

Abstract
The ability of the Fibroblast Growth Factor (FGF) 19 subfamily to signal in an endocrine fashion sets this subfamily apart from the remaining five FGF subfamilies known for their paracrine functions during embryonic development. Compared to the members of paracrine FGF subfamiles, the three members of the FGF 19 subfamily, namely FGF19, FGF21 and FGF23, have poor affinity for heparan sulfate (HS) and therefore can diffuse freely in the HS-rich extracellular matrix to enter into the bloodstream. In further contrast to paracrine FGFs, FGF 19 subfamily members have unusually poor affinity for their cognate FGF receptors (FGFRs) and therefore cannot bind and activate them in a solely HS-dependent fashion. As a result, the FGF 19 subfamily requires α/βklotho coreceptor proteins in order to bind, dimerize and activate their cognate FGFRs. This klotho-dependency also determines the tissue specificity of endocrine FGFs. Recent structural and biochemical studies have begun to shed light onto the molecular basis for the klotho-dependent endocrine mode of action of the FGF 19 subfamily. Crystal structures of FGF 19 and FGF23 show that the topology of the HS binding site (HBS) of FGF19 subfamily members deviates drastically from the common topology adopted by the paracrine FGFs. The distinct topologies of the HBS of FGF 19 and FGF23 prevent HS from direct hydrogen bonding with the backbone atoms of the HBS of these ligands and accordingly decrease the HS binding affinity of this subfamily. Recent biochemical data reveal that the αklotho ectodomain binds avidly to the ectodomain of FGFR1c, the main cognate FGFR of FGF23, creating a de novo high affinity binding site for the C-terminal tail of FGF23. The isolated FGF23 C-terminus can be used to effectively inhibit the formation of the FGF23-FGFR1c-αklotho complex and alleviate hypophosphatemia in renal phosphate disorders due to elevated levels of FGF23.
INTRODUCTION: AN OVERVIEW OF FGF-FGFR SIGNALING
The human fibroblast growth factors (FGFs) compose a family of secreted polypeptides that are encoded by 18 distinct genes (FGF1-FGF10 and FGF16-FGF23). FGFs play pleiotropic roles in human development and metabolism by binding and activating FGF receptor tyrosine kinases (FGFRs) that are encoded by four genes in humans(FGFR1-4).1–3 Based on sequence homology and phylogeny, the eighteen mammalian FGFs are grouped into five paracrine subfamilies and one endocrine subfamily.4–6 The paracrine subfamilies include the FGF1 subfamily comprising FGF 1, 2; the FGF7 sub family comprising FGF3, 7, 10, 22; the FGF4 subfamily comprising FGF4, 5, 6; the FGF8 subfamily comprising FGF8, 17, 18; and the FGF9 subfamily comprising FGF9, 16, 20. The endocrine-acting FGF19 subfamily comprises FGF 19, 21, 23. The paracrine-acting FGF1, FGF4, FGF7, FGF8 and FGF9 subfamilies play essential roles in spermatogenesis,7–9 mesoderm induction,10,11 somitogenesis,12–16 organogenesis,17–20 and pattern formation,21 whereas the FGF 19 subfamily acts in an endocrine fashion to regulate major metabolic processes including glucose,22 lipid, cholesterol and bile acid metabolism,23–25 and serum phosphate/vitamin D homeostasis.26 Based on sequence homology, four additional genes, namely FGF 11 -FGF 14, have also been considered to be members of the FGF family. Functionally speaking however, FGF 11-FGF 14 are not bona fide FGFs as they remain intracellular and lack key residues necessary for binding to FGFR.27–29
FGFs share a core homology region of about 120 amino acids consisting of twelve antiparallel β-strands arranged into three sets of four-stranded β-sheets (Fig. 1A).30–32 This globular core domain is flanked by N- and C-terminal regions that are highly divergent with respect to both length and sequence among FGFs, particularly across subfamilies.3 Moreover, even within some subfamilies the sequence identity at the N-terminus can be rather limited. The sequence identity of the N-terminal regions of FGF4 and FGF6 is only 36% compared to 69% for their core regions and that of FGF9 and FGF20 is only 38% compared to 86% for their core regions. Comparison of the crystal structures of several paracrine FGFs bound to their cognate FGFRs has shown that the FGFR binding specificity/promiscuity profile of a given FGF is principally dictated by the primary sequence of its N-terminal region.33–35 The structural data have begun to pinpoint the common primary and secondary structural elements within the N-termini of members of a given subfamily that explain their overlapping FGFR binding specificity/promiscuity profile.3
Figure 1.


An overview of FGF and FGFR structural biology. A, viewed on previous page) FGF1 is represented as a cartoon. N- and C-termini are labeled and the twelve β-sheets that compose the β-trefoil core are indicated. B, viewed on previous page) A schematic of FGFR shows its three Ig-like domains. D1 and the acid box (AB) are involved in autoinhibition, the heparan sulfate binding site (HBS) is located on D2 and alternative splicing takes place in the latter half of D3. D2 and D3 are necessary and sufficient for ligand binding. An intracellular kinase domain mediates downstream signaling. C) The formation of a 2:2:2 FGF:FGFR:HS dimer on the cell surface leads to intracellular transphosphorylation of the FGFR kinase domains and downstream signaling through PLCγ, FRS2α, and CRKL.
The prototypical FGFR is composed of three extracellular immunoglobulin domains (D1–D3), a transmembrane domain and an intracellular tyrosine kinase domain.36 (Fig. 1B). Structural studies have shown that ligand binding requires both D2 and D3 domains.34,37,38 The D1 domain and D1–D2 linker, that harbors the acid box (AB), are dispensible for ligand binding and in fact suppress FGF and HS binding affinity of the D2–D3 region.39,40 The specificity of FGFR1-3 for ligand binding is modulated by alternative splicing of mutually exclusive ‘b’ and ‘c’ exons in the second half of the D3 domain.41,42 This D3 alternative splicing event is tissue specific, with the b and c exons being preferentially used in epithelial and mesenchymal tissues respectively.41,43 Importantly, the D3 alternative splicing event elaborates the number of principal FGFRs from four to seven: FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, FGFR4. Structural studies have shown that D3 alternative splicing modulates the FGF binding specificity/promiscuity of FGFRs by switching the primary sequence of key ligand binding epitopes in D3.35
HS MODULATES FGF SIGNALING THROUGH MULTIPLE MECHANISMS
A wealth of genetic studies in mice and flies as well as cell-based studies have established that FGF signaling requires HS.44–48 HS is a highly sulfated linear polymer of alternating glucuronate (GlcA) and N-acetylglucosamine (GlcNAc) monosaccharides that undergoes heterogenous deacetylation, N-sulfation on GlcNAc, O-sulfation on both GlcA and GlcNAc, and epimerization on GlcA.49 HS impinges on FGF signaling through multiple mechanisms, including coordination/stabilization of FGF-FGFR binding and dimerization,50 control of FGF gradients in the extraceullular matrix (ECM),51 and protection of FGFs against thermal instability and proteolytic degradation.52,53
All FGFs interact with HS, albeit with differing affinities.54 The HBS of all FGFs is located within the core region and is composed of residues from the β1–β2 loop and the segment spanning the β10 to β12 strand. FGFRs also interact with HS via residues from the gA helix, the gA-βA′ loop, the βA′-βB loop, and the βB strand. Structural studies have shown that HS promotes formation of a 2:2:2 FGF-FGFR-HS cell surface signaling unit in which each ligand binds both receptors in the complex and the two receptors additionally make contact with one another.50,55 (Fig. 2A,B) Two HS molecules bind in a symmetric fashion to a positively-charged HS-binding cleft formed from the union of the HBS of the two FGFs and two FGFRs at the membrane-distal end of the dimer. By simultaneously engaging the HBS of both FGF and FGFR, HS stabilizes protein-protein contacts both within the 1:1 FGF-FGFR complex and between the two FGF-FGFR complexes in the dimer. In addition to promoting FGF-FGFR binding and dimerization, emerging data show that HS also controls the diffusion and morphogenetic gradients of paracrine FGFs in the extracellular matrix,51 and that the HS affinity of a ligand ultimately determines whether that FGF acts in a paracrine or endocrine fashion.56
Figure 2.

