

Abstract
Human immunodeficiency virus (HIV)-associated neurocognitive disorders (HAND) is a significant source of disability in the HIV-infected population. Even with stringent adherence to anti-retroviral therapy, >50% of patients living with HIV-1 will develop HAND (Heaton et al., 2010). Because suppression of viral replication alone is not enough to stop HAND progression, there is a need for an adjunctive neuroprotective therapy in this population. To this end, we have developed a small-molecule brain-penetrant inhibitor with activity against mixed-lineage kinase 3 (MLK3), named URMC-099. MLK3 activation is associated with many of the pathologic hallmarks of HAND (Bodner et al., 2002, 2004; Sui et al., 2006) and therefore represents a prime target for adjunctive therapy based on small-molecule kinase inhibition. Here we demonstrate the anti-inflammatory and neuroprotective effects of URMC-099 in multiple murine and rodent models of HAND. In vitro, URMC-099 treatment reduced inflammatory cytokine production by HIV-1 Tat-exposed microglia and prevented destruction and phagocytosis of cultured neuronal axons by these cells. In vivo, URMC-099 treatment reduced inflammatory cytokine production, protected neuronal architecture, and altered the morphologic and ultrastructural response of microglia to HIV-1 Tat exposure. In conclusion, these data provide compelling in vitro and in vivo evidence to investigate the utility of URMC-099 in other models of HAND with the goal of advancement to an adjunctive therapeutic agent.
Introduction
Despite continued advances in combination antiretroviral therapy (cART) that have turned human immunodeficiency virus-1 (HIV-1) infection into a medically manageable disease, HIV-1-associated neurocognitive disorders (HAND) persist, prompting the need for adjunctive therapy. Sixteen years after it was originally defined, HAND received a nosological update as a disease entity that reflects its changing phenotype(s) and its impact on people living with HIV-1 (Antinori et al., 2007). Although debate continues over the incidence of the mildest form of HAND (Gisslén et al., 2011), the overall prevalence in HIV-infected patients the United States has exceeded 50% (Heaton et al., 2010), which is significantly greater in the post-cART era during the medically asymptomatic stage of HIV-1 infection (Heaton et al., 2011).
Damage to synaptodendritic architecture from viral proteins and proinflammatory mediators, with failure of activity-dependent neuronal networks, are substrates for HAND (Bellizzi et al., 2005; Ellis et al., 2007). However, the Phase II, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov Trial NCT00000867) with the uncompetitive NMDA receptor antagonist memantine given with cART failed to demonstrate long-term efficacy against HAND (Zhao et al., 2010), suggesting that anti-excitotoxic agents alone are unlikely to be first-line adjunctive therapy. The search for other “druggable” targets important to neuroinflammatory events that occur upstream and downstream of excitotoxic damage to synapses in HAND led us to investigate roles for mixed-lineage kinase type 3 (MLK3; also known as MAP3K11) in preclinical models of HAND. MLK3 is expressed in neurons (Maroney et al., 2001) and CNS immune effector cells (Wang et al., 2010) and is activated by cellular and metabolic stress (Jaeschke and Davis, 2007).
In vitro studies with the nonspecific MLK inhibitor CEP-1347 had previously demonstrated protection against HIV-1 gp120-mediated neurotoxicity to rodent hippocampal and dorsal root ganglion neurons (Bodner et al., 2002, 2004). We subsequently showed that both HIV-1 Tat and gp120 induced autophosphorylation of MLK3 in rodent neuronal cultures, which could be inhibited by CEP-1347 (Sui et al., 2006). Additionally, CEP-1347 was neuroprotective under these conditions and reversed the proinflammatory phenotype of human monocytes exposed to Tat and gp120 (Sui et al., 2006). Studies with a dominant-negative MLK3 mutant confirmed the specificity of MLK3 signaling in mediating neurotoxicity after exposure to Tat (Sui et al., 2006). Subsequently, we used a murine model of HIV-1 encephalitis to demonstrate that administration of CEP-1347 prevented microgliosis, reversed macrophage production of toxic inflammatory mediators after HIV-1 infection, and restored normal synaptic architecture (Eggert et al., 2010).
Bolstered by our data supporting MLK3 as a crucial neuropathogenetic target in our preclinical models of HAND and because of limitations in the kinase specificity and CNS profile of CEP-1347, we synthesized an entirely new chemical entity for MLK3 inhibition with drug-like properties and a favorable CNS, metabolic and toxicity profile. We now report that our lead compound, URMC-099, robustly inhibits microglial release of inflammatory mediators and pathologic clearance of synaptic and axonal elements in in vitro and in vivo models of HAND.
Materials and Methods
URMC-099, a new MLK3 inhibitor: synthesis and formulations.
URMC-099, based on a pyrrolopyridine scaffold with an aryl piperazine side chain, was the result of optimization of hits discovered from a large screening campaign for inhibitors of MLK3 with nanomolar potency using BioFocus SoftFocus kinase inhibitor libraries. URMC-099 was synthesized and optimized at Califia Bio to provide the following: (1) high potency in a biochemical MLK3 inhibition assay; (2) activity in macrophages versus a relevant panel of cytokines of validated importance in HAND; (3) metabolic stability; (4) CNS penetration, as evidenced by pharmacokinetic studies in which the brain concentration against time after 10 mg/kg intravenous administration of URMC-099 in C57BL/6 mice yielded a brain area-under-the-curve of >5000 μg · kg−1 · h−1 and CNS concentrations above the in vitro IC50 for MLK3 inhibition for >6 h; and (5) confirmation of reasonable exposure on oral dosing [Compound 32, Patent WO 2010/068483 A2 (Gelbard et al., 2010)].
For all in vitro experiments, we prepared a 1000× stock solution of 100 μm URMC-099 in sterile dimethylsulfoxide (DMSO; D-8779; Sigma). For in vivo intraperitoneal injection, we dissolved 20 mg of URMC-099 in 0.5 ml of DMSO and diluted the URMC-099/DMSO solution in a mixture of 4 ml of polyethylene glycol 400 (PEG400; 91893-250-F; Sigma) and 5.5 ml of sterile saline (Hospira National Drug Code NDC0409-4888-10), resulting in a final 2 mg URMC-099/ml working solution (containing 5% DMSO). All mice treated with URMC-099 received a dose of 10 mg/kg every 12 h throughout the duration of the experiment. We pretreated mice with three doses of URMC-099 starting 36 h before stereotactic injection for immunohistochemistry (IHC), in vivo cytokine measurement, two-photon in vivo imaging, and electron microscopy experiments.
BV-2 cell line culture.
We cultured and maintained the BV-2 cell line as described previously (Marker et al., 2012). For the Western blot experiments, we plated 4 × 105 BV-2 cells on poly-d-lysine (PDL; P1149; Sigma) coated six-well plates (3516; Corning) for 36 h before treatment. For the quantitative RT (qRT)-PCR and multiplex ELISA experiments, we plated 1 × 105 BV-2 cells on PDL-coated 12-well plates (3513; Corning) for 12 h before treatment. We treated the cells with various combinations of HIV-1 Tat1–72 (purchased from Philip Ray, University of Kentucky, Lexington, KY), HIV-1 Tat101 (purchased from Philip Ray, University of Kentucky), DMSO, PBS (21-040-CV; Cellgro), and 100 nm URMC-099 in DMSO.
Western blotting.
We harvested cells by scraping in cell lysis buffer with protease inhibitors (p8340; Sigma) and phosphatase inhibitors (524629; Calbiochem). We mixed 12 μg of sample with loading dye, boiled the sample for 5 min, and ran it on a 4–15% SDS-PAGE gel (456-1086; Bio-Rad) at 100 V. We transferred the gel onto a nitrocellulose membrane at 100 V for 66 min on ice. We blocked membranes in 5% milk in 1× Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 1 h at room temperature with shaking. We applied primary antibodies overnight at 4°C as follows: rabbit anti-phospho-Jun N-terminal kinase (JNK) (4668P; Cell Signaling Technology), 1:2000 in 5% milk TBS-T and mouse anti-phospho p38 (4511; Cell Signaling Technology), 1:1000 in 5% bovine serum albumin (BSA) TBS-T. We washed membranes three times in TBS-T for 10 min each wash. We applied appropriate horseradish peroxidase-conjugated secondary antibody (170-6515 and 170-6516; Bio-Rad) at a concentration of 1:11,000 in 5% milk TBS-T for 45 min at room temperature with shaking. We washed membranes three times in TBS-T and applied enhanced chemiluminescence substrate (34076 and 34080; Thermo Fisher Scientific) for 3 min. We exposed and developed membranes on film (34091; Thermo Fisher Scientific).
To control for variations in protein loading, we stripped the membranes (2504; Millipore) and blocked them in 5% milk TBS-T for 30 min. We applied mouse anti-α-tubulin (T5168; Sigma) in 5% milk TBS-T overnight with shaking. We repeated the process of washing, secondary application, and developing to obtain the α-tubulin loading control blot. We obtained optical density measurements using NIH ImageJ.
Rodent primary hippocampal culture.
We dissected embryonic day 18 (E18) rat pups to obtain primary hippocampal neurons for our microfluidic coculture experiments. We explanted, plated, and maintained the neurons in Neurobasal media (21103-049; Invitrogen) supplemented with 3% FBS (F-0500A; Atlas Biologicals), B27 plus antioxidants (17504-044; Invitrogen), 2 mm GlutaMax (35050; Invitrogen), and 50 μm l-glutamic acid (G5889-100G; Sigma).
qRT-PCR.
