

Abstract
Matrix (M) protein mutants of vesicular stomatitis virus (VSV), such as rM51R-M virus, are attractive candidates as oncolytic viruses for tumor therapies because of their capacity to selectively target cancer cells. The effectiveness of rM51R-M virus as an antitumor agent for the treatment of breast cancer was assessed by determining the ability of rM51R-M virus to infect and kill breast cancer cells in vitro and in vivo. Several human- and mouse-derived breast cancer cell lines were susceptible to infection and killing by rM51R-M virus. Importantly, non-tumorigenic cell lines from normal mammary tissues were also sensitive to VSV infection suggesting that oncogenic transformation does not alter the susceptibility of breast cancer cells to oncolytic VSV. In contrast to results obtained in vitro, rM51R-M virus was only partially effective at inducing regression of primary breast tumors in vivo. Furthermore, we were unable to induce complete regression of the primary and metastatic tumors when tumor-bearing mice were treated with a vector expressing interleukin (IL)-12 or a combination of rM51R-M virus and IL-12. Our results indicate that although breast cancer cells may be susceptible to VSV in vitro, more aggressive treatment combinations are required to effectively treat both local and metastatic breast cancers in vivo.
Keywords: VSV, breast cancer, IL-12, tumor therapy
Introduction
Invasive breast cancer is a leading cause of cancer-related death for women worldwide (reviewed by Dodwell and Williamson1; Nicolini et al.2) Although major strides have been made in the detection and treatment of the local disease, most patients with advanced metastatic disease have fatal outcomes, mainly because of the development of resistance to standard treatments. Therefore, the challenge is to develop new and effective therapies for treatment of metastatic tumors that are unable to respond to conventional approaches. The use of genetically engineered viruses for the treatment of metastatic cancer is an attractive and promising tactic because of the natural ability of viruses to spread throughout the body and target tissues that are susceptible to infection.
Vesicular stomatitis virus (VSV) is currently being explored as an oncolytic agent in a number of tumor models3–15 because of its potent ability to induce apoptosis in infected cells.16–19 It has been proposed that the susceptibility of tumors to VSV and other oncolytic viruses is due to development of defects in antiviral responses, such as the type I interferon (IFN) response, during tumorigenesis.4,5,13,14,20 Furthermore, along with other groups, we have demonstrated that the selectivity of VSV for tumor cells versus normal cells can be enhanced either by pretreatment with IFNs or by using matrix (M) protein mutant VSV strains that induce IFN production in infected cells.3,10,11,13,14 One such recombinant strain is rM51R-M virus, which contains an arginine for methionine substitution at position 51 of the M protein sequence. This mutation renders the virus defective at inhibiting host gene expression and thus, in contrast to its isogenic recombinant wild-type counterpart (rwt virus), rM51R-M virus stimulates expression of genes involved in host innate immune responses.21 We have shown that rM51R-M virus is an effective oncolytic agent because of its ability to selectively kill tumor cells in mice without causing disease.3 Furthermore, it effectively induces maturation of several subsets of dendritic cells (DCs),22,23 thus offering the promise of promoting antitumor immunity during therapies.
Studies have shown that many breast cancer cell lines are highly sensitive to infection and killing by wt and M protein mutant VSV.7,14,24 However, survival of mice-bearing syngeneic breast tumors is not improved dramatically on treatment with VSV.7 Recombinant VSVs expressing the suicide gene product, thymidine kinase, or the cytokine, interleukin 4 (IL-4) have shown greater efficacy at treatment of local and metastatic breast cancers in vivo than treatment with wt VSV alone.24 These studies raise the question of why oncolytic therapy with VSV is not completely effective in the case of some cancers. It is possible that the immune response limits virus replication and spread, thus hindering access of the virus to the tumor cells. Alternatively, the rate of tumor growth may be faster than the ability of VSV to induce apoptosis in tumor cells. These hurdles are important to understand and overcome in order to develop effective oncolytic agents for the treatment of cancers.
In the work presented here, we determined that several breast cancer cell lines were susceptible to infection and killing by both wt and rM51R-M virus. However, breast cancer cells were no more susceptible to VSV than non-tumorigenic cell lines from normal mammary tissues. We used a syngeneic mouse model (4T1) to compare the treatment of breast cancer with rM51R-M virus with an IL-12-based immunotherapy that is currently in clinical trials. The 4T1 tumor model is known to be highly tumorigenic, invasive and nonimmunogenic.25 Our results have shown that rM51R-M virus was only partially effective at inducing regression of the primary 4T1 tumors in vivo, similar to treatment of tumor-bearing mice with a vector expressing IL-12. In addition to a delay in the growth of the primary tumor, there was a reduction in spontaneous metastases in the lungs of animals treated with rM51R-M virus, IL-12 or the combination of the two. None of the treatment regiments were successful at inducing complete regression of the primary and metastatic tumors and all animals were eventually killed because of excessive tumor burden. Therefore, although breast cancer cells may be susceptible to killing by VSV in vitro, our results suggest that oncolytic VSV therapy or immune therapies with IL-12 may be limited by the fast rate of tumor growth in vivo.