HS-dependent dimerization of the FGF-FGFR complex A) A surface representation of the FGF2-FGFR1c-heparin ternary complex, PDB ID: 1FQ9.55 FGF2 is in dark grey, the D2 and D3 domains of FGFR1c are in light grey and heparin is represented as sticks in black. B) The complex has been rotated ninety degrees around an axis parallel to the plane of the page to reveal heparin binding the HBS in the membrane distal cleft of the complex. C) The boxed region from Figure 2A is expanded to show the FGF2 residues involved in hydrogen bonding to HS. There a total of 16 hydrogen bonds between FGF2 and HS. For the sake of clarity, only those three hydrogen bonds mediated by backbone atoms are shown.
Dimerization of the extracellular domains of FGFRs juxtaposes the intracellular kinase domains, affording them with sufficient opportunity to trans-phosphorylate each other on the A-loop tyrosines. A-loop tyrosine phosphorylation increases the intrinsic kinase activity of FGFR kinase by stabilizing the active conformation of the kinase.57 Activated kinases then further trans-phosphorylate each other on tyrosines within the C-tail, kinase insert and juxtamembrane regions.58–60 The phosphorylated tyrosines in the C-tail and juxtamembrane regions of activated FGFR serve as recruitment sites for SH2 domains of PLCγ61,62 and CRKL,63 respectively. In the case of PLCγ, this recruitment serves two purposes: (i) it facilitates phosphorylation of PLCγ to increase its enzymatic activity, (ii) it brings PLCγ to the vicinity of its substrate PIP2 in the plasma membrane. Hydrolysis of PIP2 generates two second messengers: IP3 and DAG that stimulate Ca2+ release from intracellular stores and PKC activation respectively.64–66 In contrast, CRKL is an adaptor protein that lacks intrinsic enzymatic activity. Recruitment of CRKL to the phosphorylated tyrosine in the juxtamembrane region of FGFR1 and FGFR2 leads to translocation of associated Rac1/Cdc42 to the plasma membrane which culminates in cytoskeletal reorganization and cell motility.63 Lastly, activated FGFR phosphorylates FRS2α,67 another adaptor protein that, unlike PLCγ and CRKL, associates constitutively (independently of receptor phosphorylation) with the juxtamembrane region of FGFR.68–70 Phosphorylation of FRS2α by the activated FGFR generates docking sites for the SH2 domains of the adaptor protein GRB267 and the phosphatase Shp2,71 leading to activation of the Ras-MAPK and PI3K-Akt pathways (Fig. 1C).72
PARACRINE FGFs MEDIATE A MESENCHYMAL-EPITHELIAL SIGNALING LOOP WITHIN TISSUES
Historically, FGFs have been viewed as paracrine factors known for their wide ranging roles in tissue patterning and organogenesis during embryonic development and the FGF1, FGF4, FGF7, FGF8 and FGF9 subfamilies fall under this category. Members of these five FGF subfamilies have significant affinity for HS, limting their diffusion in the HS-rich ECM and accounting for their paracrine mode of action. Superimposition of crystal structures of paracrine FGFs shows that the HBS of paracrine FGFs all adopt nearly identical topologies. In these FGFs, the two HS binding regions, namely the β1–β2 loop and the segment spanning β10 to β12, are juxtaposed and form a continuous positively-charged fiat surface.32–35,37,73–76
Structure-based sequence alignment of paracrine FGFs shows that both the length and the primary sequence of the β1–β2 HS-binding loop differ across paracrine FGF subfamilies. In contrast, with the single exception of FGF5, the length of the HS binding segment spanning β10 to β12 is constant among all paracrine FGFs. Notably, all paracrine FGFs possess a prominent GXXXXGXX(T/S) motif in the HS binding β10–β12 segment. This motif, sometimes known as the glycine box,77 plays a key role in imparting the common conformation of the region between the β10 and β12 strands in paracrine FGFs (Figs. 3A, 5B,D). The first glycine of this motif makes hydrogen bonds with a conserved glycine in β3 and the second glycine hydrogen bonds with a fully conserved glycine in β7. These hydrogen bonds are essential for the formation of the β11 strand. Lastly, the side chain hydoxyl group of the threonine/serine hydrogen bonds with the backbone of the second glycine in the GXXXXGXX(T/S) motif. Crystal structures of FGF1 and FGF2 in complex with heparin oligosaccharides and SOS have demonstrated that both backbone and side chain atoms of the ligand’s HBS region partake in HS binding.55,78–80 Likewise, both sulfate groups and sugar backbone atoms of HS are engaged in FGF binding (Fig. 2C). The hydrogen bonds between backbone atoms of the HBS with HS are considered to be a key provider of binding energy, since they should incur less entropic loss upon HS binding than those hydrogen bonds involving the exposed flexible side chains.
Figure 3.
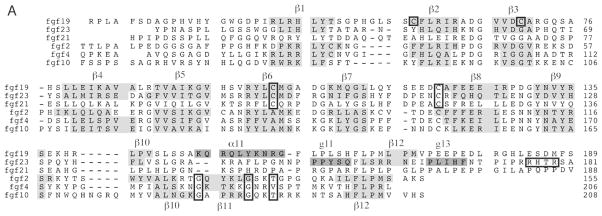

Sequence alignments of FGFs. A, viewed on previous page. Sequences of human FGF19, FGF23, FGF21, FGF2, FGF4 and FGF10 are aligned. N- and C-terminal regions of some ligands are truncated for the sake of presentation. β-sheets are highlighted with grey in the alignment and the helical secondary structures in FGF19 and FGF23 are highlighted with dark grey. Numerous important residues are enclosed in boxes to emphasize their importance: the Cys-58 and Cys-70 residues that support FGF19’s unusual β10–β12 segment structure are boxed, along with other cysteines that form disulfide bridges; the GXXXXGXX(T/S) motif in the paracrine FGFs is indicated with boxes; the RXXR motif in FGF23 is also boxed. B) Orthologs of FGF19, FGF21, FGF23 and FGF2 are aligned and the β10–β12 segment is shown for the solved structures. FGF23 exhibits greater conservation in its HBS than do either FGF19 or FGF21. The location of the GXXXXGXX(T/S) motif is indicated with arrowheads above the alignment of FGF2 orthologs.
Figure 5.

The FGF19 structure A) The FGF19 structure, PDB ID: 2P23,56 is shown as a surface and as a cartoon. The β-sheets and α-helix are labeled along with the N- and C-termini of the protein. B) The HBS of FGF19 is superimposed onto that of FGF2. Both ligands are represented as ribbons, with FGF19 in dark grey and FGF2 in light grey. The atoms of the GXXXXGXX(T/S) motif are marked with dots in the FGF2 structure and identified. Arrows indicate the positions of the Leu-145 and Leu-162 where the FGF19 Cα trace diverges from the FGF2 Cα trace. C) The FGF19 HBS is shown and the C58-C70 disulfide bridge residues are represented as sticks along with the Leu-162 and Leu-145 residues that protect them from solvent. D) The FGF2 HBS is shown, the GXXXXGXX(T/S) motif is identified, and intramolecular contacts between the β1–β2 loop and β10–β12 segment are shown. Asn-36 of the β1–β2 loop interacts with the backbone atom of Arg-129 in the β10–β12 segment and Phe-39 of the β1–β2 region engages in Van der Waals interactions with the backbone of the β10–β12 region.
