

Abstract
The fragile X mental retardation protein (FMRP) is an RNA-binding protein essential for multiple aspects of neuronal mRNA metabolism. Its absence leads to the fragile X syndrome, the most prevalent genetic form of mental retardation. The anatomical landmark of the disease, also present in the Fmr1 knock-out (KO) mice, is the hyperabundance of immature-looking lengthened dendritic spines. We used the well known continuous production of adult-born granule cells (GCs) in the mouse olfactory bulb (OB) to analyze the consequences of Fmrp loss on the differentiation of GCs. Morphological analysis of GCs in the Fmr1 KO mice showed an increase in spine density without a change in spine length. We developed an RNA interference strategy to cell-autonomously mutate Fmr1 in a wild-type OB network. Mutated GCs displayed an increase in spine density and spine length. Detailed analysis of the spines through immunohistochemistry, electron microscopy, and electrophysiology surprisingly showed that, despite these abnormalities, spines receive normal glutamatergic synapses, and thus that mutated adult-born neurons are synaptically integrated into the OB circuitry. Time-course analysis of the spine defects showed that Fmrp cell-autonomously downregulates the level and rate of spine production and limits their overgrowth. Finally, we report that Fmrp does not regulate dendritogenesis in standard conditions but is necessary for activity-dependent dendritic remodeling. Overall, our study of Fmrp in the context of adult neurogenesis has enabled us to carry out a precise dissection of the role of Fmrp in neuronal differentiation and underscores its pleiotropic involvement in both spinogenesis and dendritogenesis.
Introduction
The fragile X mental retardation protein (FMRP) is the protein whose absence, due to the silencing of the gene FMR1, leads to the fragile X syndrome (FXS) (Verkerk et al., 1991), the most common genetic form of mental retardation (Chelly and Mandel, 2001). FMRP is an mRNA-binding protein (Ashley et al., 1993; Siomi et al., 1993) expressed in neurons and localized in dendrites and spines in particular (Devys et al., 1993; Verheij et al., 1993; Feng et al., 1997). It has polyvalent neuronal functions, ranging from mRNA stability and transport to regulation of subsynaptic mRNA translation (Bassell and Warren, 2008; Melko and Bardoni, 2010). It may bind as many as 4% of fetal brain mRNAs (Ashley et al., 1993), and its most extensively studied targets comprise mRNAs involved in synaptic plasticity [e.g., CaMKIIα, ARC (activity-regulated cytoskeleton-associated protein)] or in dendritic and spine architecture (e.g., MAP1b, PSD95).
The first reported neuropathologic finding from FXS patients was the hyperabundance of dendritic spines in cortical neurons (Rudelli et al., 1985; Hinton et al., 1991; Wisniewski et al., 1991; Irwin et al., 2001). Because spines are the postsynaptic element of excitatory synapses, this suggested an enhanced synaptic excitation of FXS neurons, which could contribute to the increased incidence of epilepsy in FXS patients. However, these supernumerary spines were unusually long and thin, a morphology reminiscent of filopodia and associated with synaptic immaturity. The status of synaptic maturity of the mutated spines thus remained unclear (Pfeiffer and Huber, 2009).
Fmr1 knock-out (KO) mice have been extensively studied as a model of FXS and exhibit a spine phenotype similar to FXS patients: mature neocortical neurons display an increased density of longer spines (Comery et al., 1997; Galvez and Greenough, 2005; McKinney et al., 2005). This has been interpreted mainly as a defect in spine maturation and pruning. Indeed, a role for Fmrp in activity-dependent synapse elimination has been substantiated recently (Pfeiffer et al., 2010), as well as a role in spine stabilization (Chen et al., 2010; Cruz-Martín et al., 2010; Pan et al., 2010). However, this does not exclude a role for FMRP in early processes of spine formation. Accordingly, it has been reported that spine alterations are present in very young KO mice, although they disappear in the adolescent mice (Nimchinsky et al., 2001) and reappear in the adult (Galvez and Greenough, 2005).
Dendritic arborization defects have also been occasionally observed in the KO mice (Galvez et al., 2003; Galvez et al., 2005). However, much of the evidence for a role of Fmrp in neurite development stems from work in drosophila where dFmrp-null flies exhibit an overgrowth of axons and dendrites (Lee et al., 2003; Pan et al., 2004; Gatto and Broadie, 2008; Tessier and Broadie, 2008).
Most of the work on mammalian Fmrp has thus been performed in the postnatal or adult brain of Fmr1 KO mice, where it is impossible to discriminate Fmrp cell-autonomous functions. Moreover, its role in the process of neuronal differentiation, in terms of spine production and morphogenesis, remains controversial (Nimchinsky et al., 2001; Galvez et al., 2003; Cruz-Martín et al., 2010; Pan et al., 2010), and little is known about its role in dendritic elaboration. In the adult olfactory bulb (OB), newly generated neurons exhibit stereotyped morphological and electrophysiological hallmarks of differentiation and incorporation into synaptic circuits (Petreanu and Alvarez-Buylla, 2002; Carleton et al., 2003). Thus, to determine the role of Fmrp in neuron differentiation, we designed a strategy to knock down Fmr1 in the newly generated neurons of the adult OB. Here, we show that (1) Fmrp cell-autonomously regulates spine density and morphology, (2) the morphological changes seen in spines do not lead to alteration of synaptic inputs received by mutated adult-born neurons, (3) Fmrp cell-autonomously downregulates the level and rate of early spine production and limits their overgrowth, and (4) Fmrp is necessary for activity-dependent remodeling of dendritic architecture.
Materials and Methods
Animals.
Two-month old male mice of Swiss or C57BL/6J background (CERJanvier) were used for injections of lentiviruses. The Fmr1 knock-out mice and conditional KO were provided by Dr. B. Oostra (Mientjes et al., 2006).
Experiments involving animals were approved by the Regional Ethic Committee for Animal Handling (Comité Régional d'Ethique pour l'Expérimentation Animale Île-de-France).
shRNA plasmid contructions.
Potential siRNA specific to mouse Fmr1 mRNA (shFMR1) were designed on the http://i.cs.hku.hk/∼sirna/description.html website (shFMR1 target sequence: TGAGAAAAGTGTCCCACAAGA). Control siRNA sequence (shNeg) was generated using the standard control siRNA sequence from Qiagen called AllStars Negative Control, which does not recognize mammalian RNAs. Twenty-one-nucleotides sequence in both sense and antisense orientations separated by a nine-nucleotides spacer (5′-TTCAAGAGA-3′) followed by five thymidines as an RNA polymerase III transcription termination signal were synthesized chemically as two complementary DNA oligonucleotides (Eurofin MWG Operon). BbsI and BamHI restriction sites were incorporated at the 5′ and 3′ ends respectively. These DNA templates were annealed and inserted between the BbsI/BamHI sites of the U6-shRNA vector (kindly provided by Dr. R. Vogel, Centre National de la Recherche Scientifique (CNRS) UMR 7091, Paris, France) downstream from the U6 promoter. The complete shRNA expression cassettes (the U6 promoter plus the siRNA template) were excised from the corresponding recombinant U6-shRNA vector by MluI and BamHI digestion. These cassettes were transferred into the modified plasmid pTRIP-siRNA-PGK-GFP-WPRE vector (gift from Dr. R. Vogel) by substitution of a MluI/BamHI fragment. This vector also expresses the green fluorescent protein (GFP) as a reporter gene under the control of the PGK promoter. Plasmids were purified by using the EndoFree Plasmid Maxikit (Qiagen), and all constructs were verified by sequencing before use.
Lentiviruses.
We used the shRNA-expressing lentiviruses described above, lentiviruses expressing the GFP alone driven by the CMV or PGK promoters, or the Cre recombinase and the GFP (Addgene, plasmid 20781, GFP expression driven by UbC (human ubiquitin C) promoter; Cre expression driven by PGK promoter). They were produced by the École des neurosciences de Paris-Île-de-France (Paris, France) viral production facility.
Lentiviral vector production.
