

Summary
The HIV-1 reverse transcriptase (RT) has two associated activities, DNA polymerase and Ribonuclease H (RNase H), both essential for viral replication and validated drug targets. While all RT inhibitors approved for therapy target the DNA polymerase activity, the search of new RT inhibitors which target the RNase H function and are possibly active on RTs resistant to the known non-nucleoside inhibitors (NNRTI) is a viable approach for anti-HIV drug development. In this report several alizarine derivatives were synthesized and tested on both HIV-1 RT-associated activities. Alizarine analogues K-49 and KNA-53 showed IC50 values for both RT-associated functions around 10 μM. When tested on the K103N RT both derivatives equally inhibited the RT-associated functions, while when tested on the Y181C RT, only KNA-53 inhibited the RNase H function but was inactive on the polymerase function. Mechanism of action studies showed that these derivatives do not intercalate into DNA and do not chelate the divalent cofactor Mg2+. Kinetic studies demonstrated that they are non-competitive inhibitors, they do not bind to the RNase H active site or to the classical NNRTI binding pocket, even though efavirenz binding negatively influenced K-49/KNA-53 binding and vice versa. This behavior suggested that the alizarine derivatives binding site could be close to the NNRTI binding pocket. Docking experiments and molecular dynamic simulation confirmed the experimental data and the ability of these compounds to occupy a binding pocket close to the NNRTI site.
Keywords: HIV-1 ribonuclease H, RNase H, anthraquinones, alizarine, NNRTI-resistant RT inhibitors
Introduction
The Acquired Immunodeficiency Disease Syndrome (AIDS) is a pandemic infection whose biological agent is the human immunodeficiency virus type 1 (HIV-1). In the third decade of this pandemic, despite the availability of around twenty antiretroviral drugs already approved for the treatment of HIV-1 infection, current combination regimens still face several challenges [1]. In particular, newer broad spectrum anti-HIV drugs are urgently needed to improve convenience, reduce toxicity, and to provide antiretroviral activity against viral strains resistant to the presently used antiretroviral agents [1].
The HIV-1 reverse transcriptase (RT) is responsible for the conversion of the genomic plus (+) single-stranded viral RNA genome into the proviral double-stranded DNA that is subsequently integrated into the cell host chromosome by the viral coded integrase [2, 3]. The HIV-1 RT is a multifunctional enzyme which has two different, functionally related, catalytic activities: i) a DNA polymerase activity, which can be both RNA- and DNA dependent; ii) a ribonuclease H (RNase H) activity that selectively hydrolyzes the RNA strand of the RNA:DNA hybrid formed during the synthesis of the minus (−) strand DNA that uses (+)-strand RNA as template [2, 3]. The HIV-1 RNase H, similarly to all the RNase Hs and together with transposases, retroviral integrases and RuvC resolvase, belongs to the polynucleotidyl transferase family and catalyzes the phosphoryl transfer through nucleophilic substitution reactions on phosphate esters [4].
Even though both RT associated activities are essential for virus replication and, therefore, both enzyme functions are attractive targets for drug development, all compounds targeting the HIV-1 RT, either approved for treatment or under clinical evaluation, inhibit the RT RNA-dependent DNA polymerase (RDDP) associate activity while none of them inhibit its RNase H associate activity [1, 5, 6]. Hence, the HIV-1 RNase H function is a validated attractive viral target whose inhibition is worth to be pursued.
Anthraquinones (AQs) are common secondary metabolites occurring in bacteria, fungi, lichens, and higher plants where they are found in a large number of families [7]. AQ derivatives have been reported to have diverse biological properties comprising DNA intercalation ability, antitopoisomerase activity and telomerase expression induction [8-10] and are active ingredients of various Chinese traditional medicines [11]. Furthermore, AQs have been reported to have an effect against the encephalomyocarditis virus in mice [12], to inactivate enveloped viruses [13], to inhibit, in cell-based assays, human cytomegalovirus [14, 15], poliovirus [16] and hepatitis B viruses [17] and, in biochemical assays, the HIV-1 RT [18] and integrase activities [19]. In particular, the AQ derivative alizarine has been reported to inhibit the human cytomegalovirus replication [15] and the HIV-1 RT-associated RDDP and integrase activities [18, 19].
Recently, we reported that some derivatives of the AQ emodine inhibit the HIV-1 RT-associated RNase H activity without chelating the Mg2+ ion at the catalytic site [20], which is the proposed mode of action of other RNase H inhibitors such as the diketo acid (DKA) derivatives [6, 21, 22]. Continuing the search of agents that could inhibit the HIV-RT activities with new mechanism of actions, we tested a novel series of AQ derivatives based on the alizarine structure and found that some of them are inhibitors of both RT-associated functions. Mode of action studies demonstrate that these new AQ derivatives are non-competitive inhibitors that bind neither to the RNase H catalytic site, nor to the RT hybrid substrate. Interestingly, most of them were similarly active on the mutant K103N RT-associated functions, while only two analogues were able to inhibit the RNase H activity of the Y181C RT. It was hypothesized that they might bind to a site adjacent to the NNRTI pocket which was originally reported by Himmel and coworkers as the binding site for some hydrazones derivatives [23]. Hence, this binding site was investigated by docking studies and molecular dynamic (MD) simulation leading to the hypothesis that the AQs inhibition of the RNase H function may be due to a change in the RNA:DNA hybrid RT accommodation, induced by the AQs binding to this pocket, which results in a possible variation in the nucleic acid trajectory toward the RNase H catalytic site.
Results and Discussion
Inhibition of HIV-1 RT-associated RNase H activity by alizarine derivatives
We have previously reported that analogues of the AQ emodine inhibited the HIV-1 RT-associated RDDP and RNase H activities [20]. In the effort to better characterize the AQ derivative potentialities and mode of action and, eventually, to increase their potencies, we tested the AQ derivative alizarine which was previously reported to inhibit the HIV-1 RT-associated RDDP function [18] but, at our best knowledge, it was never tested on its RNase H function. Results showed that alizarine inhibited the HIV-1 RT-associated RDDP function with an IC50 value of 79 μM, as it was previously reported [18], but it was inactive on the HIV-1 RT-associated RNase H function (Table 1). With the aim to increase the alizarine potency of RT inhibition, we synthesized and assayed a series of derivatives with different substituents at position 1 and 2 of the AQ ring. As shown in table 1, when an acetophenon group was inserted in position 2 of the AQ ring, compound K-54 slightly inhibited both HIV-1 RT-associated activities. This was in agreement with what we observed for the emodine derivative K-67 which was able to inhibit both enzyme activities [20]. The further introduction of a Br atom in the phenyl ring increased the potency of inhibition of the analogue KNA-53 which showed IC50 values of 21 and 5 μM for the polymerase-independent RNase H and RDDP functions, respectively (Table 1). Interestingly, when the Br atom was substituted with a second phenyl ring, compound K-126 completely lost its inhibitory effect on the RNase H function although retaining the effect on the RDDP function. Finally, when a phenylketo group was inserted in both positions 1 and 2 of the AQ ring the analogue K-49 inhibited both enzyme functions with IC50 values of 12-13 μM.