We isolated RNA from BV-2 cells and synthesized cDNA as described previously (Marker et al., 2012). We used Invitrogen TaqMan universal PCR mix (4304437; Invitrogen) with Invitrogen TaqMan primers and probed for tumor necrosis factor α (TNFα) (Mm00443258_m1), interleukin-6 (IL-6) (Mm00446190_m1), monocyte chemoattractant protein-1 (MCP-1) (Mm00441242_m1), IL-10 (Mm00439614_m1), and 18S rRNA (Mm03928990_g1). We ran the samples on an Applied Biosystems 7300 Real-Time PCR machine. We normalized the data to the 18S rRNA internal control and determined fold change in gene expression using the Delta-Delta threshold cycle (ΔΔCT) method (Livak and Schmittgen, 2001).
Luminex multiplex ELISA.
We collected 0.5 ml of supernatant from BV-2 cells exposed to the various experimental conditions and froze the samples rapidly at −80°C. We used the Mouse Cytokine/Chemokine Magnetic Bead Panel (MCYTOMAG-70K; Millipore) to analyze the cytokine concentration of the supernatants. We used the following antibody coated beads in our analysis: TNFα (MCYTNFA-MAG), IL-6 (MCYIL6-MAG), MCP-1 (MCYMCP1-MAG), IL-10 (MIL10-MAG), and IL-4 (MIL4-MAG). We sonicated and vortexed the beads and combined them in assay buffer (L-AB) up to a total volume of 3 ml. We reconstituted the mouse cytokine standard (MXM8070-2) in ddH2O and performed a serial dilution to obtain standards of 10,000, 2000, 400, 80, 16, and 3.2 in assay buffer. We washed the plate with the included wash buffer (L-WB), added 25 μl of assay buffer to each well, and then added 25 μl of sample supernatant, protein lysate, or standard cytokine dilution to the appropriate wells. We then added 25 μl of the antibody bead mix to each well, sealed the plate, and incubated the beads at 4°C on a shaker overnight. Using the handheld magnetic separator block to retain the beads, we washed the plate twice in wash buffer. We then added 25 μl of detection antibodies (MXM1070-1) to each well, sealed the plate, and incubated at room temperature for 1 h with agitation. Finally, we added 25 μl of streptavidin–phycoerythrin (L-SAPE3; Millipore) to each well, sealed the plate, and incubated with agitation for 30 min at room temperature. We washed the beads two times with wash buffer and then resuspended them in 150 μl of sheath solution. We ran the plate on a Millipore Luminex 200 machine with xPONENT 3.1 software. Using the standard curves for each cytokine, we obtained absolute concentrations for the various experimental conditions for all cytokines measured. Note that we found no IL-4 present in any of our experimental conditions and thus excluded it from our analysis.
Latex bead phagocytosis assay.
We used a latex bead assay to determine the phagocytic activity of BV-2 cells as described previously (Marker et al., 2012). Briefly, we plated 1 × 105 BV-2 cells on PDL-coated 12-well plates 12 h before treatment and then treated the cells with 0.5 μg/ml Tat1–72, 1 μl/ml PBS, 1 μl/ml DMSO, and/or 100 nm URMC-099 with 1 μl/ml DMSO in a total of 1 ml of maintenance media. After 2, 4, 8, or 12 h had passed, we added 6 μl of a suspension of 0.8 μm deep blue dyed uncharged latex beads (L1398; Sigma) and cultured the cells for an additional 1.5 h. We washed, scraped, and collected the cells in 200 μl of PBS. We then sonicated the cell suspension and read the absorbance of the resulting solution on a spectrophotometer at 595 nm to quantify the relative number of beads that had been engulfed per well.
Microfluidic chambers.
We created microfluidic chambers based on the protocol of Park and colleagues as described previously (Park et al., 2006; Marker et al., 2012). The microfluidic chambers contain two compartments connected by 400 μm long × 10 μm wide × 3 μm high channels that allow physical and fluidic separation of axons and cell bodies. We used the chambers after plating E18 primary rodent hippocampal neurons at 0 d in vitro (DIV) in the somal compartment for experiments at 7 DIV. We imaged the axonal compartment on an Olympus IX70 inverted microscope with a 10× objective. We captured images with a PCO.edge camera controlled with the NIH acquisition software Micromanager (Edelstein et al., 2010). We then added 40 μl of a 2.5 × 106 cells/ml suspension of BV-2 cells in the neuronal cell culture media with a combination of 1 μl/ml DMSO, 1 μg/ml HIV-1 Tat101, and/or 100 nm URMC-099 with 1 μl/ml DMSO to the axonal compartment. The volume gradient (100 μl somal compartment vs 40 μl axonal compartment) ensured that no soluble treatment factors reached the soma compartment. After 18 h of coculture, we obtained posttreatment bright-field images and fixed the cells in 4% paraformaldehyde (PFA; S898-07; J.T. Baker) and 4% sucrose (8510; EM Science) in 1× PBS for 10 min at 4°C. We washed the cells with 1× PBS and stored the fixed cells at 4°C in 1× PBS for immunocytochemistry (ICC). We compared the pretreatment and posttreatment bright-field images to determine the elimination rate of the axons. We used the NIH ImageJ plugin NeuronJ to quantify the length of the axons past the microfluidic barrier before and after treatment (Meijering et al., 2004; Schneider et al., 2012).
ICC.
We performed ICC of microfluidic chambers as described previously (Glynn and McAllister, 2006; Marker et al., 2012). We used the following antibodies in our analysis: mouse anti-Tau5 at 1:500 (577801; Calbiochem), rabbit anti-Synapsin-1 at 1:500 (5297S; Cell Signaling Technology), rat anti-CD11b at 1:200 (MCA711; Serotech), and various Alexa Fluor-conjugated secondary antibodies at 1:500 (Invitrogen). We used the Prolong Gold mounting agent with DAPI (P36935; Invitrogen) to mount the coverslips and prevent photobleaching. We imaged the sections with a Hamamatsu ORCA-ER camera on an Olympus BX-51 upright microscope with Quioptic Optigrid optical sectioning hardware. We controlled the acquisition with the Volocity 3DM software (PerkinElmer Life and Analytical Sciences).
Bone marrow chimeras.
We engrafted CD45.1 mice with heterozygous CX3CR1/GFP+/− bone marrow as described previously (Lu et al., 2011). Briefly, we lethally irradiated 8-week-old CD45.1 host mice with 9 Gy of gamma radiation from a cesium source. We head shielded all of the mice to prevent any radiation-induced neuroinflammation or damage (Ajami et al., 2007; Mildner et al., 2007). We killed 8- to 12-week-old CX3CR1/GFP+/− donor mice by CO2 asphyxiation and collected the bone marrow from the femurs and tibias in Dulbecco's PBS (dPBS; 21-030-CV; Cellgro). We lysed the red blood cells with ACK lysing buffer (10-548E; Lonza), washed twice in sterile dPBS, and resuspended the cells at a concentration of 5 × 107 cells/ml. We injected 200 μl of this cell suspension into the tail veins of the irradiated host mice. We allowed the mice to recover for 4 weeks before performing any experimental manipulations.
In vivo Tat exposure model.
We performed stereotactic injections with 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 in sterile PBS as described previously (Marker et al., 2010; Lu et al., 2011). We used a mix of both male and female mice for all in vivo experiments, except for the chimera IHC experiments in which we used only male mice. For chimera IHC experiments, in vivo cytokine measurement experiments, and immunoelectron microscopy experiments, we performed a stereotactic injection at the coordinates 1.0 mm posterior to bregma, 1.0 mm lateral left of bregma, and 0.7 mm ventral to the pial surface. We used a 35 gauge needle with a 10 μl Hamilton syringe, which was coated with Sigmacote (SL-2; Sigma) to prevent the Tat from sticking to the inside of the syringe, controlled by a micro-syringe pump (Micro4; World Precision Instruments). We injected at a flow rate of 80 nl/min to minimize injury occurring as a result of the injection pressure. For two-photon thin-skull cortical window (TSCW) experiments, the exact injection coordinates varied within motor and somatosensory cortex but were always 3.0 mm away from the imaging site in visual cortex on the ipsilateral side of the brain and were always 0.7 mm ventral to the pial surface.
IHC.
We killed animals by pentobarbital overdose and transcardially perfused them with periodate-lysine–3.5% PFA (PLP) fixative (McLean and Nakane, 1974). We postfixed the brains in 3.5% PLP overnight and then stored them in 1× PBS at 4°C. We cut the brains into 40-μm-thick sections on a vibratome (Leica V1000) and stored them in a cryoprotectant mixture of 30% PEG300 (PX1286A-2; EMD Millipore), 30% glycerol (56-81-5; Alfa Aesa), 20% 0.1 m phosphate buffer, and 20% ddH2O at −20°C. We performed free-floating-section IHC as described previously (Lu et al., 2011) with modification. We washed the sections three times for 30 min in PBS to remove the cryoprotectant. We prepared the primary antibody mixture in 1.5% BSA (A3294; Sigma), 3% normal goat serum (NGS; S1000; Vector Laboratories), 0.5% Triton X-100 (H5142; Promega), and 1.8% NaCl in 1× PBS. We used the following antibodies in these experiments: rabbit anti-Iba1 at 1:1000 (019-19741; Wako Biochemicals), mouse anti-microtubule-associate protein 2 (Map2) at 1:500 (M4403; Sigma), rat anti-7/4 at 1:1000 (AB53547; Abcam), and rabbit anti-Synapsin-1 at 1:400 (5297; Cell Signaling Technology). We incubated the sections in the primary antibody mixture for 2 d at room temperature with agitation and an additional 5 d at 4°C with agitation if the staining included Map2 or Synapsin-1. We then washed the sections three times for 30 min in 1× PBS with 1.8% NaCl and prepared the Alexa Fluor-conjugated antibodies (1:500; Invitrogen) in the same staining mixture described above. We incubated the sections in secondary antibody overnight at room temperature with agitation. Finally, we washed the sections three times with 1× PBS, mounted the sections on slides, and coverslipped the slides with #1.5 cover glass and Prolong Gold mounting agent with DAPI. For sections that received costaining against Iba1 and GFP, we first performed primary and secondary staining against Iba-1 as described above. To label GFP, we performed two additional 30 min washes in 1× PBS with 1.8% NaCl after the secondary antibody incubation. We then blocked the sections for 30 min in 1.5% BSA, 3% NGS, 0.5% Triton X-100, and 1.8% NaCl in 1× PBS with the addition of 3% normal rabbit serum (S5000; Vector Laboratories). We then incubated the sections overnight in the above mixture plus a 1:1000 concentration of the rabbit anti-GFP–Alexa Fluor 488-conjugated antibody (A11122; Invitrogen). We washed and mounted the sections normally after these additional steps. We imaged the sections as described in above (see ICC).