Generally, paracrine FGF subfamilies exhibit tissue-specific expression patterns and are expressed in either epithelial or mesenchymal compartments within organs. The epithelially-expressed FGFs typically show specificity for FGFRc isoforms expressed in the mesenchyme and vice versa, resulting in the establishment of an epithelial-mesenchymal signaling loop (Fig. 4).81–85 It is well documented that FGF7 and FGF10, which are expressed exclusively in the mesenchyme, specifically activate FGFR2b to mediate the mesenchymal-to-epithelial signaling required for the development of multiple organs and glands including lung, thyroid, pituitary, lacrimal and salivary glands. 84,86–90 In contrast, the members of the FGF4, FGF8 and FGF9 subfamilies are expressed in the epithelium and activate the mesenchymal FGFRc isoforms to govern patterning and morphogenesis of multiple tissues and organs, including brain, lung, heart, kidney, eye, limb and ear.7,20,91,92
Figure 4.

Paracrine FGFs mediate an epithelial-mesenchymal signaling loop. Ligands expressed in the epithelium signal through receptors expressed in the mesenchyme and vice versa.
ENDOCRINE FGFs REGULATE KEY METABOLIC PROCESSES IN A KLOTHO-DEPENDENT FASHION
The perception of FGFs as paracrine factors acting mainly during embryonic development has been overturned by the discovery that members of the FGF 19 subfamily are humoral factors that regulate key metabolic processes. The involvement of FGF23 in phosphate homeostasis was discovered simultaneously by clinical studies of autosomal dominant hypophosphatemic rickets (ADHR) and tumor-induced osteomalacia (TIO), two human phosphate wasting disorders. The ADHR consortium identified mutations in FGF23 affecting either of two arginines at a RXXR motif that lies at the boundary between its core region and its 72 residue-long C-terminal tail (Fig. 3A).93 Later studies showed that the ADHR mutations interfered with the natural process of proteolytic inactivation of FGF23, leading to an increase in the serum concentration of FGF23 that in turn induces phosphate wasting.56,94,95 Shimada and colleagues showed that FGF23 secreted from the tumors of TIO patients was capable of causing hypophosphatemia.96 FGF21’s role in metabolism was originally discovered by Kharitonenkov and colleagues through experiments on FGF21 transgenic mice and murine models of diabetes,22 and FGF 19 was first identified as an important regulator of energy metabolism through studies of FGF19 transgenic rats by Tomlinson and colleagues.25
The endocrine FGFs require α/βklotho proteins as coreceptors in order to exert their metabolic actions. Klotho proteins are single-pass transmembrane proteins with an extracellular domain consisting of two tandem glycosidase-like domains, termed KL1 and KL2.97 FGF19 and FGF21 require βklotho,98–103 and FGF23 requires αklotho in order to bind and activate their cognate FGFRs.104,105 Klotho-dependency restricts signaling of FGF19 subfamily members to those tissues where klotho proteins are expressed. αklotho was originally discovered as an aging suppressor gene in mice and its requirement for FGF23 signaling was inferred from the phenotypic similarity between FGF23 and αklotho knockout mice.106,107 Likewise, mice deficient for FGF15 (the mouse ortholog of human FGF19), βKlotho-knockout mice, and FGFR4 knockout mice all have overlapping defects in bile acid metabolism, a correlation that facilitated the identification of βklotho as a coreceptor for FGF19.108–110
Each of the FGF 19 subfamily members participates in an endocrine signaling axis that is critical for maintaining homeostasis. The bone-kidney axis mediated by FGF23, FGFR1c (its main cognate FGFR)111 and αklotho plays a vital role in serum phosphate regulation. In response to rising serum phosphate, FGF23 is secreted from bone and activates FGFR1c in the kidney in an αklotho-dependent fashion,104,105,111 thus promoting phosphate excretion and suppressing vitamin D biosynthesis.112–118
FGF 19, FGFR4 (its main cognate FGFR) and βklotho are essential components of an intestine-liver axis that form a postprandial negative feedback loop to regulate bile acid synthesis and release. FGF 19 is secreted from intestinal epithelium in response to bile acid release into the intestinal lumen upon food intake.108,119 FGF19 reaches the liver via the hepatic portal vein where it activates hepatic FGFR4 in a βklotho-dependent fashion,99,100,103 thereby suppressing expression of the CYP7A1 gene that encodes the rate-limiting enzyme for bile acid synthesis.24 Additionally, FGF19 acts to promote gallbladder filling through FGFRs other than FGFR4.120
The liver-fat axis mediated by FGF21, FGFR1c (its main cognate FGFR), and βklotho is critical for inducing metabolic adaptation in response to fasting. FGF21 is secreted from the liver121 upon fasting,122–125 and it activates FGFR1c in adipocytes in a βklotho-dependent fashion98,99,101,102 to stimulate gluconeogenesis, ketogenesis and fatty acid oxidation.22,126–128
Interest in the FGF19 subfamily of ligands has been stimulated not only by their fascinating biology but also by their potential for the treatment of a variety of human diseases that have placed a major burden on health care. FGF21 agonists hold promise for the treatment of Type 2 diabetes and obesity.129 FGF19 antagonists hold promise for the treatment of hypercholesterolemia, gallstone disorders, hepatocellular carcinoma and colon cancer. FGF23 has been implicated in the pathogenesis of many congenital diseases, including FGF23 gain-of-function disorders such as ADHR,93 TIO,130,131 FGF23 loss-of-function disorders such as familial tumoral calcinosis (FTC),132–135 and X-linked hypophosphatemia (XLH),130,131 a disorder involving increased FGF23 levels that results from loss of function mutations in the metalloproteinase PHEX that is thought to cleave FGF23 at its RXXR motif. More recently, elevations in FGF23 serum concentration have been correlated with the progression of chronic kidney disease (CKD).136 FGF23 antagonists could be used to alleviate hypophosphatemia in inherited and tumor-induced phosphate wasting disorders as well as hypophosphatemia associated with other conditions such as organ transplantation and parenteral iron therapy. Conversely, FGF23 agonists could be used to correct hyperphosphatemia in FTC patients.
The recent crystal structures and biochemical studies of FGF 19 and FGF23 have begun to elucidate the molecular basis for these ligands’ endocrine behavior and the mechanism of action of their klotho coreceptors. Advances in the structural biology of FGF23 are already being translated towards the discovery of drugs for renal phosphate wasting disorders.
Structure-Function Relationships of Endocrine FGFs
Relative to the five paracrine-acting FGF subfamilies, the FGF 19 subfamily exhibits the least sequence identity amongst its members. The pairwise sequence identity between the core regions of members of FGF19 subfamily ranges between 33% for FGF21 and FGF23 and 38% for FGF 19 and FGF21 (Fig. 3A). In comparison, the identity between the core regions of members of paracrine FGF subfamilies is significantly higher and ranges between 88% for FGF9 and FGF16 to 54% for FGF7 and FGF10. Most of the sequence divergence between FGF19 subfamily members stems from the HBS regions, namely the β1–β2 loop and the segment between the β10 and β12 strands of these ligands. The identity between the HBS of FGF 19 subfamily members is at best 13%. Exclusion of the HBS from the alignment improves the identity between FGF 19 subfamily members to above 40%. In paracrine FGFs, however, the degree of sequence divergence at the HBS region is much less and is comparable with the degree of divergence in other regions of the trefoil core.
The HBS is the site of greatest divergence between the core regions of FGF19 subfamily ligands and those of other paracrine FGFs. The HS binding segment between β10 and β12 in endocrine FGFs is shortened and lacks the critical GXXXXGXX(T/S) motif, suggesting that this region cannot adopt the same conformation as in paracrine FGFs. Moreover, the β1–β2 loop of all three members of the FGF19 subfamily is longer than those of paracrine FGF subfamilies. Consistent with these major primary sequence divergences between the FGF 19 subfamily and other FGF subfamilies, the crystal structures of FGF 19 alone and of FGF23 bound to the heparin analogue sucrose octasulfate (SOS)56 revealed that the HBS of FGF 19 and FGF23 take on completely different conformations than that seen in paracrine FGFs. Notably, the conformations of the HBS seen in these two endocrine FGFs are incompatible with the hydrogen bonding of HS to backbone atoms in the HBS regions of these endocrine FGFs. This finding provides a molecular basis for the weak binding of FGF 19 subfamily members for HS and explains the subfamily’s ability to act in an endocrine fashion.