Table 1. Inhibition of the wt HIV-1 RT-associated activities by AQ derivatives.
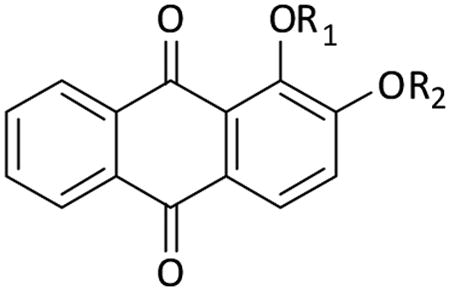
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R1 | R2 | a IC50 (μM) | |
|
| ||||
| RNase H | RDDP | |||
| Alizarine | H | H | > 100 (92%)b | 79 ± 8 |
| K-56 |
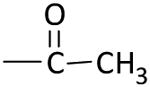
|
|
> 100 (100%) | 82 ± 9 |
| K-57 |
|
|
> 100 (100%) | > 100 (80%) |
| K-45 |
|
|
> 100 (88%) | > 100 (95%) |
| KNA-26 |
|
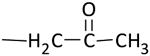
|
> 100 (90%) | > 100 (59%) |
| K-52 |
|
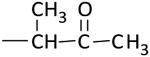
|
> 100 (100%) | 61 ± 8 |
| K-53 |
|
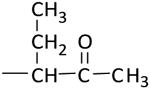
|
> 100 (100%) | > 100 |
| K-54 |
|
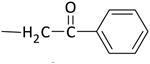
|
39 ± 6 | 60 ± 5 |
| KNA-53 |
|
|
21 ± 5 | 5 ± 2 |
| K-126 |
|
|
100 ± 8 | 6 ± 1 |
| K-61 |
|
|
> 100 (91%) | 14 ± 3 |
| K-111 |
|
|
> 100 (80%) | > 100 (100%) |
| K-49 |
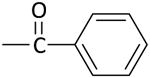
|
|
13 ± 3 | 12 ± 3 |
| EFV | > 10 | 0.003 ± 0.001 | ||
aCompound concentration required to reduce by 50% enzyme activity ± S.D.
bPercentage of enzyme activity at 100 μM compound concentration.
Characterization of the mechanism of HIV-1 RT inhibition by alizarine derivatives
Since it has been reported that the HIV-1 RNase H activity could be influenced by the sequence of the RNA:DNA template utilized in the biochemical assay [24], we also used a different, previously described [22], RNA:DNA hybrid substrate to assess the effect of the alizarine analogues on the HIV-1 RNase H function. Results showed that, also with this substrate, the newly synthesized derivatives inhibited the HIV-1 wt RT-associated polymerase-independent RNase H function with IC50 values comparable to the one shown in Table 1. In particular, compound K-49 inhibited the wt RNase H activity with an IC50 value of 7 μM without affecting the RNase H cleavage pattern (Fig. 1). The DKA derivative RDS1643 was used as positive control and showed an IC50 value of 13 μM [22]. Subsequently, considered that the hydrolysis of the Poly(dC)-Poly(rG) hybrid substrate catalyzed by the HIV-1 RNase H is a processive reaction which can be monitored according to the Michaelis-Menten kinetic assumptions, we determined the inhibition kinetics of the wt RT-associated polymerase-independent RNase H function by K-49. In this system, the Km and kcat values were of 1.5 nM and 0.82 sec−1, respectively, and K-49 resulted a non-competitive inhibitor of the polymerase-independent RNase H activity with a Ki value of 7 μM (Fig. 1). Similar results were also obtained for the KNA-53 analogue (data not shown). In addition, since it has been shown that some AQ derivatives bind non-covalently to dsDNA [9] and given that the reaction substrates used in our biochemical assays were RNA:DNA hybrids, we asked whether the observed enzyme inhibition by the newly synthesized AQ analogues could be due to intercalation into the hybrid substrate. Therefore, as previously described [20], we evaluated the ability of K-49 and KNA-53 analogues to bind to calf thymus DNA in solution and found that they are not able to intercalate into nucleic acids (data not shown). Furthermore, since it has been reported that the DKA derivatives inhibit the HIV-1 RNase H function by chelating the RT metal cofactor [5, 6], in order to verify whether also the alizarine analogues might interact with the metal ions, we measured their visible spectrum in the absence or in the presence of 6 mM MgCl2 observing that the cation addition did not significantly alter the alizarine derivatives maximum absorbance (data not shown).
Fig. 1. Inhibition of the wt HIV-1 RT-associated polymerase-independent RNase H activity by K-49.

a) Inhibition curve of the RNase H function by K-49 using Poly(dC)-[3H]Poly(rG) as reaction substrate, reactions were carried out as described in “Materials and methods”. Data represent mean values from three independent determinations; b) PAGE analysis of the RNase H function inhibition by K-49 using the tC5U/p12 hybrid as substrate. Reactions and PAGE analysis were carried out as described in “Materials and methods”. Four major bands were resolved as reaction products, each of them coming from a single cleavage event of the 32mer substrate. Lane 1 without RT; lane 2 plus RT, lanes 3-8 plus RT and K-49 (100, 33, 11, 3.3, 1.1, 0.33 μM); lanes 9-14 plus RT and RDS 1634 (100, 33, 11, 3.3, 1.1, 0.33 μM); c) Lineweaver-Burk plot of the inhibition of the HIV-1 RNase H activity by K-49. Reactions were performed as described in “Materials and methods”. HIV-1 RT was incubated in the absence (○) or in the presence of 35 μM (+), 10 μM (∇), 20 μM (◊), 40 μM (Δ) of K-49. Inset, replot of the line slopes obtained in the Lineweaver-Burk plot against the K-49 concentration to calculate the Ki value.