Materials and methods
Cells and viruses
The human mammary epithelial cell lines, immortalized human mammary epithelial (HME), HME expressing large T antigen (HMLE) and HMLE transduced with a puromycin resistance and Ras-expressing vector (HMLE-PR), were kindly provided by Dr Griffith Parks (Wake Forest University School of Medicine) and described by Elenbaas et al.26 Cells were used at low passage numbers to preserve their original phenotypes. MCF7, MCF10A, TM40D and 4T1 cells were obtained from the American Type Culture Collection (Manassas, VA). The recombinant viruses, rwt and rM51R-M, were isolated from infectious VSV complementary DNA clones18 and were grown on BHK cell monolayers. Supernatants containing progeny virions were harvested, titrated and stored at −80 °C.
Reagents
The mouse IL-12 DNA expression vector, pUMVC3-mIL-12, was obtained from Aldevron (Fargo, ND) and is described in Mahvi et al.27 Briefly, the IL-12 p35 and p40 subunits in this plasmid are separated by an internal ribosomal entry site and driven by a single cytomegalo-virus promoter (National Gene Vector Laboratory, University of Michigan, Ann Arbor, MI).
35S radiolabeling of infected cells
To analyze protein synthesis during virus infections, HME, HMLE and HMLE-PR cells (1 × 106 cells) were infected with rwt or rM51R-M virus at an multiplicity of infection (MOI) of 0.1 and 10 plaque-forming unit (PFU) per cell in RPMI containing 10% fetal bovine serum. At different times post-infection, cells were labeled with a 15-min pulse of [35S]methionine (100 μCi ml−1) in a total volume of 0.3 ml of methionine-free medium. Cells were washed with phosphate-buffered saline and harvested in radioimmunoprecipitation assay buffer (0.15M NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 10mM Tris and pH 7.4). Extracts normalized for protein levels (by the Lowry protein assay) were electrophoresed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were analyzed by phosphorescence imaging as described previously.18
Growth curves
HME, HMLE and HMLE-PR were infected with rwt or rM51R-M viruses in six-well dishes at the indicated MOI in RPMI containing 10% fetal bovine serum. Cells were washed at 1-h post-infection and recultured in 3ml of fresh media. At 24-h post-infection, 100 μl of medium was removed from each dish and stored at −70 °C. The yields of virus were determined by plaque assays on BHK cells and were expressed as PFUml−1.
Cell viability assay
In all, 4T1 cells were infected with wt and mutant viruses at an MOI of 10 or 0.1PFU per cell. At different times post-infection, live cells were measured by an MTT assay (Cell Proliferation Kit 1; Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions.
Tumor treatment
In total, 4T1 cells were harvested from semi-confluent cultures, and cell viability was determined using Trypan blue exclusion. Cells with > 90% viability were suspended at 1 × 106 cells per 0.2 ml of phosphate-buffered saline (PBS) and injected subcutaneously in the flanks of BALB/c wt or nude mice (Charles River Laboratories, Wilmington, MA). Animals were monitored for tumor development daily by palpation of the injection site. Animals with palpable tumors had their tumor volume measured by calipers and the volume calculated using the formula Volume = (width)2 × length/2. At 12 days after tumor cell injection, the tumor-bearing animals were randomly separated into experimental groups. Animals were injected in the tumor with 1 × 107PFU of rM51R-M virus at days 1, 3 and 5, 50 μg of IL-12 plasmid at days 1 and 3, or PBS alone as a negative control. Tumor volume was measured daily with calipers, as described above, and animal mass was also measured. If the animals showed irreversible symptoms of VSV infection (usually around 6 days post-infection) or signs of end-stage illness as indicated by IACUC guidelines of Wake Forest University Health Sciences, they were killed, and the tumor and selected tissues (brain, lungs, spleen and liver) were harvested for histological analysis.
IL-12 and IFNγ enzyme-linked immunosorbent assays
Tumors and spleens from tumor-bearing mice were harvested and homogenized in 0.5 ml sterile PBS containing 0.1% Triton X-100. Supernatants were removed after centrifugation of homogenized tissue solutions and frozen at −20 °C. Supernatants were assayed for the presence of IL-12 p40 and IFNγ using OptiEIA ELISA kits (BD Pharmingen, San Diego, CA).