The cell line used for lentiviral vector production was 293T, derived from embryonic human kidney, cultured at 37°C in a humidified chamber containing 5% CO2, and maintained in DMEM. Lentivirus vector particles were produced by calcium phosphate transient cotransfection of 293T cells by the vector plasmid, an encapsidation plasmid (p8.7), and a pVSV (vesicular stomatitis virus expression plasmid) as described previously (Zennou et al., 2001). The supernatants were treated with DNase I (Roche Diagnostic) and subjected to ultracentrifugation. The resulting pellet was resuspended in PBS, aliquoted, and frozen at −80°C until use. The human immunodeficiency virus (HIV) p24 Gag antigen was quantified for each stock with an ELISA kit (Beckman Coulter). In addition, GFP-expressing vectors were titered by infecting cells with serial dilutions of each lentiviral stock. The percentage of transduced cells, identified by GFP fluorescence, was estimated by flow cytometry 72 h after transfection and used to determine the number of transducing units (TU) per unit volume. The titers of the viral preparations used in the experiments described were 1.14 × 1010 TU/ml for shNeg and 3.6 × 1010 TU/ml for shFMR1.
Experimental treatments
The subventricular zone (SVZ) of the experimental mice was stereotaxically injected with lentiviruses (500nl/injection site) using a Kopf stereotaxic apparatus (Harvard Apparatus). The coordinates were as follows: anteroposterior (AP) +0.5 mm from bregma, mediolateral (ML) ±1.1 mm from bregma, dorsoventral (DV) −1.7 mm from pial surface and AP +1 mm from bregma, ML ±1 mm from bregma, and DV −2.3 mm from pial surface. Mice were then killed at 14, 21, and 28 d after injection (dpi). In odor-enriched mice, a group of animals was daily exposed, from the first day of the surgery until the day of killing, to different natural fragrances that were placed in a tea ball hanging from the top of the breeding cages, as described by Rochefort et al. (2002). For olfactory deprivation, unilateral naris occlusion was achieved by inserting nose plugs in the right naris as described by Mandairon et al. (2006). Odor-enriched and odor-deprived mice were killed at 21 dpi.
Immunofluorescence.
Anesthetized mice were fixed by intracardiac perfusion of 4% paraformaldehyde in PBS. Tissues were dissected and postfixed during 24 h with the same fixative. Coronal olfactory bulb sections were prepared (50 μm thick) on a vibratome (Leica). Sections were saturated in blocking solution (PBS 0.3% Triton X-100, 10% normal goat serum) and incubated for 48–72 h at 4°C with the following primary antibodies: a rabbit anti-Fmrp (1:1000, rAM1, provided by C. Bagni, University of Rome, Rome, Italy), or a mouse anti-Fmrp (1:200, 7G1–1, gift from J. Darnell, Rockefeller University, New York, NY), or a mouse anti-PSD95 (1:1000, ThermoFischer). The mouse anti-Fmrp was used in initial experiments (see Fig. 1C) to double-check the specificity of the rabbit anti-Fmrp staining. When using the mouse anti-Fmrp, an antigen-retrieval step was performed before the saturation step, which consisted of incubation in 0.01 m sodium citrate, pH 6.0, at 95°C for 15 min. The secondary antibodies used were Cy3-conjugated donkey anti-rabbit or anti-mouse (1:2000, Jackson ImmunoResearch) and Cy2-conjugated donkey anti-goat (1:1000, Jackson ImmunoResearch). Sections were then mounted in fluoromount G (SouthernBiotech).
Figure 1.

Adult-born granule cells of the olfactory bulb express Fmrp. A, Scheme of a sagittal section of the mouse forebrain. The SVZ of the lateral ventricle (LV) continuously produces new neurons, which migrate along the RMS and differentiate as interneurons in the OB. Subpopulations of young neurons can be genetically manipulated through stereotaxic injections of viruses into the SVZ. B, Example of newly generated GCs in the OB, 21 dpi of a GFP-expressing lentivirus into the SVZ. Immunostaining for Fmrp (red) shows its ubiquitous expression in the granular cell layer (GCL), mitral cell layer (MCL), and dendrites of the EPL. The GFP-labeled newborn GC displays the characteristic morphology of these neurons, with a cell body in the GCL, basal dendrites, and a long apical primary dendrite, which arborizes in the EPL. The apical dendritic tree has numerous spines (arrowheads), which form dendrodendritic reciprocal synapses with mitral cells. Scale bar, 25 μm. C, A newly born 21 dpi GFP-positive GC is Fmrp immunoreactive; Fmrp is localized both in its cell body and apical dendrite (arrowheads). Scale bar, 10 μm.
Electron microscopy.
Anesthetized mice were fixed by intracardiac perfusion of 4% paraformaldehyde and 0.25% of glutaraldehyde. After cryoprotection in 25% saccharose, coronal sections of the OB were freeze/thawed three times in dry-ice cooled isopentane. They were then incubated with a rabbit anti-GFP antibody (1:1000, Millipore) and processed for electron microscopy immunogold labeling as described by Trembleau et al. (1994). Following routine processing as previously described (Treloar et al., 2002), thin sections were examined on a JEOL 1200 Electron Microscope. Images were captured at 10,000–20,000× and digitized at 1200 dpi for analysis.
Electrophysiology.
C57BL/6J males were injected with 200 nl of shNeg or ShFMR1 lentiviruses in the rostral migratory stream (RMS) at the following coordinates in each hemisphere: AP +3.3 mm from bregma, ML ±0.82 mm from bregma, and DV −2.9 mm from pial surface. They were killed 14 d later, which is the approximate equivalent of 21 dpi for injections into the SVZ.
For patch-clamp recordings, mice were deeply anesthetized with isoflurane (Mundipharma) and swiftly decapitated. The OB and frontal cortices were rapidly dissected and placed in ice-cold artificial CSF (ACSF; containing in mm: 124 NaCl, 3 KCl, 1.3 MgSO4, 26 NaHCO3, 1.25 NaHPO4, 20 glucose, 2 CaCl; ∼310 mOsm, pH 7.3 when bubbled with a mixture of 95% O2 and 5% CO2; all chemicals from Sigma), before being glued to a block of 4% agarose and placed, submerged in ice-cold ACSF, in the cutting chamber of a vibratome (Leica). Horizontal slices (300 μm thick) of the OB were placed in bubbled ACSF in a warming bath at 35°C for 30 min and then at room temperature (i.e., 22 ± 1°C). For whole-cell recordings, individual slices were placed in a chamber mounted on a Zeiss Axioskop upright microscope, and continuously perfused (1.5 ml/min) with room-temperature bubbled ACSF. Slices were visualized using a 40× water-immersion objective, a halogen light source, differential interference contrast filters (Zeiss), and a charge-coupled device camera (Hamamatsu C7500).
We obtained whole-cell patch-clamp recordings from visually targeted granule cells (GCs). Patch pipettes, pulled from borosilicate glass [outer diameter (OD) 1.5 mm, inner diameter (ID) 1.17 mm, Harvard Apparatus; P-87 Flaming/Brown micropipette puller, Sutter Instrument), had resistances of 6–10 MΩ and were filled with a cesium gluconate-based solution (in mm: 126 Cs-gluconate, 6 CsCl, 2 NaCl, 10 Na-HEPES, 10 d-glucose, 0.2 Cs-EGTA, 0.3 GTP, 2 Mg-ATP, 0.2 cAMP, with 0.15% biocytin; 280–290 mOsm, pH 7.3). Rhodamine (0.1–0.4%) was included in the internal solution for GFP+ cell recordings. All voltages reported in the text are corrected for a liquid junction potential of +10 mV between our internal and external solutions.
Recordings were obtained via an Axopatch 1D amplifier. Signals were Bessel filtered at 2 kHz, digitized, and sampled at intervals of 20–450 μs (2.2–50 kHz) according to the individual protocols. Compensation for fast capacitance in cell-attached mode was ∼80% (12–13pF, 3–5 μs); we also compensated for membrane capacitance (Cm), but not series resistance (Rs), after rupture. With Cm compensation inactivated, values of Rs and membrane resistance (Rm) were estimated using peak and steady-state currents, respectively, observed in response to a 5 mV membrane step. Any experiments in which Rs changed by >20% were discarded from our analyses.