Inhibition of HIV-1 K103N and Y181C RT-associated RNase H activity by alizarine derivatives
To date, four NNRTIs (nevirapine, delavirdine, efavirenz, and etravirine) have been approved for clinical use in combination with other antiviral agents [1]. It is well known that treatment with NNRTI selects for HIV drug resistant strains mutated in RT. In particular, the mutations K103N and Y181C in the RT are the most worrying, as they lead to resistance to many different NNRTIs as a result of overlapping resistance profiles [1]. In fact, new antiviral agents that may inhibit HIV-1 strains mutated in these residues are actively pursued [1]. Therefore, in order to assess the effect of the AQ analogues on the mutant enzymes, all the compounds even weakly active at least on one HIV-1 wild type (wt) RT-associated function were tested on both enzyme activities of the K103N and Y181C RTs (Table 2). Interestingly, when tested on the K103N RT, the alizarine derivatives mainly showed potencies of inhibition similar to the ones shown on the wt RT with three exceptions: i) the K-54 analogue completely lost its ability to inhibit the polymerase-independent RNase H activity, but retained its effect on the RDDP activity; ii) the K-126 analogue, which was slightly active on the wt RT RNase H function, increased by 6-fold its potency of inhibition of the RNase H function while it retained its potency of inhibition on the polymerase function; iii) the K-61 analogue showed a 4-fold reduction of its RDDP activity potency of inhibition. Differently, when the AQ derivatives were tested on the Y181C RT, results showed that only KNA-53 and K-126 analogues retained their ability to inhibit the RT-associated RNase H function with the same IC50 values observed for the K103N RT, while all the other compounds were inactive (Table 2).
Table 2. Inhibition of the mutant HIV-1 RT-associated activities and wt HIV-1 replication by AQ derivatives.
| Compd | a IC50 (μM) | b EC50 (μM) | c CC50 (μM) | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| wt RT | K103N RT | Y181C RT | HIV-1 | MT2 | ||||
|
|
||||||||
| RNase H | RDDP | RNase H | RDDP | RNase H | RDDP | |||
| Alizarine | > 100 (92%) d | 79 ± 8 | > 100 (58%) | 68 ± 5 | > 100 (100%) | > 100 (100%) | NDe | ND |
| K-56 | > 100 (100%) | 82 ± 9 | > 100 (100%) | 100 ± 8 | > 100 (100%) | > 100 (100%) | ND | ND |
| K-52 | > 100 (100%) | 61 ± 8 | > 100 (100%) | 100 ± 9 | > 100 (100%) | > 100 (100%) | ND | ND |
| K-54 | 39 ± 6 | 60 ± 5 | >100 (98%) | 35± 4 | >100 (86%) | >100 (74%) | >100 | ~ 40 |
| KNA-53 | 21 ± 5 | 5 ± 2 | 21 ± 4 | 9 ± 3 | 22 ± 4 | > 100 (92%) | >100 | >100 |
| K-126 | 100 ± 8 | 6 ± 1 | 16 ± 3 | 9 ± 2 | 16 ± 2 | > 100 (100%) | ND | ND |
| K-61 | > 100 (91%) | 14 ± 3 | > 100 (100%) | 64 ± 8 | > 100 (100%) | > 100 (72%) | ND | ND |
| K-49 | 13 ± 3 | 12 ± 3 | 16 ± 4 | 28 ± 4 | >100 (79%) | >100 (87%) | >100 | ~ 100 |
| EFV | > 10 | 0.003 ± 0.001 | ND | 0.68 ± 0.2 | ND | 0.40 ± 0.1 | ||
Compound concentration required to reduce by 50% enzyme activity ± S.D.
Compound concentration required to reduce the HIV-1 induced cytopathic effect in MT-2 cells by 50%.
Compound concentration required to reduce the MT-2 cell multiplication by 50%.
Percentage of enzyme activity at 100 μM compound concentration.
Not done
Interaction of alizarine derivatives and the DKA RDS1643 on the HIV-1 RNase H activity
It has been proposed that the DKA derivatives chelate the RT metal cofactor in the active site [5, 6]. Hence, in order to ascertain whether the alizarine derivatives could interact with the HIV-1 RT RNase H active site, even without chelating the cofactor metal ion, we determined the effect of the interaction between the AQ analogue K-49 and the DKA analogue RDS1643 on the HIV-1 RT-associated polymerase-independent RNase H activity by using the Yonetani-Theorell model, as revised by Yonetani [25]. This graphical method allows to determine whether two inhibitors of a certain enzyme compete for the same binding site or act on two non overlapping binding sites and it was already used to dissect the effect of the interaction between RNase H and RDDP inhibitors [20, 26]. In this revised model, the plot of the reaction velocity reverse (1/v) observed in the presence of different concentrations of the first inhibitor, in the absence or in the contemporaneous presence of the second inhibitor, leads to a series of lines which are parallel if the two inhibitors compete for the same binding site, whereas they intersect if the inhibitors bind to different enzyme sites [25]. Therefore, the HIV-1 RT RNase H activity was measured in the presence of increasing concentrations of both K-49 and RDS1643 and analyzed with the Yonetani-Theorell plot (Fig. 2). Results showed that both slope and intercepts of the plots of 1/v versus K-49 concentration increased as linear function of RDS1643, indicating that the two compounds do not bind to overlapping sites.
Fig. 2. Yonetani-theorell plot of the interaction between AQ derivatives and other RT inhibitors.

a) Yonetani-Theorell plot of the combination of K-49 and RDS 1643 on the HIV-1 RT polymerase-independent RNase H activity. HIV-1 RT was incubated in the presence of different concentrations of K-49 and in the absence (●) or in the presence of 3 μM (▼) or 10 μM (▲) of RDS1643; b) Yonetani-theorell plot of the interaction of K-49 and efavirenz on the HIV-1 RT RDDP activity. HIV-1 RT was incubated in the presence of different concentrations of efavirenz and in the absence (●) or in the presence of 1.9 μM (○), 3.7 μM (▼), 7.5 μM (Δ) of K-49; c) Yonetani-theorell plot of the interaction of KNA-53 and efavirenz on the HIV-1 RT RDDP activity. HIV-1 RT was incubated in the presence of different concentrations of efavirenz and in the absence (●) or in the presence of 1 μM (▼), 2 μM (Δ), 4 μM (■) of KNA-53. Reactions were performed as described in “Materials and methods”.
In order to further investigate the possibility that the AQ derivatives could bind to the RNase H active site, the ability of K-49 and KNA-53 to inhibit the enzyme activity of the isolated RNase H domain (p15) was assessed [27]. In this system, the AQ derivatives were not able to inhibit the RNA degradation (data not shown).