FGF19 Structural Findings
The first crystal structure of FGF19 was reported by the Blundell laboratory.137 In this structure, the HS binding segment between the β10 and β12 strands is disordered, leading the authors to suggest that the HBS of FGF 19 is inherently flexible. It was proposed that the HBS region of FGF 19 assumes an ordered conformation upon HS binding and that the entropic penalty associated with HS binding was what caused the ligand’s reduced HS binding affinity.137 In the second FGF19 crystal structure published 3 years later, however, both FGF 19 copies in the asymmetric unit of the crystal displayed a well ordered β10–β12 region (Fig. 5A). Since the β10–β12 region of these two FGF 19 molecules are in different crystal packing environments, it can be argued that crystal packing did not bias the conformation of the HBS region in the FGF19 structure.56
As anticipated based on the lack of sequence homology between FGF 19 and paracrine FGFs at the HBS region between β10 to β12, the conformation of this region deviates completely from the common conformation that is seen in paracrine FGFs. The Cα trace of FGF 19 in the region of the HBS begins to diverges from that of paracrine FGFs at Leu-145 and converges again at Leu-162 (Fig. 5B). Residues 149 to 158 in the β10–β12 region of FGF19 form an α-helix (α11) that bulges out from the β-trefoil core. The atypical conformation of the β10–β12 region of FGF19 is accompanied by other structural differences between FGF19 and paracrine FGFs in the core. In FGF 19, the conserved β3 glycine of paracrine FGFs is replaced by a cysteine (Cys-70). In the FGF19 structure, Cys-70 forms a disulfide bond with Cys-58 (also unique to FGF19) in β2 that packs against Leu-145 and Leu-162 at the divergent and convergent ends of the β10–β12 region. This hydrophobic interaction helps to partially shield the disulfide bridge from solvent (Fig. 5C).
The β1–β2 loop of FGF19 is the longest among the FGFs and extends out from the β-trefoil core in the same direction as the α11 helix. In contrast to paracrine ligands where the β1–β2 loop and β10–β12 segment are juxtaposed and form a contiguous HBS, in FGF19 there is a spatial separation between the two regions as they do not engage in any intramolecular interactions (Fig. 5A, 5D).
FGF23 Structural Findings
Attempts to crystallize the full length FGF23 were unsuccessful, likely because of the flexibility of its (73-amino acid long) C-terminus. The core domain was thus crystallized in complex with SOS (Figs. 6A,B). Consistent with the major primary sequence divergence at the β10–β12 segment between FGF23 and paracrine FGFs, the Cα trace of FGF23 diverges from that of paracrine FGFs at Leu-138 and converges again at Pro-153. (Fig. 6C). Predictably, the conformation of this region is also completely different from that of FGF19 (Fig. 6D) since the two ligands share no homology in their β10–β12 regions. Like FGF19, FGF23 does not exhibit the β11 strand of paracrine FGFs and instead PROCHECK assigns a g-helix to the C-terminal region of the β10–β12 region in FGF23. Again as for FGF19, the altered conformation of the β10–β12 region in FGF23 is accompanied by changes in other regions of the core. Compared to paracrine FGFs, FGF23 has a very short β9–β10 loop which is needed to accommodate the novel HBS topology of FGF23. Structural analysis shows that even a one residue insertion in the β9–β10 loop of FGF23 would interfere with the β10–β12 conformation seen in FGF23. The HBS of FGF23 contains a cleft similar to that in FGF19 between the β1–β2 loop and β10–12 region (Fig. 6D). The SOS molecule binds with its sulfated fructose ring facing down into the cleft in the FGF23 HBS and it engages in hydrogen bonds with Arg-140 and Arg-143 in β10–β12 and Arg-48 and Asn-49 in β1–β2 (Fig. 6B). Hence, based on the crystal structure these four residues of FGF23 mediate binding of FGF23 to HS.
Figure 6.

The FGF23 structure A) The FGF23 structure, PDB ID: 2P39,56 is shown as a surface and as a cartoon. The β-sheets and α-helix are labeled along with the N- and C-termini of the protein. The SOS molecule that is bound to FGF23 is represented as sticks in dark grey. B) An expansion of the box in Figure 6A showing a close-up of the SOS molecule and the residues in the HBS of FGF23 with which it binds. C) An overlay of the HBS of FGF23 with that of FGF2 showing their divergence. FGF2 is in light grey and FGF23 is in dark grey. The residues of the GXXXXGXX(T/S) motif are labeled and the Leu-138 and Pro-153 residues where the FGF23 Cα trace diverges from that of FGF2 are indicated with arrows. D) The FGF19 and FGF23 HBS regions are superimposed, with FGF19 shown in light grey and FGF23 in dark grey.
THE MOLECULAR BASIS FOR THE FGF19 SUBFAMILY’S KLOTHO CORECEPTOR REQUIREMENT
Reduced Affinity of FGF19 Subfamily Members for HS
Superimposition of the FGF19 and FGF23 crystal structures onto FGF2 in the FGF2-FGFR1c-heparin ternary complex structure (PDBID; 1FQ9)55 illuminates the impact of these ligands’ altered HBS topology on their interaction with HS. This superimposition shows that HS would clash with the HBS of FGF19 and FGF23 if it attempted to bind to the ligand as it does in paracrine FGFs (Figs. 7A,B). These clashes could be avoided by translating HS away from the ligands’ core domains, but such a translation would be detrimental for the HS binding affinity since the backbone atoms of the two ligands would no longer be able to make direct hydrogen bonds with the N-sulfate group from 4GlcN(S)6O(S) or a 2-O-sulfate group from 5IdoA (Fig. 2C). Thus, the altered topology of the β10–β12 region in FGF19 and FGF23 should impart a major loss in HS binding. This lowered HS binding affinity is a prerequisite for the endocrine behavior of this subfamily of ligands since it allows them to diffuse unhindered through the HS-rich pericellular space and enter the bloodstream.
Figure 7.
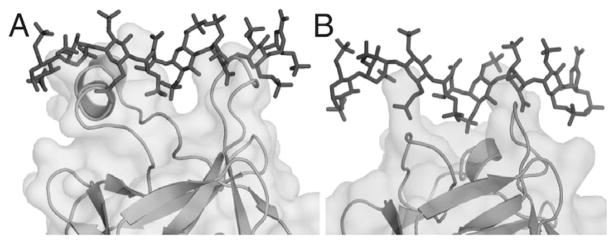
Superimposition of heparin onto the FGF19 and FGF23 structures. A) FGF19 is superimposed onto the FGF2-FGFR1c-heparin structure (PDB ID: 1FQ9)55 and the heparin from that structure is grafted onto FGF 19 to show the clashes that would occur were heparin to attempt to bind FGF 19 in the same fashion as it binds FGF2. B) Similarly for FGF23, heparin is superimposed to reveal the clashes that would occur were heparin to attempt to bind in a FGF2-like fashion.
The orientation of SOS bound to FGF23 is perpendicular to the orientation of SOS bound to FGF1,80 and of heparin oligosaccharides bound to FGF1 and FGF2.55,78,79 This structural observation further corroborates the notion that HS cannot bind to FGF19 subfamily ligands as it classically binds to paracrine FGFs.
Biochemical experiments have confirmed the conclusions drawn from structural analysis. Surface plasmon resonance (SPR) experiments showed that FGF19, FGF21 and FGF23 have poor binding to heparin relative to paracrine ligands.56 Moreover, by mutating FGF19 residues Lys-149 and Arg-157 that based on the FGF19-FGFR-heparin model are predicted to mediate the residual HS binding, the affinity of FGF19 for heparin was reduced even further.56 Likewise, by mutating residues in FGF23 that were involved in binding to SOS, the affinity of FGF23 for heparin was further decreased.56 It remains to be tested if the residual HS binding affinity of the endocrine FGFs still plays a role in their signaling.