Adult-born cells were identified by either double-labeling with both GFP and rhodamine during recording, or the presence of GFP in the tip of the patch pipette after membrane rupture. GFP-targeted recordings that did not meet either of these criteria were discarded from our analyses. Synaptic responses were evoked by using a small monopolar stimulating glass pipette (3–4 μm tip diameter, pulled from 1.5 mm OD, 1.17 mm ID borosilicate glass, and filled with ACSF), finely positioned in the external plexiform layer (EPL) using a micromanipulator. Synaptic currents were analyzed with Elphy software.
Image analysis and quantification.
Images were acquired on a Leica SP5 confocal system. z-Series stacks of confocal images were acquired at 1024 × 1024 pixel resolution, with a pinhole setting of one Airy unit and optimal settings for gain and offset. For double immunolabeling quantifications, images were taken with a 63×/1.4 numerical aperture (N.A.) Plan-Apochromat, an argon laser at an excitation wavelength of 488 nm, and a diode 561 nm. The percentage of GFP-positive GCs that expressed FMRP was obtained by analyzing the colocalization in all the optical plans containing the cell body and with the help of the x–z and y–z orthogonal projections. Lateral and z-axis resolutions were 240 and 500 nm, respectively. For analysis of spine maturity, GFP-positive spines that expressed PSD95 were analyzed as previously described, but lateral and z-axis resolutions were 80 and 250 nm, respectively. Pictures of apical dendrites and their arborizations were taken with a 40×/1.25 N.A Plan-Apochromat. Lateral and z-axis resolutions were 380 nm and 1 μm, respectively. Dendritic spine images were taken with a 63× objective, with lateral and z-axis resolutions of 80–120 and 155 nm, respectively.
Images were deconvoluted using a maximum likelihood estimation method after calculation of the theoretical point spread function (Huygens, Scientific Volume Imaging). Dendritic processes and dendritic spines were then analyzed using NeuronStudio software (Rodriguez et al., 2008). This software allows for automated detection of three-dimensional (3D) neuronal morphology (dendrites and spines) from confocal z-series stacks on a spatial scale. Because spine lengths were challenging for the automated detection in our model, spine identities were manually verified with the help of the 3D reconstruction. Moreover, spine neck lengths were also manually measured in the 3D using the measurement tool.
Statistical analysis.
Morphological data (spine density, spine length, and dendritic length) were compared between ShNeg/shFMR1, wild type (WT)/KO, and control/Cre animals using an unpaired-Student's t test. Validation of the shFMR1 lentiviruses was conducted using an unpaired-Student's t test. Spine distributions of shNeg/ShFMR1 neurons were compared using a χ2 test. Time-course analysis of shNeg and shFMR1 groups was assessed by a one-way (age) ANOVA, followed by Scheffé's post hoc test analysis. Electrophysiological data were compared between shNeg and shFMR1 groups using the nonparametric Mann–Whitney U test.
Results
Fmrp is expressed in newborn granule cells of the adult olfactory bulb
GCs of the OB are constantly renewed throughout adulthood. They are produced in the SVZ of the lateral ventricle, 2 mm away from their site of integration. This segregation allows for the genetic manipulation of progenitors and/or newly produced young neurons by viral injection into the SVZ and later on for following their differentiation in the OB circuits (Fig. 1A). GCs are GABAergic interneurons, which have no axons but display a long apical dendrite (Fig. 1B). This dendrite arborizes in the external EPL and displays numerous spines, which form dendrodendritic synapses with mitral cells, the OB principal neurons (Lledo and Saghatelyan, 2005). Immunocytochemical staining shows that Fmrp is expressed in the OB (Fig. 1B) and in particular in GCs, including newly produced GCs, as labeled by a GFP-expressing lentivirus injection into the SVZ (Fig. 1B,C). Fmrp is present in the cell bodies and dendrites of GFP-expressing GCs (Fig. 1C).
Newborn GCs in Fmr1 KO mice display increased spine density
To determine whether FMRP plays a role in the differentiation of these adult-born cells, we first injected a GFP-expressing lentivirus into the SVZ of Fmr1 KO mice. At 21 dpi, a stage when newborn GCs display intense spinogenesis, we measured the density and length of the spines seen in the EPL (Fig. 2A–D). We categorize as “spines” all protrusions with or without a clearly visible head, thus including filopodia. The spine density was 26% greater in KO mice than in WT (Fig. 2E) (unpaired Student's t test, p < 0.05, n = 10 dendrites). This result is similar to that described for neurons in other regions of the Fmr1 KO mouse (Comery et al., 1997; Galvez and Greenough, 2005; McKinney et al., 2005). By contrast, the spine length of new GCs was unchanged (n = 302 and 368 spines for WT and KO mice respectively, counted from 10 dendritic segments from different neurons) (Fig. 2F). Interestingly, the percentage of filopodia, defined as a protrusion without a visible head, was equally low in both conditions (<2% in both genotypes, data not shown). We also measured the length of both primary dendrite and dendritic tree of newborn GCs and found no difference between WT and KO mice (Fig. 2G,H) (n = 10 neurons).
Figure 2.
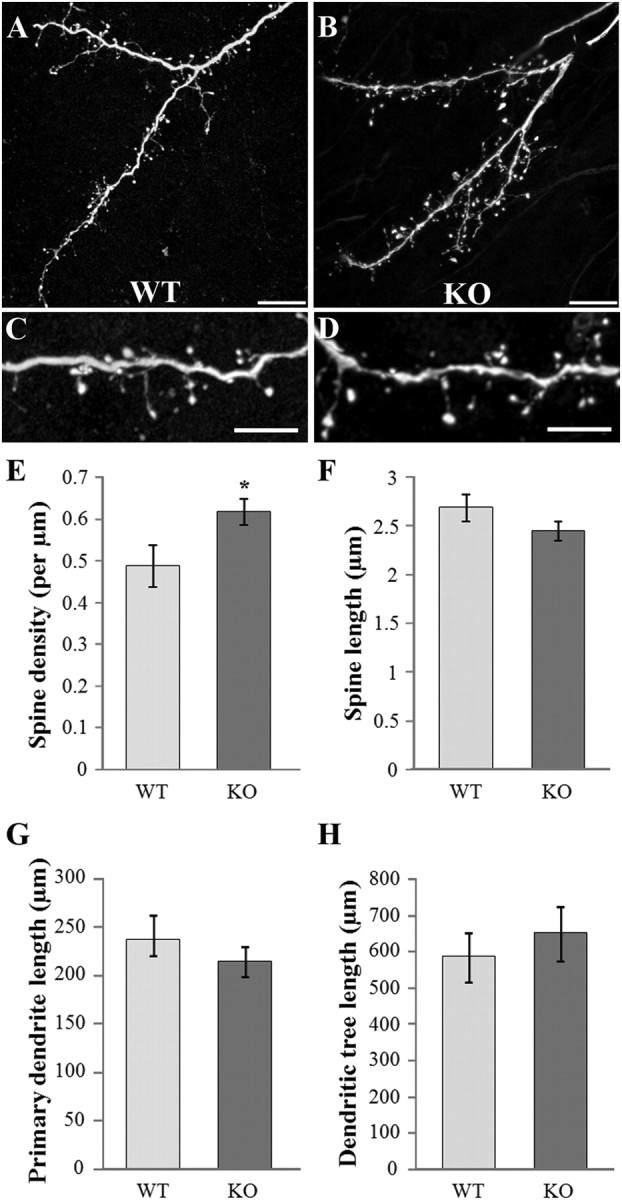
Morphology of newborn GCs in FMR1 KO mice. The SVZ of WT or Fmr1 KO mice was injected with a GFP-expressing virus, and the morphology of GFP-positive GCs was analyzed in the OB, 21 d later. A, B, Confocal acquisition of dendritic trees of newborn GCs in the EPL of WT (A) and KO (B) mice. Scale bars, 10 μm. C, D, Higher magnifications showing WT (C) and KO (D) dendritic segments with numerous spines. Scale bars, 5 μm. E, Mean spine density of newborn GCs in WT and KO mice. The spine density is significantly higher in KO GCs (t test, *p < 0.05, n = 10 dendrites). F, Mean spine length of newborn GCs in WT and KO mice (n = 302 and 368 spines for WT and KO mice respectively, spines were counted from 10 dendritic segments from different neurons). G, Mean primary dendrite length of newborn GCs in WT and KO mice (n = 10 neurons). H, Mean dendritic tree length of newborn GCs in WT and KO mice (n = 10 neurons). Errors bars, SEM.