Immunohistochemistry
Harvested tissues were fixed in 4% paraformaldehyde overnight, embedded in paraffin and sectioned at 5 μm. Sections were stained with hematoxylin and eosin for histological examination or used in immunohistochemical staining. For immunohistochemical staining, cells were fixed in descending series of ethanol washes, quenched with 0.3% peroxide in PBS and blocked in 5% goat serum. Serial sections were incubated overnight with antibodies against the viral envelope glycoprotein (rabbit anti-G, Research Diagnostics, Inc., Flanders, NJ). Secondary antibody (biotinylated anti-rabbit from BioGenex Supersensitive Kit, Biogenex, San Ramon, CA) was incubated on sections at room temperature for 30 min. Primary antibody detection was accomplished using a streptavidin alkaline phosphatase detection kit (Supersensitive Detection Kit, Biogenex). Vector Red Substrate Kit No. 1 for alkaline phosphatase (Vector Laboratories, Burlingame, CA) was used to visualize the antibody–antigen complex. Nuclei were counterstained using Mayers hematoxylin. Negative controls consisted of histologic sections processed without the addition of primary antibody, but incubated instead with 1% goat serum or mouse immunoglobulin G (Reagent Grade, 0.33mg ml−1, Sigma Chemical, St Louis, MO).
Clonogenic assay
Metastases in the lungs were determined by a clonogenic assay modified from the protocol described by Shan et al.28 Basically, lungs were harvested, washed with Hank’s balanced salt solution (HBSS) and minced into small pieces. Tissue was incubated with 2mgml−1 collagenase type IV for 2 h. After digestion, the sample was filtered through a 70 μm nylon cell strainer and centrifuged for 5 min at 1500 r.p.m. The cells in the pellet were washed with HBSS and resuspended in culture medium (1 × Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum and 60 μM 6-thioguanine). Cells were then plated onto 100mm tissue culture dishes in serial dilutions and incubated at 37 ° for 10–14 days. When colonies appeared, culture media from plates were removed and cells were fixed in methanol and stained with 0.03% methylene blue solution. Each colony represented one clonogenic metastatic cell.
Results
Non-tumorigenic and highly tumorigenic breast cancer cells are similarly sensitive to infection with VSV
A main hypothesis driving oncolytic VSV therapy is that the susceptibility of cancers to oncolytic viruses is due to development of defects in antiviral responses during tumorigenesis. To determine whether alterations involved in forming breast carcinoma lead to greater susceptibility to VSV infection, we used the cell culture system developed by Elenbaas et al.26 in which primary human mammary epithelial cells (HMECs) were sequentially transformed to carcinoma cells through the introduction of specific genes. HME cells were derived from HMECs immortalized through the loss of p16INK4a expression and the introduction of human telomerase reverse transcriptase (hTERT), and are non-tumorigenic. HME cells were transformed by introduction of the SV40 Large-T (LT) antigen to generate HMLE cells and by introduction of LT antigen together with the H-ras oncogene to generate HMLE-PR cells, which are highly tumorigenic. To determine the permissiveness of HME, HMLE and HMLE-PR cells to VSV infection, the production of infectious progeny in the supernatants of infected cells was measured by plaque assay (Figure 1). Cells were synchronously infected at a MOI of 10PFU per cell with rM51R-M virus and its isogenic wt counterpart, rwt virus, to determine the level of virus replication in a single cycle of growth. Each of the cell lines infected with rwt virus produced progeny virus between 107 and 108PFUml−1 by 24 h, while viral titers in rM51R-M virus-infected cells were slightly higher. This result is consistent with our previous data in other cell types showing that the M51RM protein mutation has little effect on the virus assembly function of M protein and that the rM51R-M virus produces slightly higher virus yields than rwt virus.21 More importantly, these results show that both non-tumorigenic and tumorigenic cells were permissive for VSV infection.
Figure 1.