The HS binding segment between β10 and β12 in FGF19 and FGF23 displays two unique conformations (Fig. 6D), both of which sterically hinder HS from hydrogen bonding with the backbone atoms of HBS in these ligands (Fig. 7). Structure-based sequence alignment of FGF21 with FGF19 and FGF23 shows that the β10–β12 region of FGF21 would adopt yet a third unique topology. There is no sequence identity between the β10–β12 segment of FGF21 and FGF19 and only 11% sequence identity between FGF21 and FGF23 at this region (Fig. 3A). These observations raise the question whether the specific conformation of the HBS of FGF19 subfamily members is important or whether their HBS needs simply to provide a steric obstacle in any way possible against HS bonding. If the former were the case, then one would expect an evolutionary pressure to preserve the sequence of the HBS in the endocrine FGFs.
To address this question, we aligned the orthologs of FGF19, FGF21, FGF23 and FGF2, a prototypical paracrine FGF (Fig. 3B). The alignments show gaps in the β10–β12 region of rat and zebrafish FGF 19 and FGF21. FGF 19 shows an additional gap in the β10–β12 of mouse (Fig. 3B). These differences between the sequences of the orthologs in FGF 19 and FGF21 indicate that the specific conformation of their β10–β12 region is not as critical as it is for FGF23, where the sequence of the protein is more conserved across the different orthologs and no gaps in the sequence are seen. In contrast to the endocrine FGFs, the sequence alignment of the FGF2 orthologs shows almost total identity in the β10–β12 region (Fig. 3B), underscoring how important the primary and secondary structure elements in the HBS are for HS binding to FGF2.
STRUCTURAL BASIS FOR THE SPECIFICITY AND AFFINITY OF ENDOCRINE FGFs’ BINDING TO FGFR
The principal cognate receptors for the FGF 19 subfamily ligands have been well characterized. FGF23 signaling in the kidney is mediated by FGFR1c, since administration of FGF23 to mice with a conditional knockdown of FGFR1 in the metanephric mesenchyme did not induce hypophosphatemia.111 FGF 19 signaling is mediated by FGFR4, since FGF15 (the mouse ortholog of human FGF 19) could not suppress CYP7A1 activity in FGFR4 knockout mice,108 FGF19-induced hepatocyte proliferation is driven through FGFR4,138 and FGFR4- and FGF 15-deficient mice both have elevated bile acid pools.108,110
The crystal structures of FGF 19 and FGF23 offer a molecular basis for the receptor binding specificity of these two ligands. Comparison of the receptor-bound crystal structures of several paracrine FGFs, including FGF1, FGF2, FGF3, FGF8 and FGF10,33–35,37–39 have shown that the extent to which β1 strand pairs with β4 plays a decisive role in the discrimination of FGFR binding specificity. Compared to β1 of FGF2, the β1 strands of the FGF7 subfamily are extended two residues N-terminally due to additional strand pairing with β4. This extension is essential for the ability of the N-termini of FGF7 subfamily members to engage in specific contacts with the alternatively spliced βC′-βE loop of FGFR2b. Conversely, the N-terminally extended β1 strand of the FGF7 subfamily clashes with the alternatively spliced loop of FGFR2c, thus actively discouraging binding of this subfamily to FGFR2c. Interestingly, akin to the FGF7 subfamily, the β1 strand of FGF19 is extended two residues longer than the β1 strand of the FGF1 subfamily (Fig. 3A) and the length of the βC′-βE loop of FGFR4, FGF19’s cognate receptor, is the same length as in FGFR2b—two residues shorter than the corresponding loop in FGFRc isoforms. Although FGF 19 does not bind to FGFR2b, we predict that the lengthened β1 strand of FGF19 confers its specificity for FGFR4. Conversely, the β1 strand of FGF23 is the same length as in FGF2, which is consistent with FGF23’s preference for FGFR1c.
The crystal structures of FGF 19 and FGF23 also afford a potential molecular basis for the poor affinity of these ligands for their cognate FGFRs. SPR, size exclusion and co-immunoprecipitation data have shown that FGF 19 subfamily members have unusually low affinity for their cognate FGFRs.3,98–101,103–105 Superimposition of FGF23 onto FGF2 in the FGF2-FGFR1c-heparin structure (PDB ID: 1FQ9)55 shows that substitution of His-117 in FGF23 for the highly conserved glutamate in the β8 strand of all other FGFs would lead to a major loss in FGFR binding affinity. In the crystal structure of FGF2 and other paracrine FGFs complexed with FGFR, this glutamate (Glu-105 in FGF2) makes direct hydrogen bonds with D3. Indeed, mutation of this glutamate to alanine in FGF2 has been shown to dramatically reduce the binding of the ligand to FGFR.139
In summary, the structural and biochemical data along with sequence analysis reveal that FGF19 subfamily members have poor HS and FGFR binding affinity. Consequently, these FGFs are incapable of binding, dimerizing and activating their cognate FGFRs in a solely HS-dependent fashion. To overcome these deficiencies the endocrine FGFs must rely on klotho proteins as coreceptors. Klotho coreceptors act by constitutively associating with the cognate FGFRs of endocrine FGFs, thereby enhancing the affinity of the FGFR for endocrine FGFs to levels sufficient for FGFR dimerization and activation.
CHARACTERIZATION OF THE BINDING OF FGF23 TO αKLOTHO
Determining why FGF 19 subfamily members depend on klotho coreceptors for signaling was an important step towards understanding the biology of this interesting FGF subfamily. The next question to be studied is how the FGF 19 subfamily members, their cognate FGFRs and klotho coreceptor interact to form ternary complexes. Through a combination of SPR, size exclusion and co-immunoprecipitation experiments, the nature of FGF23’s binding to FGFR1c and αklotho has been recently investigated.94 First, it was shown that the soluble ectodomains of FGFR1c and αklotho are sufficient to form a ternary complex with FGF23. The ability to reconstitute the ternary complex in vitro enabled a thorough characterization of the bimolecular interactions among the three components of the ternary complex, αklotho ectodomain was shown to bind to the ectodomain of FGFR1c with high affinity (72 nM). Interestingly, although FGF23 bound poorly to αklotho ectodomain, it nonetheless bound avidly to a preformed 1:1 complex of FGFR1c and αklotho ectodomains. Since FGF23 binds poorly to both FGFR1c and αklotho, these data indicate that in the context of the FGFR1c-αklotho complex a de novo site for FGF23 is generated. Importantly, truncated FGF23 lacking its C-terminal tail past the RXXR motif is unable to bind a 1:1 binary complex of FGFR 1 c-αklotho in the absence of its own C-terminal tail, revealing that the C-terminal tail of FGF23 mediates binding of FGF23 to this de novo site created at the interface of FGFR1c and αklotho.
Consistent with this finding, a SPR competition assay showed that the C-terminal tail of FGF23 was able to compete away binding of full length FGF23 from the FGFR1c–αklotho binary complex. The binding region within the FGF23 C-tail for this de novo site in the αklotho-FGFR1c binary complex was further narrowed down to the region between Ser-180 and Thr-200. Accordingly, this fragment was also able to compete away binding of full length FGF23 from the FGFR1c-αklotho binary complex, albeit with less potency than the full C-tail. Consistent with this region serving as a minimal epitope, a FGF23 ligand truncated at Thr-200 was able to elicit similar levels of FRS2α phosphorylation as the full length ligand.
PHARMACOLOGICAL IMPACT OF STRUCTURAL INSIGHTS INTO FGF19 SUBFAMILY
To date, the primary treatment for disorders of phosphate wasting has been intravenous phosphate therapy along with 1,25-dihydroxyvitamin D3 to increase phosphate absorption in the intestine. This relatively primitive therapy sometimes results in side-effects of over-shoot hyperphosphatemia, and it is also simply ineffective in cases of chronic phosphate wasting such as occur in XLH.