Fmr1-mutated GCs differentiating in a wild-type network display an increased density of lengthened spines
The stability of GC spine morphology was unexpected and we reasoned that the phenotype should be more pronounced upon integration of mutated neurons in a wild-type network. We thus cell-autonomously mutated Fmr1 by RNA interference. We produced lentiviruses expressing a shRNA directed against Fmr1 (shFMR1) or a control shRNA (shNeg) as well as the GFP, and injected them into the SVZ. In vitro and in vivo characterization of the shRNA-expressing lentiviruses demonstrated their efficiency in downregulating Fmr1 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Analysis of 21 dpi GCs showed that, indeed, the cell-autonomous mutation triggered stronger morphological changes than the ones observed in KO mice (Fig. 3A,B). Similar to Fmr1 KO mice, the spine density was increased by 29% in shFMR1 neurons, as compared with shNeg neurons (Fig. 3C) (unpaired Student's t test, p < 0.05, n = 10 dendrites). However, in sharp contrast to the observations made in KO mice, the mean spine length was also increased by 53% in shFMR1 neurons (Fig. 3D) (unpaired Student's t test, p < 0.05, n = 335 and 402 spines for shNeg and ShFMR1 neurons respectively, counted from 10 dendritic segments from different neurons). Moreover, the distribution of spine lengths differed significantly between shNeg and shFMR1 neurons (Fig. 3E) (χ2 test, p < 0.0001), with shFMR1 neurons having fewer short spines (<2 μm) and more long spines. The shFMR1 spines occasionally reached extreme lengths ranging from 6 to 12 μm, lengths that were rarely encountered in shNeg neurons (shNeg: 2.18 ± 1% of 270 spines; shFMR1: 13.75 ± 2.01% of 297 spines, p < 0.0001). Notably, there was no difference in the percentage of filopodia between shNeg and shFMR1 conditions (shNeg: 10.57 ± 1.8%; shFMR1: 10.9 ± 1.27%, n = 270 and 297 protrusions in shNeg and shFMR1 conditions, respectively). We verified the specificity of the shRNA-induced phenotype by mutating Fmr1 through an independent approach in which we injected a Cre-expressing virus into the SVZ of a floxed Fmr1 mouse (Mientjes et al., 2006). The density and length of spines from recombined newborn GCs were indeed increased similar to that seen with the shRNA approach (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 3.
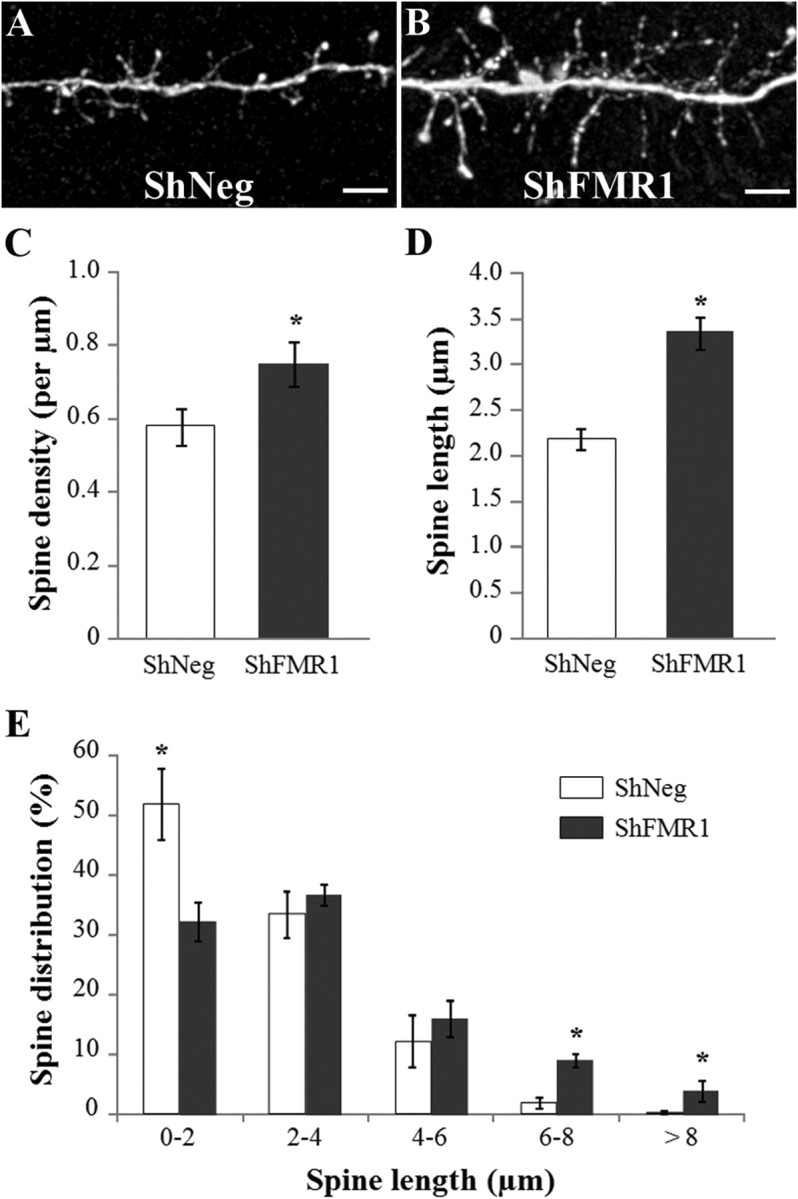
Spine morphology of Fmr1 mutated newborn GCs differentiating in a wild-type network. The SVZ of mice was injected with a lentivirus-expressing GFP and an shRNA directed against Fmr1 (shFMR1) or a control shRNA (shNeg). The spine density and morphology of GFP-positive GCs were analyzed in the EPL, 21 d later. A, B, Confocal images showing EPL dendritic segments of shNeg (A) and shFMR1 (B) newborn GCs. Scale bars, 5 μm. C, Mean spine density of shNeg and shFMR1 newborn GCs. The spine density is significantly higher in shFMR1 neurons (t test, *p < 0.05, n = 10 dendrites). D, Mean spine length of shNeg and shFMR1 newborn GCs. The spine length is significantly higher in shFMR1 neurons (t test, *p < 0.05, n = 335 and 402 spines for shNeg and ShFMR1 neurons respectively, spines were counted from 10 dendritic segments from different neurons). E, Distribution of spine lengths in shNeg and shFMR1 newborn GCs. The distribution of spine lengths is significantly different between both genotypes (χ2 test, *p < 0.0001). Errors bars, SEM.
The supernumerary spines in mutated GCs receive functional glutamatergic inputs
Long and thin spines are often considered to be immature (Irwin et al., 2000). We thus assessed the synaptic maturity of the mutated spines using immunohistochemistry for PSD95, a marker of the postsynaptic density, and indicative of a functional glutamatergic synapse (Fig. 4A–C). Interestingly, we found a similar proportion of PSD95-immunoreactive GFP spines in both genotypes (shNeg: 54.75 ± 3.25%; shFMR1: 51.5 ± 0.5%; n = 200 spines from 2 animals for both genotypes) (Fig. 4D). This suggested that the mutated spines displayed a correct functional maturation despite their abnormal morphology. To verify this, we performed an immunoelectron microscopy analysis using immunogold preembedding labeling of the GFP in shFMR1 neurons. We clearly saw that labeled GC spines in the EPL formed reciprocal dendrodendritic synapses with mitral cells. The morphological characteristics of these synapses, including the membrane specializations and polarized distributions of vesicles, were consistent with those previously described for adult-born GCs (Whitman and Greer, 2007) (Fig. 4E; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This confirmed that the loss of Fmrp did not prevent synapse formation.