In vivo cytokine analysis.
We killed animals by pentobarbital overdose and transcardially perfused them with ice-cold saline for 15 s. We then rapidly removed the brain, placed it on ice, blocked the brain with a razor blade, and rapidly froze the tissue on dry ice. We prepared a 30–40 mg section of brain that included the injection site for analysis. We homogenized the brain sections in 750 μl of a solution of 1× TBS and 0.05% Tween 20, with protease and phosphatase inhibitors (161280; Thermo Fisher Scientific) using Wheaton glass homogenizers. We freeze-thawed the lysates one time and then spun them for 10 min at 14,000 relative centrifugal force to remove the insoluble debris. Using the Bio-Rad DC protein detection assay (500-0111; Bio-Rad), we measured and equalized the protein concentration of all of the brain lysate samples to 2.1 μg/ml. We then analyzed 25 μl of the samples using the Luminex multiplex ELISA system as described in the previous section.
TSCW two-photon microscopy.
We performed TSCW microscopy as described previously (Marker et al., 2010). We imaged mice on either a custom Olympus FluoView 300 two-photon microscope with 20× water-immersion objective (Majewska et al., 2000) or on a commercial Olympus FluoView 1000 two-photon system with a 25× water-immersion objective. We imaged 8- to 12-week-old double-transgenic Thy1 yellow fluorescent protein (YFP) h-line, CX3CR1/GFP+/− mice before and 24 h after experimental treatment. We split the GFP and YFP signals using a 509 nm dichroic (Semrock) and appropriate emission filters. We captured dual-channel 40 μm z-stacks in layers I and II of visual cortex at a resolution of 1024 × 1024 pixels with a 0.138 × 0.138 × 1 μm voxel size. We performed final YFP/GFP separation digitally using the PerkinElmer Life and Analytical Sciences Volocity software.
Amira image analyses.
We used the Amira image analysis software to determine the morphologic characteristics of microglia imaged with TSCW two-photon microscopy. We imported single-channel z-stacks of fields of microglia and ran the Median Noise Reduction function to remove the pixelated noise associated with the use of photo-multiplier tubes. We then segmented and labeled the individual microglia via manually guided thresholding. We used the Center Line Tree algorithm function to create skeleton trees of the labeled microglia. Finally, we obtained volume measurements from the labeled microglia via the Material Statistics function and process measurements from the skeleton trees via the Spatial Graph Statistics function.
Immunoelectron microscopy.
We anesthetized CX3CR1/GFP+/− mice (8–12 weeks of age) for electron microscopy analyses with sodium pentobarbital and perfused them through the aortic arch with 3.5% acrolein, followed by 4% PFA (Tremblay et al., 2010b). We killed groups of animals at both 24 h and 7 d with and without URMC-099 treatment after Tat or PBS (control) injections. We cut transverse sections of the brain (50 μm thick) in ice-cooled PBS with a vibratome, immersed them in 0.1% sodium borohydride for 30 min at room temperature, washed them in PBS, and processed the free-floating sections following a preembedding immunoperoxidase protocol as described previously (Tremblay et al., 2010b). Briefly, we rinsed sections in PBS, preincubated them for 2 h at room temperature in a blocking solution of PBS containing 5% NGS and 0.5% gelatin, incubated them for 48 h at room temperature in rabbit anti-Iba1 antibody (1:1000 in blocking solution; 019-19741; Wako), and then rinsed them in PBS. We then incubated the sections for 2 h at room temperature in goat anti-rabbit IgG conjugated to biotin (Jackson ImmunoResearch) and then with streptavidin–horseradish peroxidase (Jackson ImmunoResearch) for 1 h at room temperature in blocking solution. We revealed the labeling with diaminobenzidine (0.05 mg/ml) and hydrogen peroxide (0.03%) in buffer solution (Vector Laboratories). We then postfixed the sections flat in 1% osmium tetroxide, dehydrated them in ascending concentrations of ethanol (some sections were treated with 0.2% uranyl acetate in 70% ethanol during the dehydration steps), treated them with propylene oxide, impregnated them in Durcupan (Electron Microscopy Sciences) overnight at room temperature, mounted them between ACLAR embedding films (Electron Microscopy Sciences), and cured them at 55°C for 48 h.
We mounted tissue from selected regions of visual or somatosensory cortex, located between 2 and 3 mm from the injection site, onto plastic blocks and collected ultrathin sections on copper grids (bare square mesh or pioloform-coated single slots). We enhanced the section contrast with lead citrate staining. We examined the grids using a Hitachi 7650 or a JEOL 1011 electron microscope. The immunoperoxidase labeling showed good penetration into the tissue, so we were able to sample material away from the tissue–resin interface. We collected data using two different methods, both blind to the experimental conditions. First, we obtained a series of lower-power, 20,000× magnification images randomly from the block. We took the images from randomly selected regions of the neuropil, taking care only to avoid the tissue resin interface or other ultrastructural flaws. Second, we obtained a series of higher-power, 40,000× magnification images. For these images, we attempted to include immunolabeled elements in the image without consideration of what the elements were. We obtained images in the superficial cortical layers using a Gatan Erlangshen ES1000W or Gatan 785 camera. We converted the images to TIFF files using Gatan Digital Micrograph software.
We analyzed a total surface area of ∼2 × 103 μm2 in each animal. We identified microglia, astrocytes, oligodendrocytes, and neurons according to defined criteria (Peters et al., 1991; Oster-Granite et al., 1996; Turmaine et al., 2000; Peters and Sethares, 2004; Yang et al., 2008; Tremblay et al., 2010a).
Statistical analyses.
We performed all of our statistical analyses using the GraphPad Prism statistical software. We defined significance as p < 0.05. For experiments with more than two experimental conditions at a single time point or a single condition with multiple time points, we used the one-way ANOVA with Newman–Keuls post hoc test. For experiments with multiple conditions over multiple time points, we used the two-way ANOVA with Bonferroni's post hoc test.
Results
URMC-099 exerts anti-inflammatory effects on cultured microglia
MLK3 exerts a well-characterized effect on activation of JNK (Gallo and Johnson, 2002). Therefore, we tested whether URMC-099 effectively inhibited MLK3 regulated pathways in BV-2 microglial cells by measuring the phosphorylation levels of JNK and p38 MAP kinases in Tat exposed cultures, with and without 100 nm URMC-099 treatment. We used a 100 nm concentration of URMC-099 in our in vitro models because this concentration is 10 times higher than the Ki of URMC-099 for MLK3 and should provide complete blockade of MLK3 signaling in vitro. A 30 min exposure to HIV-1 Tat101 resulted in a rapid induction of both p46 and p54 phospho-JNK (Fig. 1A–C). Cotreatment with URMC-099 resulted in a statistically significant, 70% reduction in JNK activation, as assessed by quantitation of both p46 and p54 phospho-JNK (Fig. 1B,C). Tat treatment significantly increased phosphorylation of p38 and URMC-099 cotreatment only resulted in a small 10% reduction in p38 phosphorylation (Fig. 1D,E). These results are consistent with previous studies of MLK3−/− fibroblasts, which showed that genetic ablation of MLK3 prevented JNK activation both in response to TNFα and after exposure to stress-promoting free fatty acids (FFAs) but had no effect on TNFα- or FFA-mediated activation of p38 MAP kinase (Gallo and Johnson, 2002; Brancho et al., 2005).
Figure 1.