Furthermore, since the RNase H domain contains one tryptophan and six tyrosine residues as intrinsic fluorophores, it has been reported that when the p15 domain is excitated at the wavelength of 290 nm the contribution of tryptophan to the fluorescence signal is maximized while the fluorescence energy transfer from the tyrosine residues to the tryptophan residue is minimized [27]. Therefore, compounds interacting with the HIV-1 RT-associated RNase H active site are able to quench the intrinsic protein fluorescence of the isolated HIV RNase H domain [27]. In fact, as described [27], the 2-hydroxy-1,2,3,4-tetrahydroisoquinoline-1,3-dione, used as positive control, was able to reduce the p15 intrinsic fluorescence with an IC50 of 52 μM, while only a small reduction (less than 30%) in the p15 intrinsic fluorescence was observed in the presence of the highest (100 μM) K-49 or KNA-53 concentration (data not shown). Overall, these data support the hypothesis that AQ derivatives do not inhibit the RT catalysis by primarily binding to the RNase H active site.
Interaction of alizarine derivatives with the NNRTI efavirenz on the HIV-1 RDDP activity
As the NNRTI-binding site is at a close spatial distance from the substrate (dNTP)-binding site, the NNRTIs have been shown to interfere with the polymerase catalytic site impeding the normal RDDP performance. Within the NNRTI-binding site, the amino acid residues lysine (Lys103) and tyrosine (Tyr181) interact with many NNRTIs. The observation that the AQ analogues K-49 and KNA-53 inhibited both wt and K103N RT-associated RDDP function while they were inactive on the Y181C RT-associated RDDP function raised the question whether they could actually bind to the NNRTI-binding site, possibly with low affinity. In order to answer to this question we measured the effect of the interaction between K-49, or KNA-53, and the NNRTI efavirenz on the wt RT RDDP activity by using the Yonetani-Theorell plot [25]. Results showed that when the HIV-1 RT RDDP activity was measured in the presence of increasing concentrations of one of the two AQ derivatives and efavirenz, and analyzed with the Yonetani-Theorell plot, the slope and intercepts of the two plots of 1/v versus efavirenz concentration increased as linear function of the AQ derivatives concentrations, indicating that the AQ analogues and the NNRTI efavirenz do not bind to overlapping sites (Fig. 2). However, it is worth to note that the Yonetani-Theorell plot allows also to calculate an interaction constant between the two tested inhibitors termed α [25]. When the two compounds bind to the same site, α is equal to ∞; when the two compounds are strictly independent, α is equal to 1; while when the two compounds interact repulsively in the enzyme-two inhibitors complex, the α value is > 1. Hence, when we calculated the α value for the K-49/efavirenz and KNA-53/efavirenz interactions we found that the α values were 3.5 and 3.0, respectively, indicating that the binding of an AQ derivative results in a reduction of efavirenz binding and, vice versa, the binding of efavirenz leads to a reduction of the AQ binding.
Docking studies
These results demonstrated that the AQs and NNRTI binding sites are strictly functionally related. It has been reported that some hydrazone derivatives which can inhibit both RT-associated functions bind to a site near to the NNRTI binding pocket [23], therefore we wanted to verify whether also the AQ derivatives could bind to this site. To this purpose, and to obtain a deeper understanding of the RT–ligand interactions, QM polarized (QMPL) docking [28] has been carried out. QMPL docking workflow combines docking with ab initio for ligand charges calculation within the protein environment. This methodology has showed to perform significantly better than docking alone enabling the modeling of biomolecular systems at a reasonable computational effort while providing the necessary accuracy [29]. Blind docking experiment put in evidence the existence of five possible binding areas that are shown in Figure 3. However, from energetic analysis it appears that only two of them are the most favorable: one located close to the RNase H catalytic cavity and the other close to the NNRTI binding pocket. Since experimental data deriving from testing the compounds on the isolated RNase H portion seem to suggest that the first binding pocket is not primarily responsible for AQ activity, more detailed analysis of K-49, KNA-53, K-54 and K-126 putative binding mode was carried out, in both wt and mutants RTs, exploring the binding site for RNase H inhibitors described by Himmel as second binding site [23] (Fig. 4). The analysis of the best poses of K-49/wt RT highlighted that the planar rings system guarantees a significant influence of the π–π stacking interaction with Trp229 on the orientation of the ligand in the binding site (Fig. 5a). The interactions between the benzoic moiety and the hydrophobic residues in the NNRTI pocket (Leu100, Val106, Tyr188, Phe227, Leu234 and Tyr318) allowed a sterically favorable allocation of this bulky portion inside the pocket. These contacts appear to be essential for the RDDP inhibition activity. In the case of KNA-53, the bulkier moiety 1-(4-bromophenyl)-2-oxyethanone does not allow the compound to enter further in the cavity (Fig. 5b). Several residues in the NNRTI and in the second binding pocket are involved in hydrophobic contacts that stabilize the complex.
Fig. 3. Representation of the overall structure of the HIV-1 RT heterodimer with binding areas of alizarine derivatives found after blind docking experiment.

The most favorable binding sites are highlighted in blue.
Fig. 4. Schematic representation of the overall structure of the HIV-1 RT heterodimer.

The p66 subunit (top) is displayed in colored scheme for the individual sub-domains, whereas the p51 subunit is shown with the same color. The surface represents the position of the new binding pocket for RNase H inhibitors. Close-up of the binding cavity colored according to lipophilicity: light blue for hydrophilic and pale yellow for hydrophobic residues.
Fig. 5. K-49 and KNA-53 (in sticks) best docked pose.
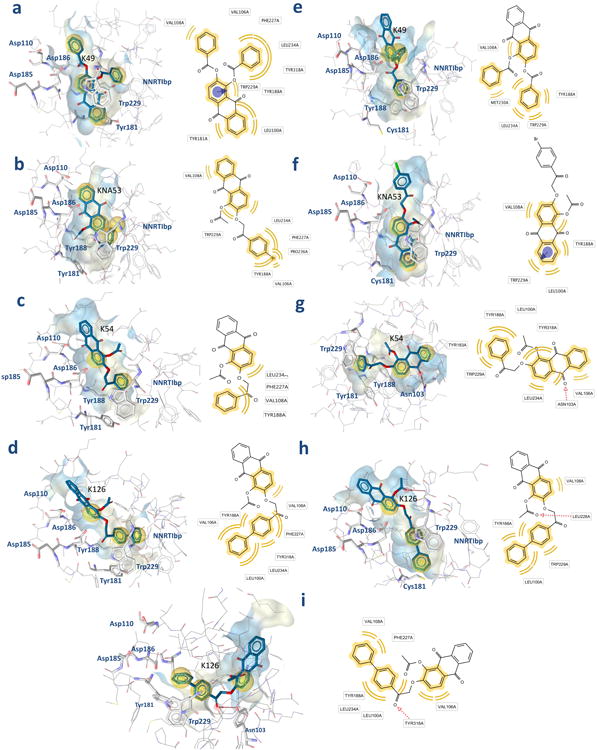
Compounds interactions with the RT were analyzed with Ligandscout: yellow spheres show the hydrophobic contacts. Binding pocket surfaces are drawn as solid and color in accordance to lipophilicity: pale yellow indicates lipophilic and light blue hydrophilic residues. a-b-c-d) K-49, KNA-53, K-54, K-126 binding mode into wt RT and respective 2D depiction of interactions; e-f-h) K-49, KNA-53, K-126 binding mode into Y181C RT and respective 2D depiction of interactions; g-i) K-54 and K-126 binding mode into K103N RT and respective 2D depiction of interactions.