Figure 4.

Synaptic maturity of mutated spines using morphological criteria. The SVZ of mice was injected with shFMR1 or shNeg lentiviruses, and the synaptic maturity of GFP-positive spines in the EPL was analyzed 21 d later. A–C, Confocal microscope images of a GFP-positive (A) shFMR1 spine (arrows) immunolabeled with PSD95 (B). Scale bar, 2 μm. D, Mean (+/− SEM) percentage of GFP-positive spines colabeled by PSD95 in shNeg or shFMR1 conditions. E, Electron micrograph showing a dendritic spine, immunostained for GFP with gold particles, from an shFMR1-infected granule cell. It establishes reciprocal dendrodendritic synapses with mitral cell secondary dendrites in the external plexiform layer. The polarity of the asymmetrical synapse from the mitral cell dendrite (MCd) is indicated with a black arrow and that of the reciprocal symmetrical synapse from the labeled spine (sh+) is indicated with a white arrow. Note the specificity of the gold label within the granule cell spine. A clathrin-coated vesicle is seen to the lower left of the white arrow in the labeled spine, consistent with the functional release/turnover of vesicles in the spines of mutated granule cells. The MCd also makes an asymmetrical synapse (black arrow) with a granule cell dendritic spine that is unlabeled (sh−).
To further test the hypothesis that the shFMR1 GC spines could establish functional connections, we performed an electrophysiological analysis of mutated neurons, using whole-cell patch-clamp recordings of adult-born GCs in acute OB slices. We first compared the membrane properties of shNeg and shFMR1 cells. Input membrane resistance and amplitude of voltage-dependent Na+ channels were not different between both genotypes (supplemental Table 1, available at www.jneurosci.org as supplemental material). However, membrane capacitances were significantly higher in shFMR1 cells, consistent with the increase of spine density and length. We then examined the synaptic integration of the cells by recording their synaptic glutamatergic inputs. Spontaneous EPSCs (sEPSCs) (Fig. 5A) and synaptic responses evoked by electrical stimulation delivered in the EPL (Fig. 5B) were analyzed in new GCs. Amplitude of spontaneous or evoked EPSCs were not significantly different between shNeg and shFMR1 cells; however, the frequency of sEPSCs was dramatically increased in shFMR1 neurons (shNeg: 0.87 ± 0.32 Hz, n = 10; shFMR1: 2.38 ± 0.46 Hz, n = 12 neurons; p < 0.05 with Mann–Whitney) (Fig. 5A,C). Analysis of the paired-pulse ratios (PPR) of evoked EPSCs suggested no difference in the probability of glutamate release (shNeg: PPR = 1.36 ± 0.13, n = 7; shFMR1: PPR = 1.24 ± 0.12, n = 8 neurons) (Fig. 5B,C). These results strongly suggest an increase in the number of functional glutamatergic input synapses received by shFMR1 GCs. Collectively, these morphological and functional data show that the supernumerary spines produced by mutated neurons at 21 dpi receive functional glutamatergic inputs, despite their abnormal morphology.
Figure 5.

Synaptic maturity of mutated spines using electrophysiological criteria. The RMS of mice was injected with shFMR1 or shNeg lentiviruses, and electrophysiological analysis was performed 14 d later. A, Representative traces of sEPSCs recorded at −70 mV (calibration: 20 ms/10 pA). B, Average traces of evoked EPSCs. A glass electrode was positioned in the EPL and two electrical stimuli were given with an interval of 50 ms (calibration: 10 ms/10 pA). C, Histograms representing the amplitudes of sEPSCs (shNeg: 11.61 ± 2.03 pA, n = 10; shFMR1: 13.92 ± 2.67 pA, n = 11; p = 0.42 with Mann–Whitney), frequencies of sEPSCs (shNeg: 0.87 ± 0.32 Hz, n = 10; shFMR1: 2.38 ± 0.46 Hz, n = 12; *p = 0.016 with Mann–Whitney), and evoked paired-pulse ratio of EPSCs (PPR = amplitude S1/amplitude S2; shNeg: PPR = 1.36 ± 0.13, n = 7; shFMR1: PPR = 1.24 ± 0.12, n = 8; p = 0.613 with Mann–Whitney). Errors bars, SEM.
To further challenge the functional integration of mutated neurons in a wild-type network, we exposed shRNA-injected animals to an enriched olfactory environment consisting of a new odor every day (Rochefort et al., 2002), thus exposing mutated neurons to a more active network during their integration. Morphological analysis of 21 dpi neurons showed that, whereas the increased olfactory activity had no effect on shNeg spine length, it triggered a 22% lengthening of shFMR1 spines (standard conditions: 3.35 ± 0.17 μm, enriched conditions: 4.08 ± 0.23 μm, unpaired Student's t test, p < 0.05, n > 300 spines for both genotypes, counted from 10 dendritic segments from different neurons). Because mutated spines respond to a change in synaptic activity, this suggested again a functional integration of mutated neurons, accompanied by an increased sensitivity to activity changes as compared with control neurons.
Fmrp regulates the rate and level of spine production and their morphogenesis
We next addressed the time course for the appearance and evolution of the morphological defects of mutated neurons. To be certain that the mutation did not trigger a delay in the initial migration or maturation of GCs, we verified the stages of differentiation of 8 dpi neurons and determined that there was no difference between shNeg and shFMR1 neurons (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We thus analyzed morphological defects at 14 dpi when GCs start to elaborate spines, at 21 dpi as above, and at 28 dpi, when GCs are fully integrated in the network (Fig. 6A–F).
Figure 6.

Time-course analysis of the spine defects. The SVZ of mice was injected with shFMR1 or shNeg lentiviruses. The density and morphology of GFP-positive spines in the EPL were analyzed 14, 21, or 28 d later. A–F, Representative confocal images of dendritic segments from shNeg (A–C) or shFMR1 (D–F) GCs at 14 dpi (A, D), 21 dpi (B, E), and 28 dpi (C, F). Scale bar, 5 μm. G, Three-dimensional reconstruction of a confocal stack by the software NeuronStudio, showing an extremely abnormal 28 dpi-mutated spine with one neck and six heads. H, Mean spine density of shNeg and shFMR1 newborn GCs at 14, 21, and 28 dpi. (n = 10 dendrites, *p < 0.05, ANOVA followed by Scheffé's post hoc test). I, Mean spine length of shNeg and shFMR1 newborn GCs at 14, 21, and 28 dpi (n > 300 spines per time point, *p < 0.0001, ANOVA followed by Scheffé's post hoc test, spines were counted from 10 dendritic segments from different neurons). Errors bars, SEM.
As shown in Figure 6H, shNeg neurons displayed a progressive increase in spine density between 14 and 28 dpi (n = 10 dendrites, p < 0.05, ANOVA followed by Scheffé's post hoc test) (Fig. 6A–C,H). By contrast, as early as 14 dpi, shFMR1 spine density was already maximal, and subsequently did not change (Fig. 6D–F,H). This suggests that the higher spine densities seen in shFMR1 neurons might result from an accelerated rate of spine production.
The length of shNeg spines remained constant between 14 and 28 dpi (n > 300 spines per time point, one-way ANOVA followed by Scheffé's test) (Fig. 6I). shFMR1 spines were longer than shNeg spines at each time point analyzed (unpaired Student's t test, p < 0.05, n > 300 spines for both genotypes at each time point, counted from 10 dendritic segments from different neurons) (Fig. 6I). Their length remained constant overtime and were at maximum level as soon as 14 dpi. This suggested a dysregulated spine morphogenesis in mutated neurons, with a role for Fmrp in limiting spine overgrowth.