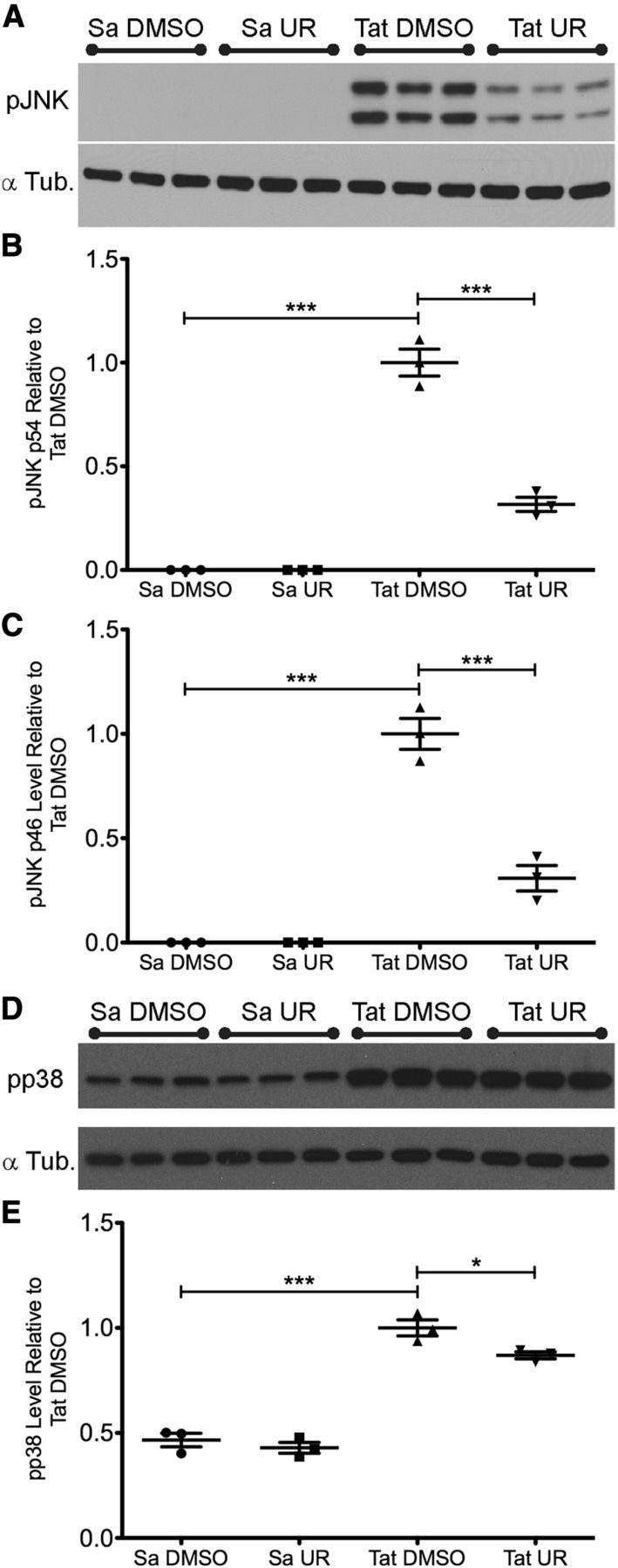
URMC-099 blocks phosphorylation of the MLK3 target JNK in BV-2 cells after HIV-1 Tat exposure. We pretreated BV-2 cells for 1 h with 1 μl DMSO/ml maintenance media or 100 nm URMC-099 in 1 μl DMSO/ml maintenance media. We then treated the cells with either 1 μl/ml saline or 1 μg/ml HIV-1 Tat101 for 30 min. We collected the cell lysate and performed Western blotting to determine levels of phospho-JNK and phospho-p38. A, Representative blot for phospho-JNK. There was no basal level of phospho-JNK in either saline condition. Tat treatment significantly induced both p54 (B) and p46 (C) phospho-JNK isoforms. URMC-099 treatment significantly decreased the levels of phosphorylation of both isoforms. D, Representative blot for phospho-p38. E, Tat treatment significantly induced p38 phosphorylation at this time point, and URMC-099 treatment slightly reduced levels of phospho-p38 in the Tat-treated condition. *p < 0.05, ***p < 0.001. We analyzed B, C, and E by one-way ANOVA with Newman–Keuls post hoc test. Error bars indicate SEM. Sa, Saline; Tub., tubulin; UR, URMC-099.
To determine whether URMC-099 had potential as a treatment for HAND-associated neuroinflammation, we measured the production of various HAND-associated cytokines in microglia exposed to HIV-1 Tat with and without URMC-099 treatment. We used the BV-2 cell immortalized mouse microglia line for these experiments, because these cells have been shown to act similarly to primary microglia in culture (Bocchini et al., 1992). We found that URMC-099 treatment significantly decreased the production of TNFα, IL-6, and MCP-1 after various durations of Tat exposure at both the transcript and protein levels (Fig. 2A–C, respectively). URMC-099 had no effect on the expression of IL-10, a cytokine with classical anti-inflammatory effects (Fig. 2D). Although 100 nm URMC-099 significantly reduced the levels of Tat induced proinflammatory cytokine expression, it did not normalize the cytokine production to control levels.
Figure 2.

URMC-099 reduces in vitro microglial activation in response to HIV-1 Tat1–72. We treated microglial BV-2 cells with HIV-1 Tat (0.5 μg/ml) for 4, 8, and 12 h and measured the production of TNFα, IL-6, MCP-1, and IL-10 (A–D, respectively), with and without URMC-099 (100 nm) treatment. We measured cytokine mRNA using qRT-PCR (ΔΔCT), normalized the transcript to 18S rRNA, and displayed the data as fold change relative to the saline control. We measured the soluble cytokine protein in the supernatant using the Luminex ELISA method and displayed the data as an absolute concentration. Control bars represent conditions with 12 h of treatment with either PBS (1 μl/ml cell culture media) or DMSO (1 μl/ml cell culture media). We quantified data from three independent experiments. E, HIV-1 Tat induces microglial phagocytosis. We treated BV-2 cells with HIV-1 Tat for 0, 2, 4, 8, and 12 h and then exposed the cells to 0.8 μm latex beads for 90 min to measure the phagocytic activity of the cells. We quantified the data from three independent experiments. F–H, URMC-099 blocks HIV-1 Tat-induced increase in phagocytosis. F, G, Representative bright-field images of BV-2 cells treated with HIV-1 Tat and HIV-1 Tat plus URMC-099 for 12 h and then exposed to 0.8 μm latex beads for 90 min, respectively (20× magnification). H, We treated BV-2 cells with saline, DMSO, URMC-099, HIV-1 Tat, and HIV-1 Tat plus URMC-099 for 12 h and then exposed the cells to 0.8 μm latex beads for 90 min. We quantified data from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. We analyzed A–D by two-way ANOVA, no sample matching with a Bonferroni's post hoc test and E and H by one-way ANOVA with Newman–Keuls post hoc test. Error bars indicate SEM. UR, URMC-099.
We then examined the effect of Tat and URMC-099 treatment on microglial phagocytosis. For our initial studies, we used a basic colored latex bead assay to measure the phagocytic activity of microglia exposed to Tat for various durations. By 4 h of Tat exposure, BV-2 cells significantly increase their level of phagocytic activity in this model system (Fig. 2E,F). For subsequent experiments, we examined the 12 h exposure time point, because this exposure led to the greatest increase in phagocytic activity. Treatment of Tat-exposed microglia with 100 nm URMC-099 significantly decreased the phagocytic activity of the BV-2 cells to saline-treated control levels (Fig. 2G,H).
We used a microfluidic chamber microglia/neuronal coculture system developed by our laboratory to determine whether the reduction in cytokine release and phagocytosis would potentially have a neuroprotective effect. We exposed axons of 7 DIV neuronal cultures to BV-2 cells that received treatment of one of the following conditions: (1) DMSO alone (Fig. 3A,B,G,H), (2) HIV-1 Tat101 and DMSO (Fig. 3C,D,I,J), or (3) HIV-1 Tat101, 100 nm URMC-099 in DMSO (Fig. 3E,F,K,L) for 18 h. We found that axonal fields exposed to DMSO-treated BV-2 cells remained nearly wholly intact (Fig. 3A,B), with BV-2 cells growing on top of healthy axons and axon remodeling around BV-2 cells (Fig. 3H). Conversely, axons exposed to Tat-treated BV-2 cells were almost completely eliminated (Fig. 3C,D). This elimination was accompanied by the presence of axonal marker-positive inclusions inside microglia, indicating phagocytosis (Fig. 3J). URMC-099 treatment preserved the majority of the axonal field, even in the presence of Tat and activated microglia (Fig. 3E,F). Some phagocytosis of neuronal components remained in the URMC-099-treated group, despite the overall protection of the axonal field (Fig. 3L). When we quantified the percentage of preserved axons (Fig. 3M) and the length of the remaining axons (Fig. 3N), we found that URMC-099 was able to protect axons from the Tat-exposed microglia to the level of control conditions (p < 0.001; Fig. 3, legend).
Figure 3.
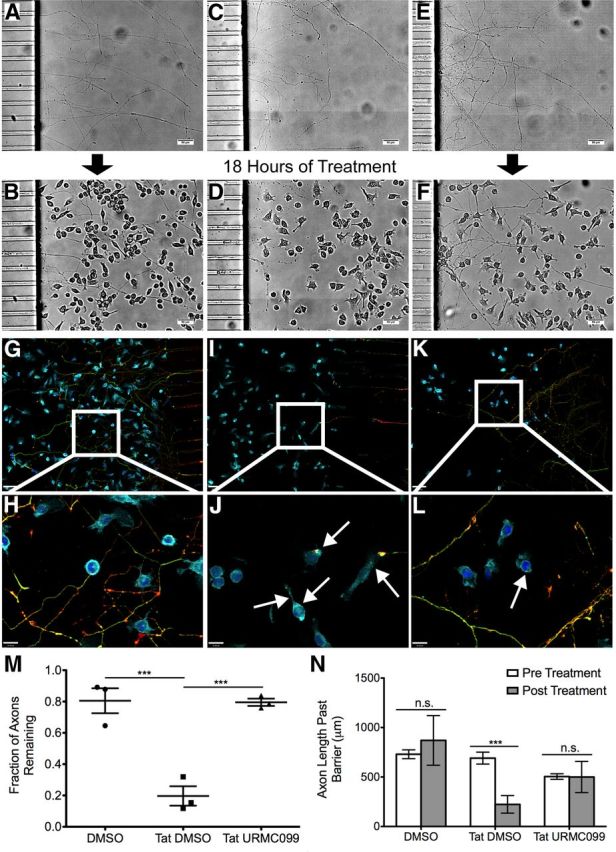
URMC-099 prevents microglial phagocytosis of axons in vitro. We grew primary rat hippocampal neurons in microfluidic chambers for 7 d, during which time they established a robust axonal arbor (A, C, E). We then plated BV-2 cells on top of the axonal arbor and treated the cells and axons with DMSO (1 μl/ml) (B), HIV-1 Tat101 (1 μg/ml) and DMSO (D), or HIV-1 Tat and URMC-099 (100 nm) in DMSO (F) for 18 h. Scale bars, 50 μm. We fixed and stained microfluidic chambers treated with DMSO (G, 10× magnification; H, 40× magnification), Tat and DMSO (I, J), and Tat and URMC-099 (K, L) for DAPI (dark blue), the microglia marker CD-11b (light blue), and the axonal markers Tau-5 (green) and Synapsin-1 (red). Arrows highlight neuronal positive inclusions inside microglia. 10× magnification images shown are a single z-plane; 40× magnification images are an accumulative z-projection of a 4 μm z-stack with a 0.2 μm step. Scale bars: 10× images, 60 μm; 40× images, 16 μm. We quantified the percentage of axons remaining (M) and the average length of the axons past the microfluidic barrier (N) in the three conditions from three independent experiments. ***p < 0.001. We analyzed data depicted in M by one-way ANOVA with Newman–Keuls post hoc test and data depicted in N by two-way ANOVA, no sample matching with Bonferroni's post hoc test. Error bars indicate SEM.