While the Lys103 mutation in Asn did not produce any effect on the RNase H inhibition by K-49 and KNA-53, confirming that their binding does not involve this portion of the NNRTI pocket, the Tyr181 mutation in Cys led to total loss of activity for K-49. In order to better explain this observation, we studied the behavior of the complex K-49/wt RT in aqueous environment running 5ns of MD simulation with Desmond [30] keeping the whole enzyme free of move into explicit solvent. During QMPL docking the receptor is treated as being rigid, therefore the phenomena of induced fit cannot be observed. The analysis of the trajectory highlighted that Tyr181 rotated to better accommodate the ligand and interacted with the lower phenyl ring adding an important π–π stacking (Fig. 6). Plots for potential energy and RMSD fluctuations involving the complex are depicted in Figs. 6c-d, the analysis shows that the structure reached the equilibrium and the low fluctuations support the stability of the intermolecular interactions. When Tyr181 is mutated in Cys electronic and sterical modifications occur. The binding mode of K-49 in the Y181C RT is different, in particular the contribution of π–π stacking interaction is lost. Furthermore, the low number of good contacts with RT leads to instability of the complex and the compound can then be easily washed off the substrate cavity or displaced (Fig. 5e). Differently, from a deep insight of the best KNA-53 docked pose into the Y181C RT (Fig. 5f), we could observe that the enlarged cavity allowed KNA-53 to go deeper and, at the same time, the bulky substituent in position 2 did not interact with many NNRTI pocket residues leading to the loss of RDDP activity while the RNase H activity was retained. Other two compounds exhibited a significant difference in their RNase H inhibition effects on mutant RTs: K-54 and K-126. In particular, K-54 was characterized by loss of activity versus the RNase H function and increased inhibition of the RDDP function in K103N RT. This behavior suggests that K-54 can bind NNRTI pocket with higher affinity when this mutation occurs. It is known that the NNRTI pocket is high flexible and shows induced fit during NNRTIi binding [31], therefore, we excluded that our compounds can adopt a “butterfly like” geometry as many NNRTI (e. g nevirapine and efavirenz) [32] by docking simulation (data not shown). Differently, on one side, as previously shown by Himmel [23], RT/DHBNH and RT/CP-94,707 complexes have similar conformation (root mean square (RMS 0,57)), on the other side, in the crystal reported by Pata and coworkers the NNRTI binding pocket most closely resembles the RT unliganded conformation [33]. Therefore, the AQ derivatives were docked into the K103N RT in this conformation. Under these conditions, we observed that K-54 not only could enter into the NNRTI pocket but was also stabilized by hydrogen bond interaction with Asn103. Furthermore, the HB between Tyr188 and Asn103 keeps the position of the Tyr residue in a conformation which allows its better fit (Fig. 5g). As highlighted for CP-94,707, even if this bond needs to be broken before ligand entrance, it could be reformed immediately after as the compound do not interfere with it [33]. Thus, this could explain the preference of this binding mode when in K103N RT. In wt RT, the steric and electrostatic effects of the Lys hamper the formation of the same interactions of this compound and the binding showed in fig 5c is therefore favorite.
Fig. 6.

Superimposed structures of 5ns MD simulations frames of K49-RT complex colored by timestep: initial (red), final (blue) along with intermediate structures snapshots. a) overall structure of the HIV-1 RT heterodimer; b) Close-up of the binding cavity. c) RMSD fluctuations of complex during 5ns trajectory; d) Potential energy of the complex during the MD simulation.
Finally, when analyzing the K-126 docking results we could observe that, due to the bulkiness of its diphenyl substituent, in the Y181C RT the bigger binding pocket can better host the compound and a higher RNase H inhibition is then observed (Fig 5h). Furthermore, also in the case of the K103N RT, for the same reasons arisen above for K-54, the K-126 can enter in the pocket and act allosterically (Fig5i).
Conclusions
Targeting new RT drug binding pockets that may inhibit one or even both RT-associated enzymatic functions is an attractive approach to allosterically inhibit the HIV-1 reverse transcription process. We have identified a new series of AQ derivatives which inhibit both HIV-1 RT-associated functions in the micromolar range in biochemical assays, even though they are not able to inhibit the viral replication in cell culture, possibly due to a lack of cell membrane penetration (Table 2 and data not shown). Beside this, however, some AQ derivatives showed a unique profile of RT inhibition resulting able to inhibit the RT-associated RNase H function also of mutant K103N and Y181C RTs. Experimental results and modeling simulations induced us to suppose that the binding pocket lying between the polymerase catalytic triad and NNRTI pocket [23] may be involved in the AQs binding to RT and in their ability to inhibit both RT-associated RDDP and RNase H activities. This conclusion is also in agreement with the very recent demonstration, obtained by X-ray crystallography, that a naphthyridinone derivative which is able to inhibit the HIV-1 RT-associated RNase H function binds to the same pocket adjacent to the NNRTI site [34]. However, given that the blind docking experiments indicated the existence of five possible binding pockets, we can not completely exclude the possibility that the AQ derivatives may additionally bind to other RT pockets and that the binding stoichiometry of the compounds to RT could also be different according to the RT mutations and the relative compound-RT affinities.
The position of the proposed major AQs binding site raises the question of the distance between this RNase H inhibitor binding site and the RNase H catalytic site. In this respect, it is worth noting that it has been previously reported that the NNRTIs binding to RT lead to an increase of the RNase H activity and, in some cases, to an alteration of the nucleic acid cleavage pattern [26, 35]. Furthermore, mutations in the primer grip, which are essential for nucleic acid binding in either the polymerase domain [36] or in the RNase H domain [35-39], alter the RNase H cleavage position of the RNA:DNA hybrid. In addition, a subset of polymerase domain primer grip residues (Phe227, Trp229, Leu234, and His235) also line the NNRTI-binding pocket, while the mutant Y181C RT mutations showed an altered RNase H cleavage kinetics [40, 41].