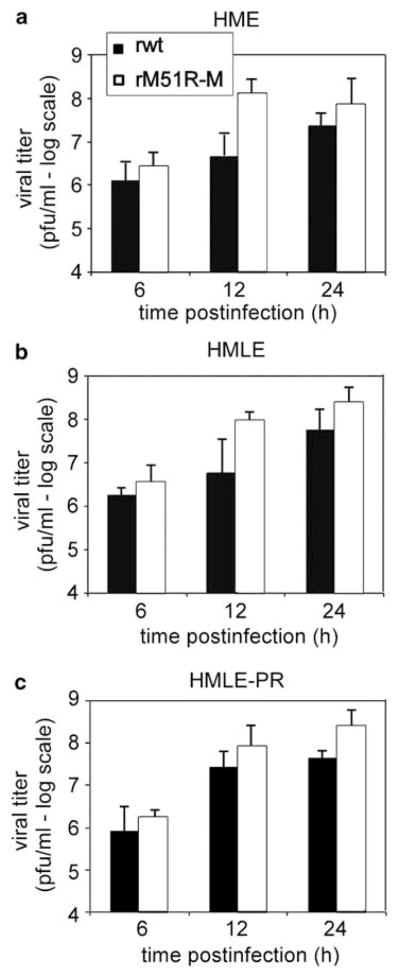
Single-cycle growth analysis of wt and M protein mutant viruses in non-tumorigenic and tumorigenic breast cancer cells. HME (a), HMLE (b) and HMLE-PR (c) cells were infected with rwt and rM51R-M viruses at a multiplicity of 10PFU per cell. A small aliquot of the supernatant was removed at the indicated times post-infection to determine the amount of progeny virus by plaque assay. Data are the average of two independent experiments.
The permissiveness of HME, HMLE and HMLE-PR cells to virus infection was further tested by determining rates of viral protein synthesis. Cells were infected with rwt or rM51R-M viruses at a multiplicities of 10PFU per cell and pulse labeled with [35S]methionine for 15 min at different times post-infection. Proteins were solubilized, and equivalent amounts of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and phosphorescence imaging. Representative images are shown in Figure 2a and the results of quantification of three independent experiments are shown in Figures 2b and c. rwt virus effectively inhibited host protein synthesis in HME, HMLE and HMLE-PR cells as compared with mock-infected controls. This is clearly seen in the regions of the gel that are devoid of viral proteins (Figure 2a and quantified Figure 2b). rM51R-M virus also inhibited host protein synthesis in each of the cell lines, but shut-off of host genes was slightly delayed as compared with that observed in rwt virus-infected cells. Again, the shut-off of host protein synthesis is typical of cells that are highly permissive for VSV. Viral proteins were synthesized at high levels in all three cell types infected with rwt virus peaking at 12-h post-infection and declining thereafter (Figure 2c). This is a typical pattern of viral protein synthesis in highly permissive cells. Levels of viral protein synthesis were slightly less in cells infected with rM51R-M virus, even though higher levels of infectious virus were produced (Figure 1), which is also typical of this virus.
Figure 2.

Rates of host and viral protein synthesis in VSV-infected HME, HMLE and HMLE-PR cells. Cells were infected with rwt and rM51R-M viruses at a multiplicity of 10 or 0.1PFU per cell, or were mock infected as a control. Cells were labeled with a 15-min pulse of [35S]methionine (100 μCi ml−1) at 6-, 12- and 24-h post-infection. Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE) and labeled proteins were quantitated by phosphorimaging. (a) Representative images of HME, HMLE and HMLE-PR cells infected with rwt and rM51R-M viruses at a MOI of 10PFU per cell. Positions of viral proteins are indicated on the left. (b) Rates of host protein synthesis (MOI = 10PFU per cell) were quantitated from images similar to that in panel (a). Results are shown as a percentage of the mock-infected control. Rates of viral protein synthesis were determined by quantitating labeled M proteins in cells infected at multiplicities of 10PFU per cell (c) and 0.1PFU per cell (d) and are expressed as the percentage of the rwt M protein in HME cells labeled at 12-h post-infection (MOI = 10PFU per cell). Data are the mean ± standard error of four experiments.
The data in Figures 2a–c indicate that the three cell lines are similarly permissive for rM51R-M virus as rwt virus in a single-cycle infection. Rates of viral protein synthesis were also evaluated under conditions of multiple-cycle virus growth (Figure 2d). In these experiments, cells were infected at a multiplicity of 0.1PFU per cells so that only 10% of cells were initially infected and infection of the remaining cells depended on spread of virus through the culture. Under these conditions, viral protein synthesis in cells infected with rwt virus was not detected above the background of host protein synthesis until 24-h post-infection. Similarly, viral protein synthesis in cells infected with rM51R-M virus was only observed at 24 h, but levels were lower than those detected in rwt virus-infected cells. This result is likely due to antiviral responses induced by rM51R-M virus that delay spread of the virus to uninfected cells.