Fmrp is necessary for experience-dependent dendritic remodeling
Ultimately, we investigated whether Fmrp had cell-autonomous functions in GCs dendritogenesis. We thus measured the length of both primary dendrite and dendritic tree of newborn shNeg and shFMR1 GCs at 21 dpi (Fig. 7A,B,E,F), and observed no difference between both genotypes. Furthermore, Fmrp has been proposed to play a role in morphological changes during activity modification (Irwin et al., 2005; Tessier and Broadie, 2008). Also, the length and complexity of the dendritic arbor of adult-born GCs have been reported to be sensitive to olfactory deprivation (Saghatelyan et al., 2005). We thus performed unilateral naris closure from the time of lentivirus injection to 21 dpi to investigate whether Fmrp was necessary for the expression of this dendritic plasticity. We observed that a sensory deprivation triggered a 49% reduction of the dendritic tree length in shNeg neurons (standard conditions: 587.93 ± 138.21 μm, deprived conditions: 298.8 ± 72.6 μm, n = 10 neurons, unpaired Student's t test, p < 0.05) (Fig. 7C--F). In contrast, shFMR1 neurons showed no morphological plasticity upon deprivation (standard conditions: 502.1 ± 62.2 μm, deprived conditions: 621.2 ± 52.6 μm, n = 10 neurons). This reveals an unexpected function of FMRP in detecting the overall activity of the host circuit to adjust the total dendritic length of newborn GCs.
Figure 7.
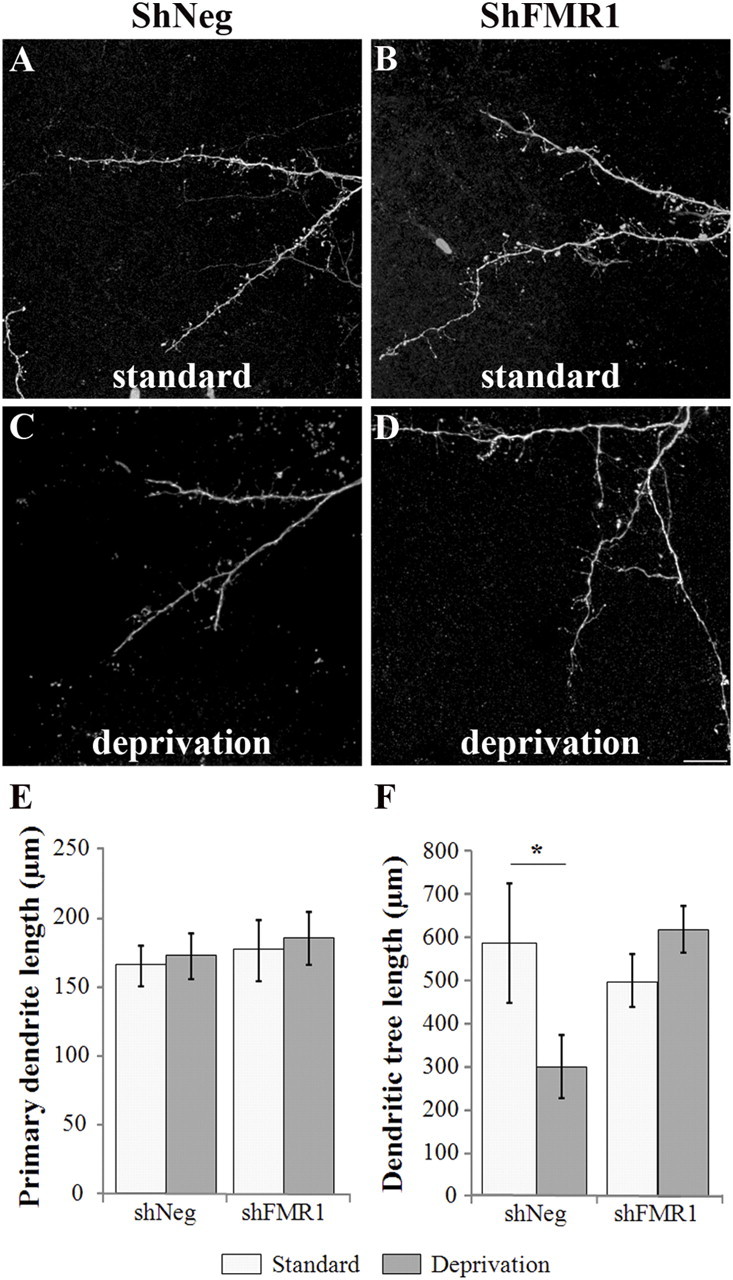
Cell-autonomous effects induced by Fmrp loss on the dendritogenesis of GCs. The SVZ of mice was injected with shFMR1 or shNeg lentiviruses. The mice were subsequently raised in a standard or deprived olfactory environment. Dendritic morphology was analyzed at 21 dpi. A–D, Representative confocal images of the dendritic trees of shNeg (A, C) or shFMR1 (B, D) GCs of mice raised in a standard (A, B) or deprived (C, D) olfactory environment. Scale bar, 10 μm. E, Mean primary dendrite length of shFMR1 or shNeg neurons (n = 10 neurons). F, Mean dendritic tree length of shFMR1 or shNeg neurons (*p < 0.05, t test, n = 10 neurons). Errors bars, SEM.
Discussion
Here, we have developed, through analysis of the differentiation of adult-born GCs, an original system to investigate Fmrp cell-autonomous function in neuronal differentiation. We show that spine abnormalities induced by Fmrp loss are more pronounced in a WT network than in KO mice, but that spine defects do not prevent them from receiving normal glutamatergic synaptic inputs. The time-course analysis of the differentiation of GCs shows that Fmrp regulates the rate and level of early spine production as well as morphogenesis. Finally, we report a new role for Fmrp as a key regulator of experience-dependent dendritic remodeling.
Methodological considerations
GCs maturation and synaptic integration have been well characterized using morphological and electrophysiological methods (Petreanu and Alvarez-Buylla, 2002; Carleton et al., 2003). As they integrate into the adult OB network, new GCs display intense spinogenesis and synaptogenesis (Kelsch et al., 2010). This integration takes place within a relatively short time frame, the first spines appearing ∼14 dpi and the maximum density of spines being reached at 28 dpi (Petreanu and Alvarez-Buylla, 2002; Whitman and Greer, 2007). This period also represents a critical period for the survival of GCs, when they are particularly susceptible to sensory deprivation (Yamaguchi and Mori, 2005), and coincides with a time of intense formation of glutamatergic synapse impinging onto GCs spines (Kelsch et al., 2008). For these reasons, we have focused the present analysis within this precise time period.
From a methodological point of view, it is notable that, in our model, spine length and density of nonmutated newborn GCs vary according to the genetic backgrounds. In a C57BL/6J inbred background (Fig. 2; supplemental Fig. 2, available at www.jneurosci.org as supplemental material), the spine density of 21 dpi GCs was <0.5 spine/μm and the spine length ∼2.5 μm. In a Swiss outbred background (Figs. 3, 6), spine density at 21 dpi was ∼0.6 spine/μm and spine length closer to 2 μm. In addition, the effects of Fmr1 mutation also appear to be sensitive to the genetic background (Irwin et al., 2002; Galvez et al., 2005). Consistent with this notion, the extent of the increase in spine density and length seen in mutated GCs differs between Swiss (Fig. 3) and C57BL/6J mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Fmrp and spine architectural defects
We first analyzed 21 dpi GCs morphology in Fmr1 KO mice (C57BL/6J background). Prior analysis using these mice have largely confirmed the spine phenotype observed in the neocortex of FXS patients (Rudelli et al., 1985; Hinton et al., 1991; Wisniewski et al., 1991; Irwin et al., 2001), with an increased density of longer spines in adult neocortical neurons (Comery et al., 1997; Galvez and Greenough, 2005; McKinney et al., 2005). Newborn 21 dpi GCs in the OB of Fmr1 KO mice similarly displayed an increased density of spines, but these spines were of normal length. Obvious differences exist between GCs of the OB and neocortical pyramidal neurons, in addition to the fact that GCs are constantly replaced throughout adulthood and axonless neurons. Notably, GCs spines in the EPL differ from pyramidal neurons spines, in that they form very special reciprocal dendrodendritic synapses with mitral cells and are thus at the same time presynaptic and postsynaptic compartments. It is thus possible that the Fmrp mRNA targets potentially involved in their production, maturation, and stabilization are different from those in pyramidal neurons spines.