URMC-099 treatment has no effect on Tat-induced peripheral immune cell recruitment to the CNS but does significantly reduce Tat-induced inflammatory cytokine production in the CNS
We next examined the effect of URMC-099 treatment in mice receiving a single cortical injection of HIV-1 Tat protein. Our previous work with hippocampal Tat exposure showed a massive influx of peripheral monocytes and neutrophils coupled with microglial activation (Lu et al., 2011). The first goal of our current study was to determine whether URMC-099 had any effect on Tat-mediated recruitment of immune cells into the CNS. We examined the immune response with URMC-099 treatment 24 h, 7 d, and 28 d after Tat or PBS injection in CD45.1 mice with allografted CX3CR1/GFP+/− bone marrow. We found that URMC-099 treatment had no effect on neutrophil infiltration in the brain parenchyma (Fig. 4A,D,G), as well as no effect on peripheral monocyte recruitment 7 d (Fig. 4B,E,H) or 28 d (Fig. 4C,F,G) after injection.
Figure 4.

URMC-099 does not alter the peripheral cellular immune response to cortical HIV-1 Tat injection. We performed bone morrow transplantation in lethally irradiated 8-week-old CD45.1 mice with CX3CR1/GFP+/− donor bone marrow and allowed the mice to recover for 4 weeks. We then injected either 3 μl of sterile PBS (A–C) or 3 μl of 3 μg/μl HIV-1 Tat 700 μm deep into somatosensory cortex. The mice receiving Tat were either not treated (D–F) or were treated with intraperitoneal injections of 10 mg/kg URMC-099 twice a day before and after Tat exposure (G–I). We killed the mice 24 h, 7 d, and 28 d after Tat injection and collected the tissue for immunohistochemical analysis. We stained for the neutrophil marker 7/4 (white), GFP (green), which was present only in monocytes derived from the peripheral bone marrow, and the monocyte/microglia marker Iba-1 (red). Images shown were acquired using a 20× air objective and are shown as an accumulative z-projection of a 16 μm z-stack with a 0.5 μm z-step. Scale bars, 32 μm.
We then measured whether URMC-099 treatment reduced inflammatory cytokine production in the CNS of mice receiving a single cortical Tat injection. At the 24 h time point, we found that Tat exposure caused significantly increased production of TNFα, MCP-1, and IL-6 compared with saline-injected control animals (Fig. 5A–C, respectively). Cotreatment with URMC-099 significantly reduced the production of MCP-1 and normalized the production of TNFα and IL-6 to saline control levels. Neither Tat nor URMC-099 treatment had any effect of IL-10 levels (Fig. 5D). These results indicate that, although Tat-induced peripheral immune recruitment is not affected by URMC-099 treatment, the inflammatory response of resident CNS immune cells and peripheral immune cells that infiltrate the CNS after exposure to Tat is inhibited by URMC-099 treatment.
Figure 5.
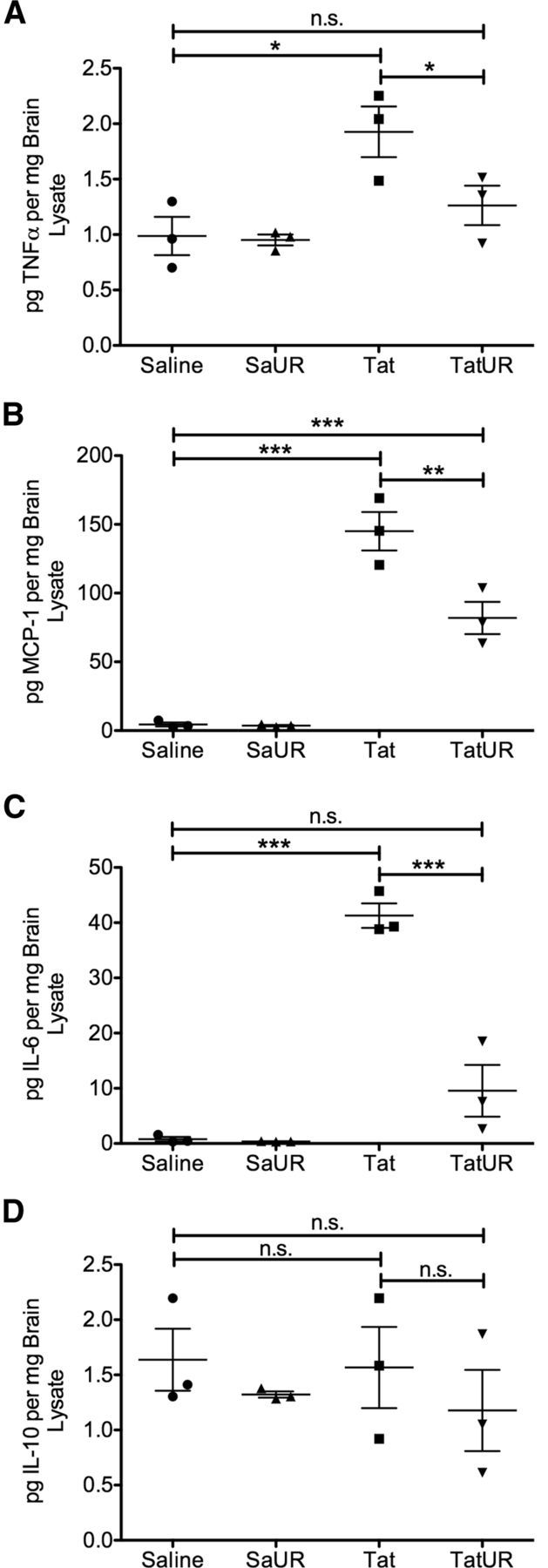
URMC-099 reduces Tat-induced inflammatory cytokine production in vivo. We stereotactically injected either 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 700 μm deep into somatosensory cortex. We treated half of the mice receiving saline and half of the mice receiving Tat with twice a day peritoneal injections of 10 mg/kg URMC-099. The other mice were left untreated. After 24 h, we rapidly dissected and flash froze total brain tissue from around the injection site and prepared protein lysates for Luminex protein analysis. We measured the levels of TNF-α (A), MCP-1 (B), IL-6 (C), and IL-10 (D) present in 50 μg of total brain lysate for each animal. Tat exposure significantly increased levels of TNF-α, MCP-1, and IL-6 above saline control. URMC-099 treatment significantly decreased the production of these cytokines, and, in the case of TNFα and IL-6, reduced production to saline control levels. URMC-099 had no effect on the levels of cytokines in saline-treated animals. Tat exposure did not significantly induce production of IL-10, and URMC-099 treatment had no effect on IL-10 levels. Results are expressed as picograms of cytokine detected per milligrams of total brain protein lysate analyzed. *p < 0.05, **p < 0.01, ***p < 0.001. We analyzed data depicted in A–D by one-way ANOVA with Newman–Keuls post hoc test. Error bars indicate SEM. Sa, Saline; UR, URMC-099.
URMC-099 is neuroprotective in mice exposed to HIV-1 Tat
Next, we examined neuronal integrity to determine whether URMC-099 was neuroprotective. We examined the 28 d time point, because our pilot experiments showed that this was the time point of greatest Tat-induced neuronal damage in untreated animals. We first examined the integrity of Map2. The loss of Map2 immunoreactivity has been shown to be one of the major correlates of HAND severity, and furthermore, HIV-1 Tat directly interferes with Map2 function (Masliah et al., 1997; Aprea et al., 2006). Animals receiving PBS injection have no alteration in Map2 immunostaining directly at the injection site (Fig. 6A), as shown by the dense filamentous staining. Conversely, animals exposed to HIV-1 Tat lose almost all of the filamentous Map2 immunoreactivity around the injection site (Fig. 6C). Continuous treatment with URMC-099 before and after Tat exposure partially preserved normal Map2 staining at the Tat injection site (Fig. 6E). Finally, we quantified synaptic density at the injection site by measuring puncta of Synapsin-1, a marker for axonal terminals. Tat exposure significantly decreased puncta density at the injection site, and URMC-099 treatment restored puncta density to control levels (Fig. 6B,D,F,G; p < 0.001 by one-way ANOVA with Newman–Keuls post hoc test). These results indicate that URMC-099 treatment is neuroprotective in this model.