To determine the susceptibility of HME, HMLE and HMLE-PR cells to killing by VSV, cells were infected with rwt and rM51R-M viruses, and cell viability was determined by MTT assays (Figure 3). For this experiment, cells were infected with wt and mutant viruses at both a high and low MOI in order to determine the susceptibility of cells to VSV in a synchronous infection (MOI = 10) (Figure 3a) or to determine whether antiviral responses affect the ability of virus to spread and kill surrounding cells in the culture (MOI 0.1) (Figure 3b). All cell lines were susceptible to killing by rwt virus when infected at a multiplicity of 10PFU per cell such that close to 40% of cells were viable by 48 h. However, cells were more resistant to rM51R-M virus-induced killing. When infected at the low MOI, cells were more resistant to killing by either virus, again indicating that these cells may retain intact antiviral signaling pathways to prevent spread and killing by VSV. Furthermore, our results indicate that there was no difference in the ability of VSV to kill non-tumorigenic versus highly tumorigenic cells.
Figure 3.

Viability of non-tumorigenic and tumorigenic breast cancer cells infected with rwt and rM51R-M viruses under singleand multiple-cycle infection conditions. HME, HMLE and HMLE-PR cells were infected with rwt and rM51R-M viruses at multiplicities of 10 (a) and 0.1PFU per cell (b). At 24- and 48-h post-infection, live cells were measured by MTT assay. Data are expressed as the percentage of the cell viability of mock-infected cells and represent the means ± s.d. of three experiments.
Many breast cancer cell lines are sensitive to killing by VSV
To further determine the ability of VSV to kill non-tumorigenic mammary cells versus breast cancer cells, non-tumorigenic MCF10A (Figure 4a) and highly tumorigenic MCF7 (Figure 4b) cells were infected and compared for their susceptibility with killing by rwt and rM51R-M viruses. Both cell lines were highly sensitive to infection and killing by rwt and rM51R-M viruses when infected at a MOI of 10PFU per cell as indicated by a decrease in cell viability by 24–48-h post-infection. Although killing by both viruses was delayed when cells were infected at a MOI of 0.1, cells succumbed to VSV-induced cell death over time as a result of virus replication and spread. These results further support the conclusion that tumorigenic and non-tumorigenic cells are similarly sensitive to infection and killing by oncolytic VSV. Furthermore, although there appears to be a slight delay in the ability of rM51R-M virus to kill these cells, they eventually succumb to infection and killing by both viruses.
Figure 4.
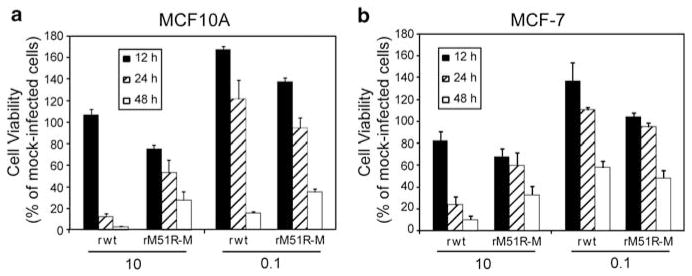
Sensitivity of human breast cancer cell lines to killing by VSV. MCF10A (a) and MCF7 (b) cells were infected with rwt and rM51R-M viruses at multiplicities of 10 and 0.1PFU per cell. At indicated times post-infection, live cells were measured by MTT assay. Data are expressed as the percentage of the cell viability of mock-infected cells and represent the means ± s.d. of three experiments.
rM51R-M virus is partially effective at killing breast cells in vivo
We used a syngeneic mouse model (4T1) to compare the treatment of breast cancer with rM51R-M virus to an IL-12-based immunotherapy that is currently in clinical trials. 4T1 mouse mammary tumor cells are highly invasive breast cancer cells with the capacity to metastasize efficiently in vivo to sites affected in human breast cancer.25,29 In cell culture, 4T1 cells were sensitive to killing by rwt and rM51R-M viruses at both high and low MOIs, and there was no difference between wt and mutant viruses in their ability to kill these cells (Figure 5a). 4T1 cells were injected subcutaneously in the flanks of wt BALB/c mice. When palpable tumors were obtained, approximately 14 days following implantation, mice were treated intratumorally with rM51R-M virus, IL-12 plasmid DNA, or the combination of rM51R-M virus and IL-12 plasmid DNA. The IL-12-encoding plasmid used in this study has been shown to induce tumor regression when administered locally or systemically in several tumor systems30,31 in the absence of local or systemic toxicity.27 Tumors were mock treated with PBS as a control, and tumor volume was measured every other day (Figure 5b). Treatment with rM51R-M virus significantly delayed the growth of 4T1 tumors as compared with mock-treated tumors. Nevertheless, tumor size continued to increase over time, indicating that therapy with rM51R-M virus was only partially effective in this model.
Figure 5.