All these observations, together with our results, led us to speculate that the AQ binding to RT may induce a variation of the RNA:DNA hybrid trajectory toward the RNase H catalytic site. According to this mechanism, the AQs inhibit the RT-associated RNase H by avoiding the correct anchorage of the primer grip to the nucleic acid, while they inhibit the RT-associated RDDP function due to a deep occupancy of the NNRTI binding pocket and hydrophobic contacts with the residues in this cavity. Further studies, providing deeper understanding of the AQ-RT interactions, will allow to confirm this hypothesis and to develop more potent RT inhibitors with new mode of actions.
Materials and Methods
Materials
His-binding resin were obtained from GE Healthcare; [γ32P]-ATP and [3H]-dGTP were purchased by PerkinElmer; G-25 Sephadex quick spin column and T4 polynucleotide kinase were from Roche. The p12 DNA oligonucleotide (5′-GTCTTTCTGCTC-3′), the tC5U RNA oligonucleotide (5′-CCCCCUCUCAAAAACAGGAGCAGAAAGACAAG-3′), and the 12mer DNA oligonucleotide oligo(G)12 were purchased by Operon. All buffer components and the other materials were obtained from Sigma-Aldrich.
HIV-1 RTs mutagenesis and purifications
HIV-1 wt RT was mutated into K103N RT and Y181C RT using the Stratagene mutagenesis kit. Heterodimeric RT was expressed essentially as previously described [20].
RT assays
For the RNase H polymerase-independent cleavage assay, when the Poly(dC)- [3H[Poly(rG) hybrid was used as reaction substrate the RNase H activity was measured as previously described [22]. When the tC5U/p12 hybrid was used as reaction substrate the RNase H activity was measured as described [20, 42]. The RDDP activity of HIV-1 RT was measured as previously described [43].
Kinetic Studies
The analysis of the kinetic of inhibition was performed according to Lineaweaver-Burke plots; v was expressed as fmoles/min, Ki was calculated by replotting the intercept values versus the inhibitor concentration using the Sigmaplot 9.0 software. The Yonetani-Theorell graphical method was performed as described [25].
HIV-1 Replication Assay
Drug-mediated inhibition of virus-induced cytotoxicity was assayed in MT-2 cells as described [44] with minor modifications [20].
Docking
The ligand structures and the corresponding receptor protein structures were prepared using utilities provided in the Schrödinger Suite [45].
Ligand preparation
The ligands were built within the Maestro platform, the geometry was optimized with MacroModel using the MMFFs force field, the GB/SA solvation model, and the Polak-Ribier Coniugate Gradient (PRCG) method converging on gradient with a threshold of 0.05kJ/(molÅ).
Protein preparation
Starting crystal coordinates of the complex RT-RNase inhibitor were downloaded from the Protein Data Bank (http://www.rcsb.org/) pdb accession code 2i5j [23]. Afterward, the protein was then prepared using the Schrödinger protein preparation wizard. Hydrogen atoms were added to the system. Partial atomic charges were assigned according to the OPLS-2005 force field. A minimization was performed to optimize hydrogen atoms and to remove any high energy contacts or distorted bonds, angles, and dihedrals. The compounds were docked with QM-polarized ligand docking protocol utilizing Glide version 4.5, QSite version 4.5, Jaguar version 7.0, and Maestro version 8.5 [28]. The QPLD workflow consists of three steps: in the first one protein–ligand complex is generated with Glide (Grid Based Ligand Docking with Energetics). The receptor van der Waals radii was scaled to 0.9 in order to avoid overemphasizing of steric repulsive interactions might otherwise be overemphasized, leading to rejection of overall correct binding modes of compounds. The enzyme was divided in 5 boxes of the same size (50Å × 50Å × 50Å) covering overall the whole enzyme and the compounds were docked into each of them. Glide uses hierarchical filters to explore plausible docking poses for a given ligand within the receptor site. It examine the complementarities of ligand–receptor interactions using a grid-based method. Conformational flexibility is handled by an extensive conformational search, improved by a heuristic screen that eliminates unsuitable conformations. Poses passed through these initial screens enter the final stage, which involves evaluation and minimization of a grid approximation to the OPLS-AA non-bonded ligand–receptor interaction energy. Final scoring is then carried out on the energy-minimized poses. Finally, the minimized poses are rescored using Schrödinger's proprietary GlideScore scoring function. In the second step, a mixed quantum mechanical/molecular mechanics (QM/MM) method is used to compute the ligand charge distribution. For QM/MM calculations the Qsite program is used. The protein is defined as the MM region, and the ligand is defined as the QM region. Polarizable ligand charges were determined at 6-31G*/LACVP* basis sets with the B3LYP density functional and Ultrafine SCF accuracy level. In the third step, the ligands are submitted to another Glide docking run where the ligand charges are substituted with the new charge sets calculated in the second step. The extra-precision mode of Glide, which has a higher penalty for unphysical interactions, was used for both first and third step [46]. For each ligand Glide was allowed to return up to 10 most energetically favorable poses.
MD simulation
The best scored pose of complex K-49/RT was used as starting point for 5ns MD simulation in Desmond [47] keeping everything free of move into aqueous solvent. The complex was solvated with an orthorhombic box with a buffer of 10 Å TIP3P water [48] and counter ions were added to neutralize the system net charge. The solvated models were relaxed and minimized, and subsequently the Martyna-Tobias-Klein isobaric-isothermal ensemble (MTK_NPT) was employed. The default stages in the relaxation process for the NPT ensemble comprehend two minimizations, and four simulation steps. During the minimizations, two runs of 2000 steps were processed using the steepest descent method: during the first run, the protein structure was fixed by a force restraint constant of 50 kcal/(molÅ) and in the second all restraints were released. With the first simulation, in the volume and temperature constant (NVT) ensemble, the system reached a temperature of 10 K, while in the following three simulations in the NPT ensemble, the system was heated up to 300 K and the pressure was kept constant at 1 bar using the Berendsen thermostat–barostat. During the production phase, temperature and pressure were kept constant using the Nosè-Hoover thermostat-barostat. The energy and trajectory were recorded every 4.8 ps and every 10.2 ps, respectively. For multiple time step integration, RESPA [49] was applied to integrate the equation of motion with Fourier-space electrostatics computed every 6 fs, and all remaining interactions computed every 2 fs. All chemical bond lengths involving hydrogen atoms were fixed with SHAKE [50]. Short range cut-off was set to 9 Å and the smooth particle mesh Ewald method (PME) [51] was used for long range electrostatic interaction. The analysis of the trajectories was performed with Desmond simulation analysis event and VMD [52].
Y181C and K103N mutant enzymes
The wt RT residue 181 was mutated to Cys and the 103 residue in Asn. Then, the enzymes were minimized using OPLS 2005 force field, the GB/SA solvation model, and the PRCG method converging on gradient with a threshold of 0.05kJ/(molÅ) allowing maximum 10000 iterations.