Figure 6.
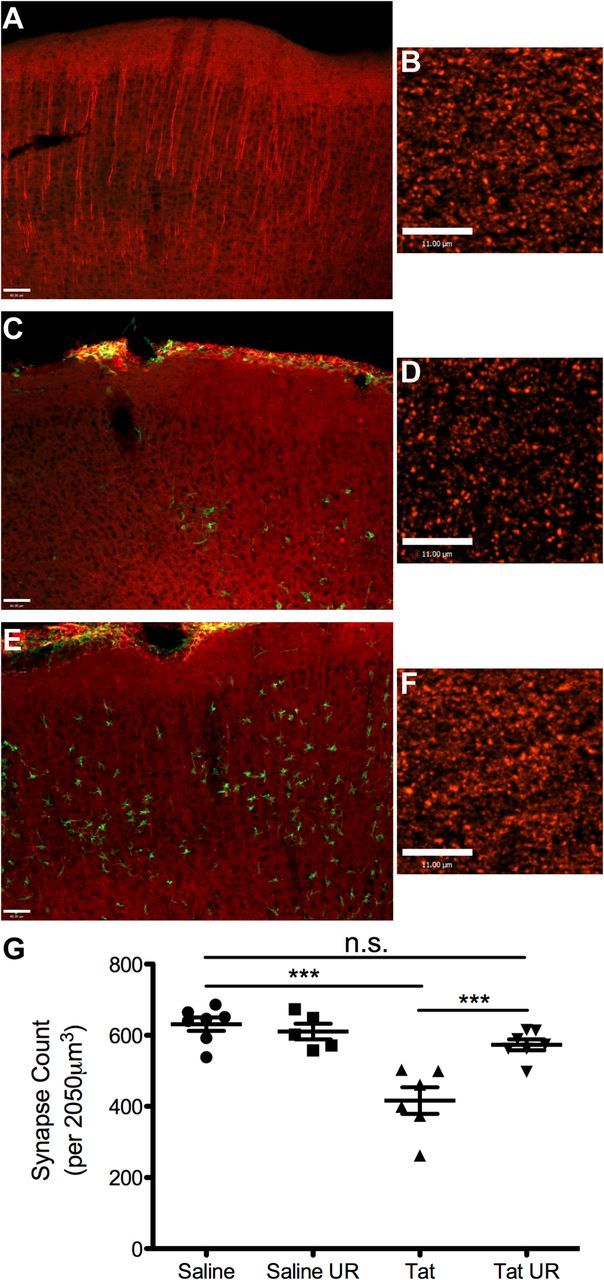
URMC-099 protects cortical neurons from the effects of HIV-1 Tat exposure. We performed bone marrow transplantation in lethally irradiated 8-week-old CD45.1 mice with CX3CR1/GFP+/− donor bone marrow and allowed the mice to recover for 4 weeks. We then stereotactically injected either 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 700 μm deep into somatosensory cortex. We treated half of the mice receiving saline and half of the mice receiving Tat with twice a day peritoneal injections of 10 mg/kg URMC-099. The other mice were left untreated. We killed the mice 28 d after stereotactic injection and collected the tissue for immunohistochemical analysis. We stained tissue from mice receiving PBS (A), Tat alone (C), or Tat and URMC-099 treatment (E) for the dendritic marker Map-2 (red) and the peripheral monocyte marker GFP (green). Mice receiving Tat and URMC-099 treatment had preserved filamentous Map-2 staining when compared with Tat alone. Scale bars, 60 μm. We then stained the tissue from the same animals for Synapsin-1 (B, D, F, red staining) and quantified the number of Synapsin puncta per volume at the injection site (G). Scale bars, 11 μm. ***p < 0.001. We analyzed data depicted in G by one-way ANOVA with Newman–Keuls post hoc test. n = 5–7 animals per group. Error bars indicate SEM. UR, URMC-099.
URMC-099 alters the morphology and neuronal interactions of microglia exposed to HIV-1 Tat
We then examined the effect of URMC-099 on microglia and microglia–neuronal interactions in vivo, as a potential contributing factor for the preserved neuronal architecture. Using TSCW two-photon microscopy, we observed the morphology of microglia in the same cortical area before and after either PBS or Tat injection. We found that microglia exposed to Tat (Fig. 7B) simplified their process arbor, as measured by total process number (Fig. 7E) and total process length (Fig. 7F), compared with PBS-treated controls (Fig. 7A). We found that treatment with URMC-099 had no effect on microglia in untreated animals (Fig. 7D–F, Pre Injection). Tat-exposed microglia treated with URMC-099 maintained control levels of process number and process length. However, these microglia were not morphologically identical to control treated microglia, because they were significantly larger in volume than either PBS or Tat alone treated microglia (Fig. 7D). Although the microglia are not morphologically identical to control microglia, the normalization of the process arbor may indicate maintenance of normal microglial function or reduction in pathologic microglial activity in the presence of Tat in URMC-099-treated animals.
Figure 7.
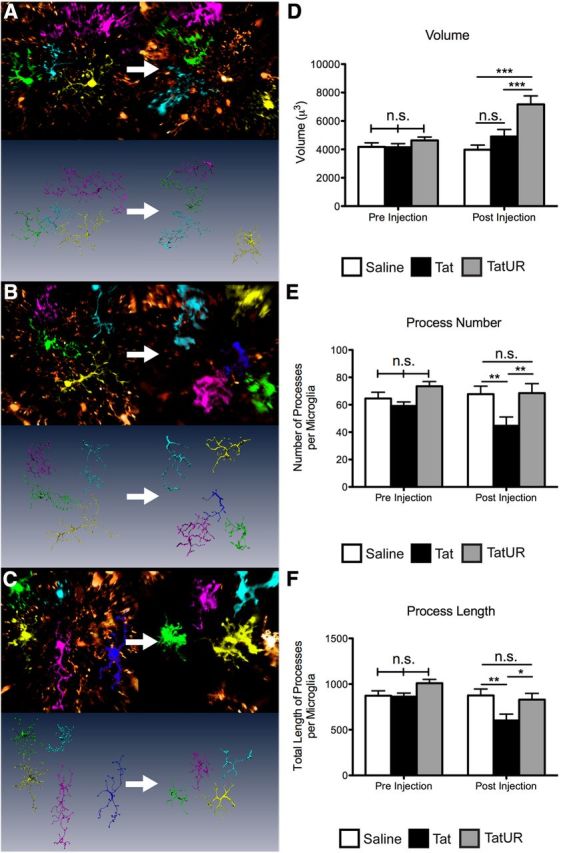
URMC-099 treatment alters Tat-induced morphologic changes in microglia in vivo. We performed TSCW two-photon microscopy on double-transgenic mice (Thy1 YFP h-line and CX3CR1/GFP+/−) before and after receiving a stereotactic injection of 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 700 μm deep into cortex with or without URMC-099 treatment (10 mg/kg twice a day, i.p.). We imaged multiple areas 3 mm away from the injection site immediately before injection and imaged the same areas 24 h after injection. We used the Amira image analysis program to segment, label, and skeletonize microglia from the animals receiving PBS (A; n = 28 microglia preinjection; n = 24 microglia postinjection), Tat alone (B; n = 26 preinjection; n = 27 postinjection), and Tat plus URMC-099 treatment (C; n = 30 preinjection; n = 26 postinjection) before and 24 h after injection. Images depict representative fields from each treatment group of the same area before and 24 h after treatment. Using these data, we quantified microglia volume (D), process number (E), and total length of the process arbor (F) before and after injection with Tat or vehicle control. *p < 0.05, **p < 0.01, ***p < 0.001. We analyzed data depicted in D–F by two-way ANOVA, no sample matching with Bonferroni's post hoc test. Error bars indicate SEM. UR, URMC-099.
Finally, we examined the effect of URMC-099 on the in vivo interaction between microglia and neurons. Recent studies by our group and collaborators have demonstrated microglial contact and engulfment of neuronal components during normal plasticity, as well as in neuroinflammation associated with Tat exposure (Tremblay et al., 2010a, 2011; Lu et al., 2011). We hypothesized that URMC-099 treatment might reduce some of these pathologic interactions. Using TSCW two-photon microscopy, we found that Tat exposure caused extended duration of microglia–neuronal contact (Fig. 8A). Furthermore, we found that Tat exposure led to a significant increase in the rate of dendritic spine loss (Fig. 8B,C; p < 0.05 by one-way ANOVA with Newman–Keuls post hoc test). URMC-099 treatment decreased the rate of spine elimination to that of PBS-treated controls, again confirming its neuroprotective properties.
Figure 8.

URMC-099 treatment reduces cortical dendritic spine elimination after HIV-1 Tat exposure. We performed TSCW two-photon microscopy on double-transgenic mice (Thy1 YFP h-line and CX3CR1/GFP+/−) receiving a stereotactic injection of either 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 700 μm deep into cortex with and without URMC-099 treatment (10 mg/kg twice a day, i.p.). We imaged multiple areas 3 mm away from the injection site immediately before injection and imaged the same areas 24 h after injection. A, We observed extended microglial contact with dendritic spines 24 h after Tat injection (microglia in green, dendrites in orange; 10 min between frames; scale bars, 14 μm). We examined dendritic fields before and 24 h after experimental injection and quantified the number of eliminated spines. B, Representative images of a dendritic branch before and after experimental exposure in an animal receiving a Tat injection with no URMC-099 treatment. Red triangles indicate spines that were eliminated 24 h after Tat exposure. C, We quantified the fraction of spines that were eliminated in mice receiving a stereotactic injection of PBS alone (n = 3), PBS with URMC-099 treatment (n = 2), Tat alone (n = 3), and Tat with URMC-099 treatment (n = 3). We analyzed a minimum of 50 spines per animal to determine the elimination rate. *p < 0.05. We analyzed data depicted in C by one-way ANOVA with Newman–Keuls post hoc test. Error bars indicate SEM. UR, URMC-099.