Treatment of 4T1 tumors with M protein mutant VSV and IL-12. (a) 4T1 cells were infected with rwt and rM51R-M viruses at multiplicities of 10 and 0.1PFU per cell. Cell viability was measured at different times post-infection. Data are expressed as the cell viability of mock-infected cells. (b) 4T1 cells were injected subcutaneously in the flanks of BALB/c mice. Animals with palpable tumors were randomly separated into five experimental groups (5–10 animals per group) and were injected in the tumor with 1 × 107 PFU of rM51R-M virus at days 1, 3 and 5, 50 μg of IL-12 plasmid at days 1 and 3, or PBS alone as a negative control (mock). Tumor volume was measured daily with calipers. Results are expressed as the change in tumor volume on treatment of mice with rM51R-M virus and/or IL-12. Data represent two separate experiments and are shown as the percentage of original tumor size on day 1 (mean ± s.e.). (c) Viral antigen staining in sections of 4T1 tumors from untreated mice or mice treated with rM51R-M virus and rM51R-M + IL-12.
Treatment of mice with IL-12 alone also delayed tumor growth as compared with mock-treated animals but was no more effective than treatment with rM51R-M virus. Furthermore, the addition of IL-12 to virus therapy had no additional benefit.
Immunohistochemical analysis of tumors from mice at day 7 post-treatment was carried out with antibodies against the viral G protein to determine the ability of rM51R-M virus to replicate and spread in the tumor tissue (Figure 5c). We were able to detect areas of antigen-positive cells in the tumor tissue of mice treated with rM51R-M virus and rM51R-M + IL-12, corresponding to areas of necrosis. However, staining was not widespread, suggesting that the low efficacy of rM51R-M virus therapy may be due in part to the inefficient replication and spread of virus in the tumor tissue.
To determine the extent of immune stimulation in treated animals, the levels of IL-12 were assayed in the tumors and spleens (Figures 6a and b) and the levels of IFNγ, which is induced by IL-12, were assayed in the spleens (Figure 6c). Levels of IL-12 in response to treatment with virus were comparable to those produced from plasmid DNA and there was little, if any, increase from the combination treatment. However, treatment with virus was more effective in stimulating IFNγ production than treatment with IL-12 plasmid DNA.
Figure 6.
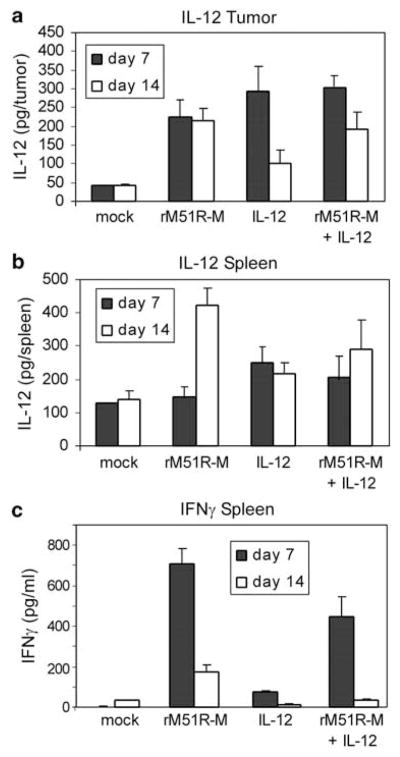
IL-12 and IFNγ levels in the tumors and spleens of tumor-bearing mice treated with rM51R-M virus or IL-12 plasmid DNA. Tumors and spleens from tumor-bearing mice were harvested at days 7 and 14 post-treatment and assayed for the presence of IL-12 p40 (a and b) and IFNγ (c) by enzyme-linked immunosorbent assay (ELISA). Data represent the average ± s.d. of a total of 3–5 mice from two experiments.
Treatment of 4T1 tumors decreases lung metastases
Previous studies have demonstrated that IL-12 treatment is effective at reducing spontaneous metastases in the lungs of 4T1 tumor-bearing mice while significantly prolonging their survival time.32 To determine whether rM51R-M virus affected tumor metastases, the lungs of treated mice were collected at day 14 post-treatment and examined for metastases. Mice injected with the control PBS showed large numbers of metastasized 4T1 cells in the lung (Figure 7a), while no tumor masses were detected in any of the treated animals. When lung metastasis was measured by a clonogenic assay, the number of metastasized 4T1 cells in the lungs of each of the treated mice treated was significantly reduced by a log (Figure 7b), corresponding to a decrease in lung weights (Figure 7c). We were unable to detect infectious virus in the lungs of rM51R-M virus-treated animals suggesting that immune components may contribute to anti-metastatic effects during local therapies with VSV.
Figure 7.

Reduction of spontaneous metastases to the lungs of tumor-bearing mice. Lungs of treated mice were collected at day 14 post-treatment and examined for metastases. (a) Hematoxylin and eosin (H and E) staining of lung tissue from a mock-treated mouse. (b) Number of metastatic 4T1 cells in the lungs of mice treated with rM51R-M virus or IL-12. (c) Weight of lungs from tumor-bearing mice. Data is the average ± s.d. of 4–5 mice from two experiments.