Visualization of molecular modeling results
3D models of docking and MD simulation results for visualization were created using the VMD and LigandScout software [53].
Acknowledgments
This work was supported by Fondazione Banco di Sardegna and by NIAID, NIH, grant n. AI-38204. Yung-Chi Cheng is a fellow of the National Foundation for Cancer Research while Tatyana Kharlamova was visiting professor at the University of Cagliari. The wt P6HRT-prot and p15 plasmids was kindly provided by Dr. S. Le Grice (NCI at Frederick). We thank Elias Macaronis and Stefano Alcaro for helpful discussions, Vito Lipppolis and Claudia Caltagirone for assisting in the fluorescent studies. Francesca Esposito and Luca Zinzula were supported by RAS fellowships, co-financed with funds of PO Sardinia FSE 2007-2013 and of LR 7/2007, projects CRP2_683 and CRP2_682, respectively.
Abbreviations
- AIDS
acquired immunodeficiency disease syndrome
- AQ
Anthraquinone
- DKA
diketo acid
- HIV-1
human immunodeficiency virus type 1
- MD
molecular dynamic
- NNRTI
non-nucleoside reverse transcriptase inhibitors
- QM/MM
quantum mechanical/molecular mechanics
- QMPL
quantum mechanical polarized
- RDDP
RNA-dependent DNA polymerase
- RNase H
ribonuclease H
- RT
reverse transcriptase
References
- 1.Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 2.Hughes SH, Arnold E, Hostomsky Z. RNase H of Retroviral Reverse Transcriptases. In: Crouch RJ, Toulme JJ, editors. Ribonucleases H. Les. INSERM; Paris: 1998. pp. 195–224. [Google Scholar]
- 3.Klarmann GJ, Hawkins ME, Le Grice SF. Uncovering the complexities of retroviral ribonuclease H reveals its potential as a therapeutic target. AIDS Rev. 2002;4:183–194. [PubMed] [Google Scholar]
- 4.Rice PA, Baker TA. Comparative architecture of transposase and integrase complexes. Nature Struct Biol. 2001;8:302–307. doi: 10.1038/86166. [DOI] [PubMed] [Google Scholar]
- 5.Tramontano E. HIV-1 RNase H: recent progress in an exciting, yet little explored, drug target. Mini Rev Med Chem. 2006;6:727–737. doi: 10.2174/138955706777435733. [DOI] [PubMed] [Google Scholar]
- 6.Tramontano E, Di Santo R. HIV-1 RT-associated RNase H function inhibitors: recent advances in drug development. Curr Med Chem. 2010;17:2837–2853. doi: 10.2174/092986710792065045. [DOI] [PubMed] [Google Scholar]
- 7.Singh R, Geetanjali R, Chauhan SMS. 9,10-Anthraquinones and other biologically active compounds from the Genus Rubia. Chem Biodiversity. 2004;1:1241–1264. doi: 10.1002/cbdv.200490088. [DOI] [PubMed] [Google Scholar]
- 8.Huang HS, Chiou JF, Fong Y, Hou CC, Lu YC, Wang JY, Shih JW, Pan YR, Lin JJ. Activation of human telomerase reverse transcriptase expression by some new symmetrical bis-substituted derivatives of the anthraquinone. J Med Chem. 2003;46:3300–3307. doi: 10.1021/jm020492l. [DOI] [PubMed] [Google Scholar]
- 9.Tan JH, Zhang QX, Huang ZS, Chen Y, Wang XD, Gu LQ, Wu JY. Synthesis, DNA binding and cytotoxicity of new pyrazole emodin derivatives. Eur J Med Chem. 2006;41:1041–1047. doi: 10.1016/j.ejmech.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrivch-Wright JR. DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev Med Chem. 2007;6:627–48. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]
- 11.Huang Q, Lu G, Shen HM, Chung MC, Onq CN. Anticancer properties of anthraquinones from Rhubarb. Med Res Rev. 2007;27:609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- 12.Sill AD, Andrews ER, Sweet FW, Hoffman JW, Tiernan PL, Grisar JM, Fleming RW, Mayer GD. Bis-basic-substituted polycyclic aromatic compounds. A new class of antiviral agents. Bis-basic ethers of anthraquinone and bisalkamine esters of anthraquinonedicarboxylic acids. J Med Chem. 1974;17:965–968. doi: 10.1021/jm00255a011. [DOI] [PubMed] [Google Scholar]
- 13.Sydiskis RJ, Owen DG, Lohr JL, Rosler KHA, Blomster RN. Inactivation of enveloped viruses by anthraquinones extracted from plants. Antimicrob Agents Chemother. 1991;35:2463–2466. doi: 10.1128/aac.35.12.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnard DL, Huffman JH, Morris JLB, Wood SG, Hughes BG, Sidwell RW. Evaluation of antiviral activity of anthraquinones, anthrones and anthraquinone derivatives against human cyromegalovirus. Antiv Res. 1992;17:63–77. doi: 10.1016/0166-3542(92)90091-i. [DOI] [PubMed] [Google Scholar]
- 15.Bamard DL, Fairbaim DW, O'Neill KL, Gage TL, Sidwell RW. Anti-human cytomegalovirus activity and toxicity of sulfonated anthraquinones and anthraquinone derivatives. Antiv Res. 1995;28:317–329. doi: 10.1016/0166-3542(95)00057-7. [DOI] [PubMed] [Google Scholar]
- 16.Semple SJ, Pyke SM, Reynolds GD, Flower FLP. In vitro antiviral activity of the anthraquinone chrysophanic acid against poliovirus. Antiv Res. 2001;49:169–178. doi: 10.1016/s0166-3542(01)00125-5. [DOI] [PubMed] [Google Scholar]
- 17.Dang S, Zhang Z, Chen Y, Zhang X, Wang B, Yuan L, Cheng Y. Inhibition of the replication of hepatitis B virus in vitro by emodin. Med Sci Monit. 2006;12:302–306. [PubMed] [Google Scholar]
- 18.Higuchi H, Mori K, Kato A, Ohkuma T, Endo T, Kaji H, Kaji A. Antiretroviral activities of anthraquinones and their inhibitory effects on reverse transcriptase. Antiv Res. 1991;15:205–216. doi: 10.1016/0166-3542(91)90067-2. [DOI] [PubMed] [Google Scholar]
- 19.Farnet CM, Wang B, Lipford JR, Bushman FD. Differential inhibition of HIV-1 preintegration complexes and purified integrase protein by small molecules. Proc Natl Acad Sci USA. 1996;93:9742–9747. doi: 10.1073/pnas.93.18.9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kharlamova T, Esposito F, Zinzula L, Floris G, Cheng YC, Dutschman GE, Tramontano E. Inhibition of HIV-1 ribonuclease H activity by novel frangula-emodine derivatives. Med Chem. 2009;5:398–410. doi: 10.2174/157340609789117840. [DOI] [PubMed] [Google Scholar]