We next developed a strategy to mutate newborn GCs by RNA interference, which allowed us to analyze the differentiation of mutated neurons in a WT network (Swiss background). This approach is very similar to the MARCM (mosaic analysis with repressible cell marker) technique, which was used to cell-autonomously mutate drosophila Fmrp in isolated mushroom body neurons, and showed that mutated neurons exhibit increased axonal growth and overbranching (Pan et al., 2004). We found that the morphological defects of the mutation were stronger for 21 dpi GCs in a WT network than in the KO, with a lengthening of spines in addition to their increased density. This suggests that functional activity within the network influences the morphological defects of mutated spines. Accordingly, we observed that mutated shFMR1 neurons of mice raised in an enriched olfactory environment displayed an even greater spine lengthening, suggesting that lengthening could be a response to increased levels of synaptic activity.
Our analysis also clearly shows that Fmrp cell-autonomously regulates spine architecture. shFMR1 spines reach maximum length as soon as 14 dpi, suggesting that Fmrp is inhibitory to structural spine changes and is necessary to limit their growth. shFMR1 spines sometimes displayed spectacular structural defects with an extreme length >10 μm and spines comprising one neck and up to six heads (Fig. 6G). Spine growth and morphogenesis have been linked to regulation of spine actin cytoskeleton (Zito et al., 2004). These extreme abnormalities are thus evocative of actin misregulation, as described in drosophila-mutated neurons (Lee et al., 2003; Schenck et al., 2003; Reeve et al., 2005), the putative mRNA targets being Rac1 (Lee et al., 2003; Castets et al., 2005) and profilin (Lee et al., 2003; Reeve et al., 2005). Other mRNAs encoding some of the scaffolding proteins of the postsynaptic density could also be involved in these spine architectural defects, such as PSD95 (Todd et al., 2003; Zalfa et al., 2003; Muddashetty et al., 2007), Shank1 (Schütt et al., 2009), and Sapap3/4 (Brown et al., 2001; Kindler et al., 2004; Narayanan et al., 2007; Dictenberg et al., 2008).
Fmrp and synaptogenesis
We were particularly interested in the maturity of supernumerary lengthened spines. Because of their resemblance to filopodia, long thin spines are often considered as immature and thus not synaptically integrated (Comery et al., 1997; Irwin et al., 2000; Galvez and Greenough, 2005). This has led to the assumption that some FXS defects could be consecutive to a deficit of spine maturation and integration. Indeed, a failure in the transition from filopodia to spines in Fmr1 neurons has recently been reported in layer 2/3 somatosensory pyramidal neurons (Cruz-Martín et al., 2010). In our model system, if we define a filopodium as a protrusion without a detectable head, the percentage of filopodia that we observed in KO/ WT mice as well as in shNeg/shFMR1 neurons is low and of similar level in mutated or nonmutated neurons. However, it has been suggested that morphological criteria alone might not be sufficient to distinguish filopodia from spines (Cruz-Martín et al., 2010). We thus turned to more objective criteria of maturity through PSD95 immunolabeling and found that 21 dpi shFMR1 GCs displayed similar postsynaptic maturation as shNeg GCs. This was confirmed by electron microscopic analysis of the mutated spines, in which typical membrane specializations and vesicle distributions were found in reciprocal dendrodendritic synapses involving shFMR1 GC spines. Moreover, electrophysiological analysis of shFMR1 GCs showed an increase in the frequency of spontaneous EPSCs without a change in PPR, as compared with shNeg neurons. This likely reflects an increased number of functional glutamatergic inputs on the long supernumerary spines. Similarly, developing hippocampal Fmr1 KO neurons in culture display an increased number of PSD95-immunoreactive synapses (Pfeiffer and Huber, 2007). Therefore, loss of Fmrp and production of abnormally long spines do not prevent postsynaptic maturation. It would thus be interesting to directly test the maturity of immature-looking spines through immunohistochemistry in FXS patients, and to investigate whether an increased number of glutamatergic synapses could account for their increased rate of epilepsy.
FMRP and spine production
Although the increase in numbers and lengths of spines in Fmr1 KO mice and FXS patients has been widely documented, few studies have dealt with the differentiation defects leading to this phenotype (Nimchinsky et al., 2001; Galvez and Greenough, 2005; Cruz-Martín et al., 2010). The adult differentiation of GCs offers an effective model to address the developmental correlates of the FXS phenotype. Because Fmrp has recently been involved in regulating neural stem cells proliferation (Callan et al., 2010; Luo et al., 2010), we first tested whether the production and early maturation of young 8 dpi GCs was not altered after induction of Fmr1 mutation in the SVZ (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We then analyzed the spine phenotype of 14, 21, and 28 dpi GCs and saw that 14 dpi shFMR1 GCs, upon initiating spinogenesis, immediately reach maximum levels of spine density with an excessive length. A difference in spine density between control and mutated neurons was thus not visible anymore at 28 dpi, when control neurons reached the same plateau. This is very reminiscent of the transient spine phenotype seen in layer 5 neurons of the somatosensory cortex of Fmr1 KO mice, visible at 1 week but not 3 weeks later (Nimchinsky et al., 2001). Together with our data, this suggests that Fmrp might regulate the early rate and level of spine production. Galvez and Greenough (2005) confirmed the lack of phenotype of 4-week-old layer 5 neurons but reported the reappearance of a spine phenotype in the adult, suggesting a later role for Fmrp in spine pruning and maturation. This role in spine elimination has been recently confirmed (Pfeiffer et al., 2010).
FMRP and dendritogenesis
Much of the evidence for a role for Fmrp in neurite pruning comes from observations made in the drosophila, as dFmrp-null flies exhibit an overgrowth of axons and dendrites (Zhang et al., 2001; Morales et al., 2002; Lee et al., 2003; Pan et al., 2004). This alteration is thought to be due to loss of translational suppression of cytoskeleton regulators, either of actin filaments, as mentioned above, or of microtubules, through Map1B/Futsch (Zhang et al., 2001). Dendritic arborization defects have also occasionally been observed in the KO mice (Galvez et al., 2003; Galvez et al., 2005). We did not observe any alteration of dendritic architecture of newborn GCs, neither in the KO context nor in shFMR1 neurons. However, mutated neurons were incapable of reducing the length of their dendritic tree upon olfactory deprivation. This is very interesting as dFmrp plays a role in activity-dependent axon pruning (Tessier and Broadie, 2008), and as Fmr1 KO mice display deficits in neocortical plasticity in response to sensory deprivation (Dölen et al., 2007; Bureau et al., 2008).
Our work has allowed a precise dissection of the early defects induced by cell-autonomous loss of Fmrp in adult-born GCs. We show that abnormal immature-like spines can synaptically integrate in a wild-type network. As Fmrp is involved in the regulation of synaptic plasticity (Bear et al., 2004; Bassell and Warren, 2008), it would be interesting to test the effects of Fmrp loss on a form of plasticity of GCs that we have previously described (Nissant et al., 2009). We show new functions for Fmrp as a regulator of early spine production and as a mediator of activity-dependent dendritic remodeling. Better knowledge of the multiple functions of Fmrp in controlling neuronal architecture is essential for a better understanding of FXS.