Discussion
The selectivity of VSV as an antitumor agent is attributed to inherent defects in the antiviral response, such as the type I IFN response, in tumor cells as compared with normal cells from which they were derived.4,13 The underlying assumptions in this hypothesis are that normal cells are relatively resistant to VSV-induced cell killing, and that oncogenic transformation leads to alterations in antiviral pathways, thus promoting sensitivity to the cytopathic effects of VSV. Data presented here, comparing the ability of wt and M protein mutant VSV with infect non-tumorigenic HME cells to those that had been sequentially transformed (HMLE and HMLE-PR cells) indicates that there was little if any difference in their sensitivity to infection with either virus. Normal HME cells were sensitive to VSV-induced inhibition of host gene expression when infected synchronously at a MOI of 10PFU per cell (Figure 2). These results are similar to those obtained in primary prostate epithelial cells3 that are permissive to VSV infection, but contrast with those in rat hepatocytes8 that appear to be non-permissive for VSV growth. On the other hand, tumorigenic HMLE and HMLE-PR cells were relatively resistant to M protein mutant VSV at the low MOI (Figures 2 and 3), suggesting that these cells retain antiviral signaling to prevent spread of virus to surrounding cells. Therefore, normal mammary cells are not completely resistant to infection and killing by VSV and oncogenic transformation may not always result in the disruption of antiviral signaling pathways.
M protein mutant viruses, such as rM51R-M virus, are promising candidates as oncolytic viruses for antitumor therapies because they have been shown to selectively, and effectively kill tumor cells in vivo without causing disease in normal tissues.3,14 In addition, they offer the promise of more effectively activating DC as means of initiating tumor-specific T-cell responses.22,23 Results shown here indicate that rM51R-M virus effectively killed breast cancer cells in vitro and that there was little difference in the sensitivity of cancer cells to infection with rwt or rM51R-M virus. However, rM51R-M virus was only partially effective at killing 4T1 tumors in vivo (Figure 5b). We have shown that rM51R-M virus is non-virulent in vivo, and effectively stimulates innate and adaptive immune cells in vitro and in vivo.21–23,33 Although the combination of immune cell and viral infection within the tumor has the potential to prime antitumor immune responses, host responses may severely limit viral replication and tumor cell infection, thus limiting anticancer efficacy.34
Studies have shown that IL-12 is one of the most potent antitumoral cytokines in animal models.35 IL-12 facilitates the presentation of tumor antigens through the upregulation of class I and II major histocompatibility molecules and the generation of T helper type I immune responses. In addition, IL-12 mediates tumor regression by increasing CD8+ T cell, natural killer (NK)T cell, NK cell, and granulocyte cytotoxicity, and by inhibiting angiogenesis. 36 Several viruses engineered to express IL-12 or used in combination with IL-12 treatment, including VSV, alphaviruses and herpes simplex virus type I, have shown promise at treatment of both local tumors and metastases in a variety of tumor models.12,37–39 In the case of VSV, IL-12 expression has been shown to enhance the effect of viral therapy in murine squamous cell carcinoma.12,37 However, we did not observe an added benefit of treating 4T1 tumor-bearing animals with IL-12 in combination with rM51R-M virus (Figure 5b). Therefore, we can conclude that IL-12 treatment does not markedly enhance rM51R-M therapies in the 4T1 model system.
In immunocompetent mice there is the possibility that the effectiveness of oncolytic virus therapy is limited by immune clearance of the virus. However, we have found that there is no difference in the rate of 4T1 tumor regression during therapies in nude versus immunocompetent mice (data not shown). 4T1 breast carcinoma is a highly malignant and poorly immunogenic tumor model, which is only partially sensitive to most immune stimulation-based treatments,25 including treatment with IL-12 (Figure 5b). Several studies have demonstrated that the depletion of both CD4- and CD8-positive T cells does not impact the growth and metastases of 4T1 cells even in the presence of an elevated immune response.32,40 In fact, Tlymphocyte infiltration into the host stroma has been shown to promote the growth and metastases of 4T1 tumors, most likely because of the immune-suppressive effects of CD4 + CD25 + regulatory T cells.41 Therefore, our results are consistent with these findings and suggest that T cells may not have a major role in the efficacy of antitumor therapies with VSV. However, previous studies have shown that virus-specific and tumor-specific CD8 + T cells are induced during wt VSV therapy of B16-OVA melanoma tumors, and that the combination of virotherapy and adoptive tumorspecific T-cell therapy enhances the efficacy of tumor killing.42 Thus, breast cancers may differ from melanoma in that the potential beneficial effects of T cells may be balanced by the potential negative effects of the immune response.
