 10.1016/j.biopsych.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Tomizawa K, Iga N, Lu YF, Moriwaki A, Matsushita M, Li ST, Miyamoto O, Itano T, Matsui H. Oxytocin improves long-lasting spatial memory during motherhood through MAP kinase cascade. Nat Neurosci. 2003;6:384–390. doi: 10.1038/nn1023. [DOI] [PubMed] [Google Scholar]
- Trapella C, Pela M, Del Zoppo L, Calo G, Camarda V, Ruzza C, Cavazzini A, Costa V, Bertolasi V, Reinscheid RK, Salvadori S, Guerrini R. Synthesis and separation of the enantiomers of the neuropeptide S receptor antagonist (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68) J Med Chem. 2011;54:2738–2744. doi: 10.1021/jm200138r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–497. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK. Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol. 2007;500:84–102. doi: 10.1002/cne.21159. [DOI] [PubMed] [Google Scholar]