However, two-photon microscopy does not have the spatial resolution to confirm that microglia are actually contacting or engulfing neuronal components. To overcome this limitation, we performed immunoelectron microscopic analysis of brain tissues from Tat-exposed mice, using Iba1 as a marker for microglia. We replicated our previous findings, demonstrating microglial contacts, both phagocytic and nonphagocytic, with multiple neuronal components 24 h and 7 d after Tat injection (Fig. 9A–D). The contacted components were predominantly nondegenerating, as revealed by their light cytoplasm, intact organelles, and cytoskeletal elements. URMC-099 treatment significantly reduced the prevalence of microglial phagocytic inclusions, which prominently consisted of ultrastructural features of myelinated axons, and of direct contacts with myelinated axons in Tat-treated animals at both 24 h and 7 d after injection (Fig. 9E,F; p < 0.001 by two-way ANOVA, no sample matching with Bonferroni's post hoc test). However, URMC-099 had no effect on contacts with dendrites or astrocytes (Fig. 9G,H). These data further support the preservation of axonal integrity when URMC-099 is coadministered with Tat that we observed in the microfluidic chamber model.
Figure 9.
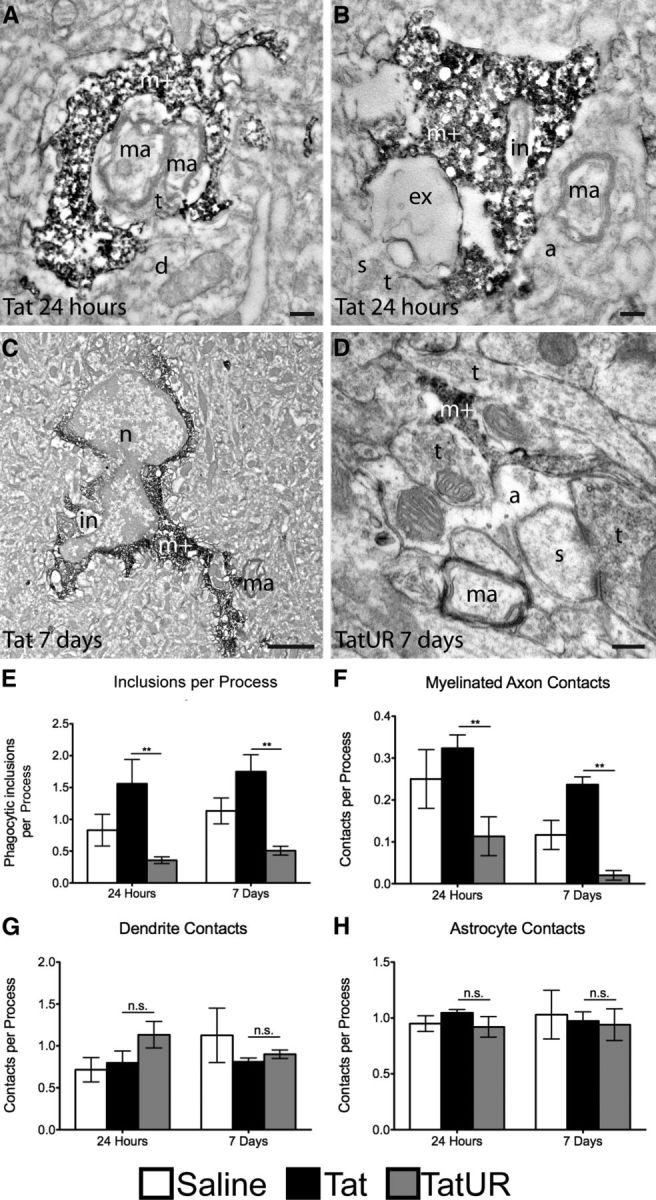
URMC-099 changes the interaction between Tat-exposed microglia and neurons at the ultrastructural level. We performed a stereotactic injection of 3 μl of sterile PBS or 3 μl of 3 μg/μl HIV-1 Tat1–72 700 μm deep into cortex with and without URMC-099 treatment (10 mg/kg twice a day, i.p.) in wild-type 8- to 12-week-old C57BL/6 mice. We killed the animals after 24 h and 7 d and prepared the tissue for analysis by immunoelectron microscopy against Iba-1. A–C, Representative electron microscopic images from mice examined 24 h and 7 d after Tat exposure without URMC-099 treatment showing frequent contacts between Iba-1-stained microglia (m+) and nondegenerating myelinated axons (ma). In A, two myelinated axons are almost completely ensheathed by the microglial process. B, The microglial process contains a phagocytic inclusion that resembles a shrunken myelinated axon in the process of becoming degraded (in). Unidentified cellular elements undergoing extracellular digestion (ex) and a myelinated axon (ma) ensheathed by an astrocytic process (a) are also observed. C, An enlarged microglia that contains a phagocytic inclusion (in), while contacting a myelinated axon, is shown at lower magnification. n = nucleus. D, Representative image from a mouse examined 7 d after Tat exposure with URMC-099 treatment. Two axon terminals and an astrocytic process receive microglial juxtaposition, while a myelinated axon remains uncontacted. Scale bars: A, B, D, 0.2 μm; C, 2 μm. d, Dendrite; s, dendritic spine; t, axon terminal. Based on the immunoelectron microscopy staining, we quantified the frequency of intracellular inclusions in Iba-1-positive processes (E), the frequency of Iba-1 contact with myelinated axons (F), the frequency of Iba-1 contact with dendritic branches (G), and the frequency of Iba-1 contact with astrocytes (H). This quantification was performed for mice receiving a PBS injection at 24 h (n = 2) or 7 d (n = 3), mice receiving a Tat injection at 24 h (n = 3) or 7 d (n = 3), and mice receiving a Tat injection with URMC-099 treatment at 24 h (n = 3) or 7 d (n = 3). **p < 0.01. We analyzed data depicted in E–H by two-way ANOVA, no sample matching with Bonferroni's post hoc test. Error bars indicate SEM. UR, URMC-099.
Discussion
Previous in vitro and in vivo studies with the MLK3 inhibitor CEP-1347 demonstrated the potential of MLK3 inhibition in various models of HAND (Sui et al., 2006; Eggert et al., 2010). Based on these studies, our group developed a novel MLK3 inhibitor, URMC-099, that has a favorable CNS profile with drug-like properties (Gelbard et al., 2010), making it suitable for treating CNS disease. In this study, we show that URMC-099 decreased the production of inflammatory cytokines in cultured microglia exposed to HIV-1 Tat and protected cultured neuronal axons from Tat-activated microglia. We further showed that in vivo treatment with URMC-099 reduced Tat-induced cytokine production in the CNS, protected synaptic architecture from Tat exposure, and altered the morphologic response of microglia to Tat. These results provide the basis for additional research into MLK3 inhibition as an adjuvant therapy for HAND and possibly other neuroinflammatory diseases with similar cytokine and chemokine profiles.
MLK3 inhibition has been studied previously in Parkinson's disease (PD), progressing to a Phase II interventional study (PRECEPT) with the pan-MLK inhibitor CEP-1347, in which the therapeutic endpoint was time to initiate dopaminergic therapy (Parkinson Study Group, 2007). Despite promising preclinical results (Maroney et al., 2001; Saporito et al., 2002; Mathiasen et al., 2004; Lotharius et al., 2005), the human study was ended early because of futility (Parkinson Study Group, 2007), with no difference between the placebo group and the groups that received up to 50 mg CEP-1347 twice a day. As noted by Wang and Johnson (2008) and others, reasons for the failure of the PRECEPT trial may include the following: (1) that MLK inhibitors cannot, alone, retard progression of PD; and (2) that CEP-1347 did not accumulate to therapeutic levels in the CNS. We believe that the second issue is an important concern because CEP-1347 was developed through modification of the naturally occurring indolocarbazole K-252a (Kaneko et al., 1997) and, as a consequence, has a large molecular weight, is polar, and has an excess of hydrogen bond donors and acceptors. These properties likely limit CNS penetrance of the drug (which was not measured in the PRECEPT trial). In contrast, URMC-099 was designed from proprietary libraries based on a pyrrolopyridine scaffold with an aryl piperazine side chain and optimized using structure–activity relationships to have a favorable CNS profile, with CNS levels rapidly reaching and persisting at therapeutic levels for hours after injection (data not shown; Gelbard et al., 2010). Our studies also support the idea that a significant amount of the neuroprotection imparted by MLK3 inhibition results from anti-inflammatory effects in immune cells and microglia. In this regard, the known inflammatory etiology of HAND may make it a significantly better candidate for MLK3-targeted therapies than PD.