Thus, the finding that the FGF23 C-terminus effectively inhibits the FGF23 interaction with FGFR1c-αklotho holds great promise for the development of therapeutics. Developing pharmacologically active C-terminal peptides, peptidomimetics or organomimetics thereof would introduce an important new tool for the treatment of phosphate wasting disorders. This method of inhibition has an advantage over neutralizing FGF23 by antibodies, since the C-terminal peptide only inhibits the αklotho-specific activity of FGF23, leaving any potential klotho-independent activity of FGF23 intact. C-terminal peptides thus may also serve as a useful tool to dissect the αklotho dependent and αklotho independent functions of FGF23.
These studies of the inhibition of FGF23 activity by a FGF23 fragment produced through proteolysis are also fascinating at the level of general biological principle. Here, we observed a hormone being inhibited by the product of its own degradation. Accordingly, in pathological hyperphosphatemic states such as FTC where serum levels of C-terminal peptides of FGF23 are high, this C-terminal fragment may be aggravating the disease by inhibiting any residual function of the full length mutant FGF23 ligand found in FTC.
CONCLUSION
Undoubtedly, solving the crystal structures of the FGF19-FGFR4-βklotho, FGF21-FGFR1c-βklotho, and FGF23-FGFR1c-αklotho complexes will be the immediate priority for this fast moving and exciting field. The structure of the FGF23-FGFR1c-αklotho complex should unveil the details of how a de novo site for FGF23’s C-tail is created when αklotho and FGFR1c ectodomain interact. It would be fascinating to see whether βklotho also utilizes the same mechanism as αklotho to promote the signaling of FGF19 and FGF21 through their respective cognate receptors, FGFR4 and FGFR1c. The structures of these ternary complexes will also inform us of the determinants of the FGFR binding specificity/promiscuity of klotho proteins. Biochemically, it will be important to find out if the residual HS binding affinity of the FGF19 subfamily is still required for signaling. If this were the case then we could envision the model shown in Figure 8 for how the cell surface signaling unit of an endocrine FGF might look like. The signaling unit will still have a 2:2:2 FGF-FGFR-HS dimer at its core that is further stabilized by interactions of klotho proteins with FGFR and endocrine FGFs.
Figure 8.
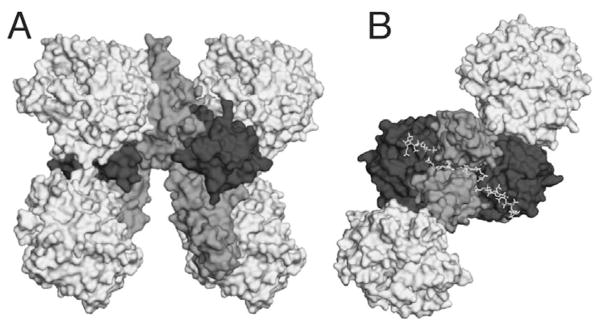
A proposed model of the FGF23-FGFR1c-heparin-αklotho complex. A) FGF23 is superimposed onto FGF2 in the FGF2-FGFR1c-heparin complex (PDB ID:1FQ9)55 and is depicted in dark grey. The FGFRs from 1FQ9 are depicted in a medium tint of grey. Myrosinase (PDB ID: 1E6S),140 a glycosidase, is used as a model for the two KL domains of αklotho and is depicted in light grey. B) The FGF23-FGFR1c-heparin-αklotho complex has been rotated 90 degrees to show the HS binding cleft in the membrane-distal portion of the complex.
The discovery that a major difference in HS binding was responsible for the endocrine mode of action of the FGF 19 subfamily suggested the corollary that subtle differences in the HS binding affinity of paracrine FGFs may also play a role in distinguishing their distinct biological activity. Indeed, a recent study has shown that differences in the HS affinity of two members of the FGF7 subfamily, FGF7 and FGF10, was responsible for differences in their branching morphogenetic potentials.51 Using a branching morphogenesis assay, it was shown that FGF10 could be transformed into a functional mimic of FGF7 through a single Arg→ Val mutation in the FGF10 HBS. This mutation reduced FGF10’s HS affinity to FGF7-like levels and thus allowed FGF 10 to diffuse through the extracellular matrix more easily and establish a gradient similar to that made by FGF7. Future studies should aim to elucidate the extent to which the differing biology of ligands within other FGF subfamilies can also be accounted for on the basis of their affinities for HS. In which case, the central role of HS affinity in differentiating the biological functions of FGFs will not be a feature only of the endocrine FGFs but of all FGFs.
References
- 1.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- 3.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16(2):107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20(11):563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237(1):18–27. doi: 10.1002/dvdy.21388. [DOI] [PubMed] [Google Scholar]
- 6.Popovici C, Roubin R, Coulier F, et al. An evolutionary history of the FGF superfamily. Bioessays. 2005;27(8):849–857. doi: 10.1002/bies.20261. [DOI] [PubMed] [Google Scholar]
- 7.Colvin JS, Green RP, Schmahl J, et al. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001;104(6):875–889. doi: 10.1016/s0092-8674(01)00284-7. [DOI] [PubMed] [Google Scholar]
- 8.Goriely A, Hansen RM, Taylor IB, et al. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41(11):1247–1252. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goriely A, McVean GA, Rojmyr M, et al. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301(5633):643–646. doi: 10.1126/science.1085710. [DOI] [PubMed] [Google Scholar]
- 10.Kato S, Sekine K. FGF-FGFR signaling in vertebrate organogenesis. Cell Mol Biol (Noisy-le-grand) 1999;45(5):631–638. [PubMed] [Google Scholar]
- 11.Niswander L, Tickle C, Vogel A, et al. FGF-4 replaces the apical ectodermal ridge and directs outgrowth and patterning of the limb. Cell. 1993;75(3):579–587. doi: 10.1016/0092-8674(93)90391-3. [DOI] [PubMed] [Google Scholar]
- 12.Baker RE, Schnell S, Maini PK. A clock and wavefront mechanism for somite formation. Dev Biol. 2006;293(1):116–126. doi: 10.1016/j.ydbio.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Dale JK, Malapert P, Chal J, et al. Oscillations of the snail genes in the presomitic mesoderm coordinate segmental patterning and morphogenesis in vertebrate somitogenesis. Dev Cell. 2006;10(3):355–366. doi: 10.1016/j.devcel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Dubrulle J, McGrew MJ, Pourquie O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell. 2001;106(2):219–232. doi: 10.1016/s0092-8674(01)00437-8. [DOI] [PubMed] [Google Scholar]
- 15.Pourquie O. The chick embryo: a leading model in somitogenesis studies. Mech Dev. 2004;121(9):1069–1079. doi: 10.1016/j.mod.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Sawada A, Shinya M, Jiang YJ, et al. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development. 2001;128(23):4873–4880. doi: 10.1242/dev.128.23.4873. [DOI] [PubMed] [Google Scholar]
- 17.Colvin JS, White AC, Pratt SJ, et al. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128(11):2095–2106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- 18.Itoh N. The Fgf families inhumans, mice and zebrafish: their evolutional processes and roles in development, metabolism and disease. Biol Pharm Bull. 2007;30(10):1819–1825. doi: 10.1248/bpb.30.1819. [DOI] [PubMed] [Google Scholar]
- 19.Lu SY, Sheikh F, Sheppard PC, et al. FGF-16 is required for embryonic heart development. Biochem Biophys Res Commun. 2008;373(2):270–274. doi: 10.1016/j.bbrc.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugi Y, Ito N, Szebenyi G, et al. Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev Biol. 2003;258(2):252–263. doi: 10.1016/s0012-1606(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 21.O’Leary DD, Chou SJ, Sahara S. Area patterning of the mammalian cortex. Neuron. 2007;56(2):252–269. doi: 10.1016/j.neuron.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145(6):2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 24.Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17(13):1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomlinson E, Fu L, John L, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143(5):1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 26.Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16(2):221–232. doi: 10.1016/j.cytogfr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Goetz R, Dover K, Laezza F, et al. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J Biol Chem. 2009;284(26):17883–17896. doi: 10.1074/jbc.M109.001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure and function. Cytokine Growth Factor Rev. 2005;16(2):215–220. doi: 10.1016/j.cytogfr.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen SK, Garbi M, Zampieri N, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278(36):34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- 30.Eriksson AE, Cousens LS, Weaver LH, et al. Three-dimensional structure of human basic fibroblast growth factor. Proc Natl Acad Sci USA. 1991;88(8):3441–3445. doi: 10.1073/pnas.88.8.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang JD, Cousens LS, Barr PJ, et al. Three-dimensional structure of human basic fibroblast growth factor, a structural homolog of interleukin 1 beta. Proc Natl Acad Sci USA. 1991;88(8):3446–3450. doi: 10.1073/pnas.88.8.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X, Komiya H, Chirino A, et al. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 1991;251(4989):90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 33.Olsen SK, Li JY, Bromleigh C, et al. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006;20(2):185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plotnikov AN, Hubbard SR, Schlessinger J, et al. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101(4):413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 35.Yeh BK, Igarashi M, Eliseenkova AV, et al. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc Natl Acad Sci USA. 2003;100(5):2266–2271. doi: 10.1073/pnas.0436500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee PL, Johnson DE, Cousens LS, et al. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245(4913):57–60. doi: 10.1126/science.2544996. [DOI] [PubMed] [Google Scholar]