- 21.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen S, Olsen D, Carroll S, Embrey E, Wai J, Miller M, Cole J. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J Biol Chem. 2003;278:2777–2780. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- 22.Tramontano E, Esposito F, Badas R, Di Santo R, Costi R, La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005;65:117–124. doi: 10.1016/j.antiviral.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold A. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol. 2006;1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hang QL, Li Y, Yang Y, Cammack N, Mirzadegan T, Klumpp K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs) Biochem Biophys Res Comm. 2007;352:341–350. doi: 10.1016/j.bbrc.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 25.Yonetani T. The Yonetani-Theorell graphical method for examining overlapping subsite of enzyme centers. Methods Enzymol. 1982;87:500–509. doi: 10.1016/s0076-6879(82)87028-6. [DOI] [PubMed] [Google Scholar]
- 26.Shaw-Reid CA, Feuston B, Munshi V, Getty K, Jrueger J, Hazuda DJ, Parniak MA, Miller MD, Lewis D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry. 2005;44:1595–1606. doi: 10.1021/bi0486740. [DOI] [PubMed] [Google Scholar]
- 27.Hang JQ, Rajendran S, Yang Y, Li Y, In PWK, Overton H, Parkes KEB, Cammack N, Martin JA, Klumpp Activity of the isolated HIV RNase H domain and specific inhibition by N-hydrosymides. Biochem Biophys Res Comm. 2004;317:321–329. doi: 10.1016/j.bbrc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 28.QM-Polarized Ligand Docking. Schrödinger Inc; Portland, OR: 2006. [Google Scholar]
- 29.Chung JY, Hah JM, Cho AE. Correlation between Performance of QM/MM Docking and Simple Classification of Binding Sites. J Chem Inf Model. 2009;49:2382–2387. doi: 10.1021/ci900231p. [DOI] [PubMed] [Google Scholar]
- 30.Desmond Molecular Dynamics System 2.0. Shaw Research, D. E.; New York, NY: 2008. [Google Scholar]
- 31.Hang JQ, Li Y, Yang Y, Cammack N, Mirzadegan T, Klumpp K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside riverse transcriptase inhibitors (NNRTIs) Biochem Biophys Res Commun. 2007;352:341–50. doi: 10.1016/j.bbrc.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Sluis-Cremer N, Temiz NA, Bahar I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr HIV Res. 2004;2:323–32. doi: 10.2174/1570162043351093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pata JD, Stirtan WG, Goldstein SW, Steitz TA. Structure of HIV-1 reverse transcriptase bound to an inhibitor active against mutant reverse transcriptases resistant to other nonnucleoside inhibitors. Proc Natl Acad Sci USA. 2004;101:10548–53. doi: 10.1073/pnas.0404151101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.So HP, Yan Y, Prasad G, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol. 2010;84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palaniappan C, Fay PJ, Bambara RA. Nevirapine alters the cleavage specificity of ribonuclease H of human immunodeficiency virus 1 reverse transcriptase. J Biol Chem. 1995;270:4861–4869. doi: 10.1074/jbc.270.9.4861. [DOI] [PubMed] [Google Scholar]
- 36.Palaniappan C, Wisniewski M, Jacques PS, Le Grice SF, Fay PJ, Bambara RA. Mutations within the primer grip region of HIV-1 reverse transcriptase result in loss of RNase H function. J Biol Chem. 1997;272:11157–11164. doi: 10.1074/jbc.272.17.11157. [DOI] [PubMed] [Google Scholar]
- 37.Rausch JW, Lener D, Miller JT, Julias JG, Hughes SH, Le Grice SF. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry. 2002;41:4856–4865. doi: 10.1021/bi015970t. [DOI] [PubMed] [Google Scholar]
- 38.Julias JG, McWilliams MJ, Sarafianos SG, Arnold E, Hughes SH. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc Natl Acad Sci USA. 2002;99:9515–9520. doi: 10.1073/pnas.142123199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Julias JG, McWilliams MJ, Sarafianos SG, Alvord WG, Arnold E, Hughes SH. Mutation of amino acids in the connection domain of human immunodeficiency virus type 1 reverse transcriptase that contact the template-primer affects RNase H activity. J Virol. 2003;77:8548–8554. doi: 10.1128/JVI.77.15.8548-8554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archer RH, Dykes C, Gerondelis P, Lloyd A, Fay P, Reichman RC, Bambara RA, Demeter LM. Mutants of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors demonstrate altered rates of RNase H cleavage that correlate with HIV-1 replication fitness in cell culture. J Virol. 2000;74:8390–8401. doi: 10.1128/jvi.74.18.8390-8401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Archer RH, Wisniewski M, Bambara RA, Demeter LM. The Y181C mutant of HIV-1 reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors alters the size distribution of RNase H cleavages. Biochemistry. 2001;40:4087–4095. doi: 10.1021/bi002328a. [DOI] [PubMed] [Google Scholar]
- 42.Esposito F, Fanti V, Marzeddu R, Randaccio P, Tramontano E, Zinzula L. Validation of a Computed Radiography device to monitor the HIV-1 RNase H activity. Nucl Instr Meth Phys Res. 2009;607:226–228. [Google Scholar]
- 43.Tramontano E, Cheng YC. HIV-1 reverse transcriptase inhibition by dipyridodiazepinone derivative: BI-RG-587. Biochem Pharmacol. 1992;43:1371–1376. doi: 10.1016/0006-2952(92)90515-k. [DOI] [PubMed] [Google Scholar]
- 44.Mellors JW, Dutschman GE, Im GJ, Tramontano E, Winkler SR, Cheng YC. In vitro selection and molecular characterization of human immunodeficiency virus-1 resistant to non-nucleoside inhibitors of reverse transcriptase. Mol Pharmacol. 1992;41:446–451. [PubMed] [Google Scholar]
- 45.Schrödinger Suite. Schrödinger Inc; New York, NY: 2007. [Google Scholar]
- 46.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 47.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable algorithms for molecular dynamics simulations on commodity cluster; Proc. ACM/IEEE Conf. on Supercomputing (SC06); Tampa, FL. 2006. [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 49.Gibson DA, Carter EA. Time-reversible multiple time scale ab initio molecular dynamics. J Phys Chem. 1993;97:13429–13434. [Google Scholar]
- 50.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 51.Darden T, York D, Pedersen L. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Wolber G, Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J Chem Inf Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]