The microfluidic chamber coculture model recently developed by our laboratory gives us the unique opportunity to study the effect of neuroinflammation on isolated neuronal components (Marker et al., 2012). This model, which we used to examine the interaction between microglia and primary neuronal axons in culture, is particularly applicable to HAND because there is consistent white matter damage even in cART-treated HAND patients (Harezlak et al., 2011). Our in vivo murine Tat exposure model also replicates several key features seen in HAND, including inflammatory cytokine production, monocyte recruitment, Map2 dystrophic staining, synapse loss, and white matter damage. Treatment with URMC-099 was successful in ameliorating much of the soluble inflammatory response and structural damage to neurons and synapses that occurs after Tat exposure. Among possible mechanisms underlying this neuroprotection, the increased phagocytosis of apparently nondegenerating neuronal components induced by Tat exposure and reversed by URMC-099 treatment suggests that microglia may contribute to eliminating viable neurons and synapses in HAND by “primary” phagocytosis executing cell death (Fricker et al., 2012; Neher et al., 2012). However, there is significant evidence in other models of brain injury for neuroprotective phagocytosis. Known as “secondary” phagocytosis, this mechanism generally involves the phagocytosis of already apoptotic or necrotic neurons and synapses, as well as other proinflammatory debris in the extracellular environment (Neumann et al., 2009; Sierra et al., 2013). The data in this manuscript do not distinguish between potential multiple mechanisms of phagocytosis in response to the presence of Tat in the CNS. Therefore, it is possible that the decrease in phagocytic inclusions seen in URMC-099-treated animals is the result of fewer degenerating neuronal structures, as opposed to the inhibition of primary phagocytosis. Future studies are needed to precisely determine the mechanism of the neuroprotection imparted by URMC-099 treatment in HAND models.
URMC-099 appeared to have no effect on Tat-mediated immune cell recruitment into the CNS. This could be for a number of reasons. First, although URMC-099 decreased the production of inflammatory and recruitment related cytokines in our in vitro and in vivo models, it did not reduce their production to control levels, most notably in the case of MCP-1, suggesting that remaining cytokine production after Tat exposure in vivo could be large enough to drive monocyte recruitment into the CNS (Weiss et al., 1999; McManus et al., 2000). It is also possible that recruitment depends on endothelial activation and adhesion factor expression, and, although there are data to support roles for MLK3 signaling in both of these processes (Muniyappa and Das, 2008; Li et al., 2012), it does not appear to play a major role in our experimental paradigm. Regardless, although leukocytes still enter the brain parenchyma after exposure to Tat, MLK3 inhibition by URMC-099 treatment alters the ability of these immune effector cells to damage synapses and axons, thus contributing to its efficacious neuroprotective profile.
In summary, we provide compelling in vitro and in vivo data to investigate the use of URMC-099 in other models of HAND with the goal of advancement to an adjunctive therapeutic agent.
Footnotes
This work was funded by National Institutes of Health Grants PO1 MH64570 (H.A.G.), RO1 MH56838 (H.A.G.), UL1 RR024160 (H.A.G.), T32 GM07356 (D.F.M.), T32 AI049815 (D.F.M.), F30 MH095664 (D.F.M.), F32 MH099913 (J.M.P.), P51 RR000165 (E.C.M.), the Fonds de Recherche en Santé du Québec (M.-È.T.), the Canadian Institutes of Health Research (M.-È.T.), Merit Award from the Office of Research and Development, Department of Veterans Affairs (E.C.M.), and the Geoffrey Waisdorp Pediatric Neurology Fund (H.A.G.). We sincerely thank Dr. Ania Majewska, whose expertise made the two-photon microscopy experiments possible, Karen Bentley and Gayle Schneider of the University of Rochester Electron Microscopy core facility, and Ann E. Casey and Richard P. Phipps for technical assistance with the Luminex Assay and use of their Luminex machine and Millipore software.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. V.S.G., S.D., and H.A.G. hold patent application WO/2010/068483, “MLK Inhibitors and Methods of Use,” but have no commercial interests.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea S, Del Valle L, Mameli G, Sawaya BE, Khalili K, Peruzzi F. Tubulin-mediated binding of human immunodeficiency virus-1 Tat to the cytoskeleton causes proteasomal-dependent degradation of microtubule-associated protein 2 and neuronal damage. J Neurosci. 2006;26:4054–4062. doi: 10.1523/JNEUROSCI.0603-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Masliah E, Gelbard HA. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest. 2005;115:3185–3192. doi: 10.1172/JCI25444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res. 1992;31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- Bodner A, Maroney AC, Finn JP, Ghadge G, Roos R, Miller RJ. Mixed lineage kinase 3 mediates gp120IIIB-induced neurotoxicity. J Neurochem. 2002;82:1424–1434. doi: 10.1046/j.1471-4159.2002.01088.x. [DOI] [PubMed] [Google Scholar]
- Bodner A, Toth PT, Miller RJ. Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol. 2004;188:246–253. doi: 10.1016/j.expneurol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25:3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter. 2010;14 doi: 10.1002/0471142727.mb1420s92. Unit14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert D, Dash PK, Gorantla S, Dou H, Schifitto G, Maggirwar SB, Dewhurst S, Poluektova L, Gelbard HA, Gendelman HE. Neuroprotective activities of CEP-1347 in models of neuroAIDS. J Immunol. 2010;184:746–756. doi: 10.4049/jimmunol.0902962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Fricker M, Neher JJ, Zhao JW, Théry C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32:2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, Dewhurst S, Goodfellow VS, Wiemann T. MLK inhibitors and methods of use. Geneva: World Intellectual Property Organization; 2010. [Google Scholar]
- Gisslén M, Price RW, Nilsson S. The definition of HIV-associated neurocognitive disorders: are we overestimating the real prevalence? BMC Infect Dis. 2011;11:356. doi: 10.1186/1471-2334-11-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, Alger J, Singer E, Campbell T, Yiannoutsos C, Cohen R, Navia B, HIV Neuroimaging Consortium Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS. 2011;25:625–633. doi: 10.1097/QAD.0b013e3283427da7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group; HNRC Group HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Saito Y, Saito H, Matsumoto T, Matsuda Y, Vaught JL, Dionne CA, Angeles TS, Glicksman MA, Neff NT, Rotella DP, Kauer JC, Mallamo JP, Hudkins RL, Murakata C. Neurotrophic 3,9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J Med Chem. 1997;40:1863–1869. doi: 10.1021/jm970031d. [DOI] [PubMed] [Google Scholar]
- Li CH, Liao PL, Shyu MK, Liu CW, Kao CC, Huang SH, Cheng YW, Kang JJ. Zinc oxide nanoparticles-induced intercellular adhesion molecule 1 expression requires Rac1/Cdc42, mixed lineage kinase 3, and c-Jun N-terminal kinase activation in endothelial cells. Toxicol Sci. 2012;126:162–172. doi: 10.1093/toxsci/kfr331. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P, Leist M. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SM, Tremblay MÈ, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJ, Majewska AK, Dewhurst S, Gelbard HA. HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One. 2011;6:e23915. doi: 10.1371/journal.pone.0023915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska A, Yiu G, Yuste R. A custom-made two-photon microscope and deconvolution system. Pflugers Arch. 2000;441:398–408. doi: 10.1007/s004240000435. [DOI] [PubMed] [Google Scholar]
- Marker DF, Tremblay ME, Lu SM, Majewska AK, Gelbard HA. A thin-skull window technique for chronic two-photon in vivo imaging of murine microglia in models of neuroinflammation. J Vis Exp. 2010;43:2059. doi: 10.3791/2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation. 2012;9:261. doi: 10.1186/1742-2094-9-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, Roos RP, Holskin BP, Wu ZL, Trusko SP, Connors TC, Maroney AC, Thomas BA, Thomas JC, Bozyczko-Coyne D. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW. Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol. 2000;156:1441–1453. doi: 10.1016/S0002-9440(10)65013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Muniyappa H, Das KC. Activation of c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: a MLK-3-MKK7-dependent mechanism. Cell Signal. 2008;20:675–683. doi: 10.1016/j.cellsig.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Brown GC. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front Pharmacol. 2012;3:27. doi: 10.3389/fphar.2012.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster-Granite ML, McPhie DL, Greenan J, Neve RL. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996;16:6732–6741. doi: 10.1523/JNEUROSCI.16-21-06732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group PI. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C. Oligodendrocytes, their progenitors and other neuroglial cells in the aging primate cerebral cortex. Cereb Cortex. 2004;14:995–1007. doi: 10.1093/cercor/bhh060. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster H. The fine structure of the nervous system: the neurons and supporting cells. Philadelphia: Saunders; 1991. [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/S0079-6468(08)70081-X. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Fan S, Sniderhan L, Reisinger E, Litzburg A, Schifitto G, Gelbard HA, Dewhurst S, Maggirwar SB. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006;177:702–711. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- Tremblay MÈ, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010a;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Riad M, Majewska A. Preparation of mouse brain tissue for immunoelectron microscopy. J Vis Exp. 2010b;41:2021. doi: 10.3791/2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MÈ, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Johnson EM., Jr Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2008;71:462. doi: 10.1212/01.wnl.0000324506.93877.5e. author reply 462–463. [DOI] [PubMed] [Google Scholar]
- Wang MJ, Huang HY, Chen WF, Chang HF, Kuo JS. Glycogen synthase kinase-3beta inactivation inhibits tumor necrosis factor-alpha production in microglia by modulating nuclear factor kappaB and MLK3/JNK signaling cascades. J Neuroinflammation. 2010;7:99. doi: 10.1186/1742-2094-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Nath A, Major EO, Berman JW. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–2959. [PubMed] [Google Scholar]
- Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer's disease. Am J Pathol. 2008;173:665–681. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Navia BA, Marra CM, Singer EJ, Chang L, Berger J, Ellis RJ, Kolson DL, Simpson D, Miller EN, Lipton SA, Evans SR, Schifitto G, Adult Aids Clinical Trial Group (ACTG) 301 Team Memantine for AIDS dementia complex: open-label report of ACTG 301. HIV Clin Trials. 2010;11:59–67. doi: 10.1310/hct1101-59. [DOI] [PMC free article] [PubMed] [Google Scholar]