- 37.Plotnikov AN, Schlessinger J, Hubbard SR, et al. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98(5):641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 38.Stauber DJ, DiGabriele AD, Hendrickson WA. Structural interactions of fibroblast growth factor receptor with its ligands. Proc Natl Acad Sci USA. 2000;97(l):49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsen SK, Ibrahimi OA, Raucci A, et al. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc Natl Acad Sci USA. 2004;101(4):935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Kan M, Yan G, et al. Alternately spliced NH2-terminal immunoglobulin-like Loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J Biol Chem. 1995;270(17):10231–10235. doi: 10.1074/jbc.270.17.10231. [DOI] [PubMed] [Google Scholar]
- 41.Johnson DE, Lu J, Chen H, et al. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11(9):4627–4634. doi: 10.1128/mcb.11.9.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miki T, Bottaro DP, Fleming TP, et al. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci USA. 1992;89(l):246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orr-Urtreger A, Bedford MT, Burakova T, et al. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158(2):475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 44.Forsberg E, Kjellen L. Heparan sulfate: lessons from knockout mice. J Clin Invest. 2001;108(2):175–180. doi: 10.1172/JCI13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin X, Buff EM, Perrimon N, et al. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development. 1999;126(17):3715–3723. doi: 10.1242/dev.126.17.3715. [DOI] [PubMed] [Google Scholar]
- 46.Ornitz DM, Yayon A, Flanagan JG, et al. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12(1):240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252(5013):1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 48.Yayon A, Klagsbrun M, Esko JD, et al. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64(4):841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 49.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105(7):2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 50.Mohammadi M, Olsen SK, Goetz R. A protein canyon in the FGF-FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol. 2005;15(5):506–516. doi: 10.1016/j.sbi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Makarenkova HP, Hoffman MP, Beenken A, et al. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal. 2009;2(88):ra55. doi: 10.1126/scisignal.2000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6(7):530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 53.Saksela O, Moscatelli D, Sommer A, et al. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. J Cell Biol. 1988;107(2):743–751. doi: 10.1083/jcb.107.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ashikari-Hada S, Habuchi H, Kariya Y, et al. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004;279(13):12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- 55.Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6(3):743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 56.Goetz R, Beenken A, Ibrahimi OA, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27(9):3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen H, Ma J, Li W, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27(5):717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen H, Xu CF, Ma J, et al. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc Natl Acad Sci USA. 2008;105(50):19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furdui CM, Lew ED, Schlessinger J, et al. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell. 2006;21(5):711–717. doi: 10.1016/j.molcel.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 60.Mohammadi M, Dikic I, Sorokin A, et al. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16(3):977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohammadi M, Dionne CA, Li W, et al. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature. 1992;358(6388):681–684. doi: 10.1038/358681a0. [DOI] [PubMed] [Google Scholar]
- 62.Peters KG, Marie J, Wilson E, et al. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358(6388):678–681. doi: 10.1038/358678a0. [DOI] [PubMed] [Google Scholar]
- 63.Seo JH, Suenaga A, Hatakeyama M, et al. Structural and functional basis of a role for CRKL in a fibroblast growth factor 8-induced feed-forward loop. Mol Cell Biol. 2009;29(11):3076–3087. doi: 10.1128/MCB.01686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Divecha N, Irvine RF. Phospholipid signaling. Cell. 1995;80(2):269–278. doi: 10.1016/0092-8674(95)90409-3. [DOI] [PubMed] [Google Scholar]
- 65.Huang J, Mohammadi M, Rodrigues GA, et al. Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem. 1995;270(10):5065–5072. doi: 10.1074/jbc.270.10.5065. [DOI] [PubMed] [Google Scholar]
- 66.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 67.Kouhara H, Hadari YR, Spivak-Kroizman T, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89(5):693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 68.Dhalluin C, Yan KS, Plotnikova O, et al. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Mol Cell. 2000;6(4):921–929. doi: 10.1016/s1097-2765(05)00087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ong SH, Guy GR, Hadari YR, et al. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol. 2000;20(3):979–989. doi: 10.1128/mcb.20.3.979-989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ong SH, Hadari YR, Gotoh N, et al. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci USA. 2001;98(11):6074–6079. doi: 10.1073/pnas.111114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hadari YR, Kouhara H, Lax I, et al. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol. 1998;18(7):3966–3973. doi: 10.1128/mcb.18.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dailey L, Ambrosetti D, Mansukhani A, et al. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16(2):233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 73.Bellosta P, Iwahori A, Plotnikov AN, et al. Identification of receptor and heparin binding sites in fibroblast growth factor 4 by structure-based mutagenesis. Mol Cell Biol. 2001;21(17):5946–5957. doi: 10.1128/MCB.21.17.5946-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalinina J, Byron SA, Makarenkova HP, et al. Homodimerization Controls the FGF9 Subfamily’s Receptor Binding and Heparan Sulfate Dependent Diffusion in the Extracellular Matrix. Mol Cell Biol. 2009 doi: 10.1128/MCB.01780-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Osslund TD, Syed R, Singer E, et al. Correlation between the 1. 6 A crystal structure and mutational analysis of keratinocyte growth factor. Protein Sci. 1998;7(8):1681–1690. doi: 10.1002/pro.5560070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Plotnikov AN, Eliseenkova AV, Ibrahimi OA, et al. Crystal structure of fibroblast growth factor 9 reveals regions implicated in dimerization and autoinhibition. J Biol Chem. 2001;276(6):4322–4329. doi: 10.1074/jbc.M006502200. [DOI] [PubMed] [Google Scholar]
- 77.Luo Y, Lu W, Mohamedali KA, et al. The glycine box: a determinant of specificity for fibroblast growth factor. Biochemistry. 1998;37(47):16506–16515. doi: 10.1021/bi9816599. [DOI] [PubMed] [Google Scholar]
- 78.DiGabriele AD, Lax I, Chen DI, et al. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature. 1998;393(6687):812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 79.Faham S, Hileman RE, Fromm JR, et al. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271(5252):1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 80.Zhu X, Hsu BT, Rees DC. Structural studies of the binding of the anti-ulcer drug sucrose octasulfate to acidic fibroblast growth factor. Structure. 1993;1(1):27–34. doi: 10.1016/0969-2126(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 81.Hogan BL, Yingling JM. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev. 1998;8(4):481–486. doi: 10.1016/s0959-437x(98)80121-4. [DOI] [PubMed] [Google Scholar]
- 82.Jin C, Wang F, Wu X, et al. Directionally specific paracrine communication mediated by epithelial FGF9 to stromal FGFR3 in two-compartment premalignant prostate tumors. Cancer Res. 2004;64(13):4555–4562. doi: 10.1158/0008-5472.CAN-03-3752. [DOI] [PubMed] [Google Scholar]