Footnotes
This work was supported by a CNRS, Université Pierre et Marie Curie (UPMC), INSERM Avenir program (R06496DS), ANR (06-NEUR-039–01), FRM (INE20071110908 and DEQ20071210561), and Fondation Lejeune Grants. The “Development and Plasticity of Neural Networks” team and the “Laboratory for Perception and Memory” are affiliated with the Paris School of Neuroscience (ENP), which provided help for the viral production. The laboratory of P.M.L. is supported by the life insurance company “Aprionis” and by the Fondation pour la Recherche Médicale (Equipe FRM). We thank the IFR 83 for imaging, animal facilities, and real time PCR, R.Vogel and C. Sarkis for help with lentivirus plasmid design and virus production, F. Frédéric for her expertise in statistical analysis, Drs. Bagni and Darnell for anti-FMRP antibodies, and Drs. A. Didier and N. Mandairon for their help with the nasal tubings for olfactory deprivation.
References
- Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan MA, Cabernard C, Heck J, Luois S, Doe CQ, Zarnescu DC. Fragile X protein controls neural stem cell proliferation in the Drosophila brain. Hum Mol Genet. 2010;19:3068–3079. doi: 10.1093/hmg/ddq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo PM. Becoming a new neuron in the adult olfactory bulb. Nat Neurosci. 2003;6:507–518. doi: 10.1038/nn1048. [DOI] [PubMed] [Google Scholar]
- Castets M, Schaeffer C, Bechara E, Schenck A, Khandjian EW, Luche S, Moine H, Rabilloud T, Mandel JL, Bardoni B. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum Mol Genet. 2005;14:835–844. doi: 10.1093/hmg/ddi077. [DOI] [PubMed] [Google Scholar]
- Chelly J, Mandel JL. Monogenic causes of X-linked mental retardation. Nat Rev Genet. 2001;2:669–680. doi: 10.1038/35088558. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci. 2010;30:10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knock-out mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci. 2010;30:7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14:926–939. doi: 10.1016/j.devcel.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17:1539–1547. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135:155–160. doi: 10.1002/ajmg.a.30709. [DOI] [PubMed] [Google Scholar]
- Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 2003;971:83–89. doi: 10.1016/s0006-8993(03)02363-1. [DOI] [PubMed] [Google Scholar]
- Galvez R, Smith RL, Greenough WT. Olfactory bulb mitral cell dendritic pruning abnormalities in a mouse model of the Fragile-X mental retardation syndrome: further support for FMRP's involvement in dendritic development. Brain Res Dev Brain Res. 2005;157:214–216. doi: 10.1016/j.devbrainres.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development. 2008;135:2637–2648. doi: 10.1242/dev.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo RA, Larsen BP, Mehta A, Alcantara CJ, Patel B, Swain RA, Weiler IJ, Oostra BA, Greenough WT. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111:140–146. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Christmon CA, Grossman AW, Galvez R, Kim SH, DeGrush BJ, Weiler IJ, Greenough WT. Fragile X mental retardation protein levels increase following complex environment exposure in rat brain regions undergoing active synaptogenesis. Neurobiol Learn Mem. 2005;83:180–187. doi: 10.1016/j.nlm.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Kelsch W, Lin CW, Lois C. Sequential development of synapses in dendritic domains during adult neurogenesis. Proc Natl Acad Sci U S A. 2008;105:16803–16808. doi: 10.1073/pnas.0807970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsch W, Sim S, Lois C. Watching synaptogenesis in the adult brain. Annu Rev Neurosci. 2010;33:131–149. doi: 10.1146/annurev-neuro-060909-153252. [DOI] [PubMed] [Google Scholar]
- Kindler S, Rehbein M, Classen B, Richter D, Böckers TM. Distinct spatiotemporal expression of SAPAP transcripts in the developing rat brain: a novel dendritically localized mRNA. Brain Res Mol Brain Res. 2004;126:14–21. doi: 10.1016/j.molbrainres.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Lee A, Li W, Xu K, Bogert BA, Su K, Gao FB. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130:5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Saghatelyan A. Integrating new neurons into the adult olfactory bulb: joining the network, life-death decisions, and the effects of sensory experience. Trends Neurosci. 2005;28:248–254. doi: 10.1016/j.tins.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandairon N, Sacquet J, Jourdan F, Didier A. Long-term fate and distribution of newborn cells in the adult mouse olfactory bulb: influences of olfactory deprivation. Neuroscience. 2006;141:443–451. doi: 10.1016/j.neuroscience.2006.03.066. [DOI] [PubMed] [Google Scholar]
- McKinney BC, Grossman AW, Elisseou NM, Greenough WT. Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am J Med Genet B Neuropsychiatr Genet. 2005;136B:98–102. doi: 10.1002/ajmg.b.30183. [DOI] [PubMed] [Google Scholar]
- Melko M, Bardoni B. The role of G-quadruplex in RNA metabolism: involvement of FMRP and FMR2P. Biochimie. 2010;2:919–926. doi: 10.1016/j.biochi.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, Rifé M, Willemsen R, Nelson DL, Oostra BA. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21:549–555. doi: 10.1016/j.nbd.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR, Nelson DL, Hassan BA. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34:961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissant A, Bardy C, Katagiri H, Murray K, Lledo PM. Adult neurogenesis promotes synaptic plasticity in the olfactory bulb. Nat Neurosci. 2009;12:728–730. doi: 10.1038/nn.2298. [DOI] [PubMed] [Google Scholar]
- Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:17768–17773. doi: 10.1073/pnas.1012496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Alvarez-Buylla A. Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J Neurosci. 2002;22:6106–6113. doi: 10.1523/JNEUROSCI.22-14-06106.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci. 2007;27:3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol. 2005;15:1156–1163. doi: 10.1016/j.cub.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Rochefort C, Gheusi G, Vincent JD, Lledo PM. Enriched odor exposure increases the number of newborn neurons in the adult olfactory bulb and improves odor memory. J Neurosci. 2002;22:2679–2689. doi: 10.1523/JNEUROSCI.22-07-02679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS ONE. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67:289–295. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- Saghatelyan A, Roux P, Migliore M, Rochefort C, Desmaisons D, Charneau P, Shepherd GM, Lledo PM. Activity-dependent adjustments of the inhibitory network in the olfactory bulb following early postnatal deprivation. Neuron. 2005;46:103–116. doi: 10.1016/j.neuron.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/s0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Schütt J, Falley K, Richter D, Kreienkamp HJ, Kindler S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem. 2009;284:25479–25487. doi: 10.1074/jbc.M109.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008;135:1547–1557. doi: 10.1242/dev.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treloar HB, Feinstein P, Mombaerts P, Greer CA. Specificity of glomerular targeting by olfactory sensory axons. J Neurosci. 2002;22:2469–2477. doi: 10.1523/JNEUROSCI.22-07-02469.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembleau A, Morales M, Bloom FE. Aggregation of vasopressin mRNA in a subset of axonal swellings of the median eminence and posterior pituitary: light and electron microscopic evidence. J Neurosci. 1994;14:39–53. doi: 10.1523/JNEUROSCI.14-01-00039.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363:722–724. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GJ, Blonden LA, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostraa BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Whitman MC, Greer CA. Synaptic integration of adult-generated olfactory bulb granule cells: basal axodendritic centrifugal input precedes apical dendrodendritic local circuits. J Neurosci. 2007;27:9951–9961. doi: 10.1523/JNEUROSCI.1633-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski KE, Segan SM, Miezejeski CM, Sersen EA, Rudelli RD. The Fra(X) syndrome: neurological, electrophysiological, and neuropathological abnormalities. Am J Med Genet. 1991;38:476–480. doi: 10.1002/ajmg.1320380267. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Mori K. Critical period for sensory experience-dependent survival of newly generated granule cells in the adult mouse olfactory bulb. Proc Natl Acad Sci U S A. 2005;102:9697–9702. doi: 10.1073/pnas.0406082102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Zennou V, Serguera C, Sarkis C, Colin P, Perret E, Mallet J, Charneau P. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat Biotechnol. 2001;19:446–450. doi: 10.1038/88115. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]