- 83.Pirvola U, Zhang X, Mantela J, et al. Fgf9 signaling regulates inner ear morphogenesis through epithelial-mesenchymal interactions. Dev Biol. 2004;273(2):350–360. doi: 10.1016/j.ydbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 84.Xu X, Weinstein M, Li C, et al. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125(4):753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]
- 85.Zhang X, Stappenbeck TS, White AC, et al. Reciprocal epithelial-mesenchymal FGF signaling is required for cecal development. Development. 2006;133(1):173–180. doi: 10.1242/dev.02175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bellusci S, Grindley J, Emoto H, et al. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124(23):4867–4878. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- 87.Hoffman MP, Kidder BL, Steinberg ZL, et al. Gene expression profiles of mouse submandibular gland development: FGFR1 regulates branching morphogenesis in vitro through BMP- and FGF-dependent mechanisms. Development. 2002;129(24):5767–5778. doi: 10.1242/dev.00172. [DOI] [PubMed] [Google Scholar]
- 88.Izvolsky KI, Shoykhet D, Yang Y, et al. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol. 2003;258(1):185–200. doi: 10.1016/s0012-1606(03)00114-3. [DOI] [PubMed] [Google Scholar]
- 89.Makarenkova HP, Ito M, Govindarajan V, et al. FGF10 is an inducer and Pax6 a competence factor for lacrimal gland development. Development. 2000;127(12):2563–2572. doi: 10.1242/dev.127.12.2563. [DOI] [PubMed] [Google Scholar]
- 90.Sekine K, Ohuchi H, Fujiwara M, et al. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21(1):138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 91.Liu A, Joyner AL. Early anterior/posterior patterning of the midbrain and cerebellum. Annu Rev Neurosci. 2001;24:869–896. doi: 10.1146/annurev.neuro.24.1.869. [DOI] [PubMed] [Google Scholar]
- 92.Sun X, Mariani FV, Martin GR. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature. 2002;418(6897):501–508. doi: 10.1038/nature00902. [DOI] [PubMed] [Google Scholar]
- 93.Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 94.Goetz R, Nakada Y, Hu MC, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shimada T, Muto T, Urakawa I, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143(8):3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- 96.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shiraki-Iida T, Aizawa H, Matsumura Y, et al. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998;424(1–2):6–10. doi: 10.1016/s0014-5793(98)00127-6. [DOI] [PubMed] [Google Scholar]
- 98.Kharitonenkov A, Dunbar JD, Bina HA, et al. FGF-21/FGF-21 receptor interaction and activation is determined by betaKlotho. J Cell Physiol. 2008;215(1):1–7. doi: 10.1002/jcp.21357. [DOI] [PubMed] [Google Scholar]
- 99.Kurosu H, Choi M, Ogawa Y, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282(37):26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin BC, Wang M, Blackmore C, et al. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282(37):27277–27284. doi: 10.1074/jbc.M704244200. [DOI] [PubMed] [Google Scholar]
- 101.Ogawa Y, Kurosu H, Yamamoto M, et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci USA. 2007;104(18):7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suzuki M, Uehara Y, Motomura-Matsuzaka K, et al. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol Endocrinol. 2008;22(4):1006–1014. doi: 10.1210/me.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu X, Ge H, Gupte J, et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J Biol Chem. 2007;282(40):29069–29072. doi: 10.1074/jbc.C700130200. [DOI] [PubMed] [Google Scholar]
- 104.Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 106.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 107.Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 109.Ito S, Fujimori T, Furuya A, et al. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J Clin Invest. 2005;115(8):2202–2208. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275(20):15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 111.Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297(2):F282–291. doi: 10.1152/ajprenal.90742.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bai XY, Miao D, Goltzman D, et al. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem. 2003;278(11):9843–9849. doi: 10.1074/jbc.M210490200. [DOI] [PubMed] [Google Scholar]
- 113.Fukumoto S. Physiological regulation and disorders of phosphate metabolism—pivotal role of fibroblast growth factor 23. Intern Med. 2008;47(5):337–343. doi: 10.2169/internalmedicine.47.0730. [DOI] [PubMed] [Google Scholar]
- 114.Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha 1(I) collagen promoter exhibit growth retardation, osteomalacia and disturbed phosphate homeostasis. Endocrinology. 2004;145(7):3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 115.Liu S, Guo R, Simpson LG, et al. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278(39):37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 116.Riminucci M, Collins MT, Fedarko NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saito H, Kusano K, Kinosaki M, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate cotransport activity and 1 alpha,25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278(4):2206–2211. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 118.Segawa H, Kawakami E, Kaneko I, et al. Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch. 2003;446(5):585–592. doi: 10.1007/s00424-003-1084-1. [DOI] [PubMed] [Google Scholar]
- 119.Lundasen T, Galman C, Angelin B, et al. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260(6):530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 120.Choi M, Moschetta A, Bookout AL, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12(11):1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 121.Nishimura T, Nakatake Y, Konishi M, et al. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492(1):203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 122.Badman MK, Pissios P, Kennedy AR, et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5(6):426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 123.Galman C, Lundasen T, Kharitonenkov A, et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008;8(2):169–174. doi: 10.1016/j.cmet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 124.Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5(6):415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 125.Palou M, Priego T, Sanchez J, et al. Sequential changes in the expression of genes involved in lipid metabolism in adipose tissue and liver in response to fasting. Pflugers Arch. 2008;456(5):825–836. doi: 10.1007/s00424-008-0461-1. [DOI] [PubMed] [Google Scholar]
- 126.Coskun T, Bina HA, Schneider MA, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149(12):6018–27. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 127.Kharitonenkov A, Wroblewski VJ, Koester A, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148(2):774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 128.Wente W, Efanov AM, Brenner M, et al. Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes. 2006;55(9):2470–2478. doi: 10.2337/db05-1435. [DOI] [PubMed] [Google Scholar]
- 129.Kharitonenkov A, Shanafelt AB. Fibroblast growth factor-21 as a therapeutic agent for metabolic diseases. BioDrugs. 2008;22(1):37–44. doi: 10.2165/00063030-200822010-00004. [DOI] [PubMed] [Google Scholar]
- 130.Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 131.Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87(11):4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 132.Araya K, Fukumoto S, Backenroth R, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90(10):5523–5527. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- 133.Benet-Pages A, Orlik P, Strom TM, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14(3):385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 134.Larsson T, Yu X, Davis SI, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90(4):2424–2427. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- 135.Lyles KW, Burkes EJ, Ellis GJ, et al. Genetic transmission of tumoral calcinosis: autosomal dominant with variable clinical expressivity. J Clin Endocrinol Metab. 1985;60(6):1093–1096. doi: 10.1210/jcem-60-6-1093. [DOI] [PubMed] [Google Scholar]
- 136.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Harmer NJ, Pellegrini L, Chirgadze D, et al. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry. 2004;43(3):629–640. doi: 10.1021/bi035320k. [DOI] [PubMed] [Google Scholar]
- 138.Wu X, Ge H, Lemon B, et al. FGF19 induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem. 2009 doi: 10.1074/jbc.M109.068783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhu H, Ramnarayan K, Anchin J, et al. Glu-96 of basic fibroblast growth factor is essential for high affinity receptor binding. Identification by structure-based site-directed mutagenesis. J Biol Chem. 1995;270(37):21869–21874. doi: 10.1074/jbc.270.37.21869. [DOI] [PubMed] [Google Scholar]
- 140.Burmeister WP, Cottaz S, Rollin P, et al. High resolution X-ray crystallography shows that ascorbate is a cofactor for myrosinase and substitutes for the function of the catalytic base. J Biol Chem. 2000;275(50):39385–39393. doi: 10.1074/jbc.M006796200. [DOI] [PubMed] [Google Scholar]