Our results have shown that treatment of the primary tumor with rM51R-M virus or IL-12 resulted in the reduction of spontaneous metastases in the lungs of mice (Figure 7). Although viral antigen-positive cells were readily detected in the primary tumor (Figure 5c), we were unable to detect viral antigen-positive cells in the lungs of virus-treated mice, suggesting that the anti-metastatic effect was either because of lower numbers of 4T1 cells migrating to the lung, or because of the induction of an immune response by VSV. Previous studies have shown that the anti-metastatic effect of IL-12 in the 4T1 system occurs independently of T cells, but involves NK cells and IFNγ.32 In fact, studies have shown IL-12 therapies promote activation of antitumor NK cells 43,44 perhaps through stimulation of IFN-α producing plasmacytoid DC.45 VSV replication and expression of the viral glycoprotein are essential for recognition and lysis of VSV-infected B16 cells by NK cells.46 Therefore, production of antitumor NK cells may have a major role in tumor regression during treatment with VSV. Our studies have shown that rM51R-M virus induces expression of IL-12 by myeloid DC and type I IFN by plasmacytoid DC.22,23 In addition, IL-12 was detected in the tumors and spleens of mice treated with rM51R-M virus (Figure 6), thus having the potential to stimulate an effective antitumor NK cell response. Future studies will determine the role of NK cells during therapies with rM51R-M virus and investigate more aggressive treatment combinations in order to improve the outcome of treating tumors that are resistant to oncolytic VSV therapies.47
Acknowledgments
We thank Griffith Parks for providing the human mammary epithelial cell lines, Hermina Borgerink for H and E staining of tissue sections and Nancy Kock for examination of tissues. This work was supported by grant BC024238 from the Department of Defense to M Ahmed and grant R01-AI032983 from the NIAID to D Lyles.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Dodwell D, Williamson D. Beyond tamoxifen: extended and late extended endocrine therapy in postmenopausal early breast cancer. Cancer Treat Rev. 2008;34:137–144. doi: 10.1016/j.ctrv.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Nicolini A, Giardino R, Carpi A, Ferrari P, Anselmi L, Colosimo S. Metastatic breast cancer: an updating. Biomed Pharmacother. 2006;60:548–556. doi: 10.1016/j.biopha.2006.07.086. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Balachandran S, Barber GN. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life. 2000;50:135–138. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
- 5.Balachandran S, Barber GN. Oncloytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or Myc function and involves the induction of apoptosis. J Virol. 2001;75:3474–3479. doi: 10.1128/JVI.75.7.3474-3479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesaire R, Oliere S, Sharif-Askari E, Loignon M, Lezin A, Olindo S, et al. Oncolytic activity of vesicular stomatitis virus in primary adult T-cell leukemia. Oncogene. 2005;25:349–358. doi: 10.1038/sj.onc.1209055. [DOI] [PubMed] [Google Scholar]
- 7.Ebert O, Harbaran S, Shinozaki K, Woo SLC. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 2004;12:350–358. doi: 10.1038/sj.cgt.7700794. [DOI] [PubMed] [Google Scholar]
- 8.Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia-Sastre A, Woo SL. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–3611. [PubMed] [Google Scholar]
- 9.Huang TG, Ebert O, Shinozaki K, Garcia-Sastre A, Woo SL. Oncolysis of hepatic metastasis of colorectal cancer by recombinant vesicular stomatitis virus in immune-competent mice. Mol Ther. 2003;8:434–440. doi: 10.1016/s1525-0016(03)00204-1. [DOI] [PubMed] [Google Scholar]
- 10.Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D, et al. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV\{delta\}M51) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- 11.Obuchi M, Fernandez M, Barber GN. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol. 2003;77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin EJ, Wanna G, Choi B, Aguila DI, Ebert O, Genden EM, et al. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope. 2007;117:210–214. doi: 10.1097/01.mlg.0000246194.66295.d8. [DOI] [PubMed] [Google Scholar]
- 13.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 14.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 15.Wollmann G, Robek MD, van den Pol AN. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J Virol. 2007;81:1479–1491. doi: 10.1128/JVI.01861-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balachandran S, Roberts PC, Kipperman T, Bhalla KN, Compans RW, Archer DR, et al. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/caspase-8 death signaling pathway. J Virol. 2000;74:1513–1523. doi: 10.1128/jvi.74.3.1513-1523.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaddy DF, Lyles DS. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J Virol. 2005;79:4170–4179. doi: 10.1128/JVI.79.7.4170-4179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopecky SA, Willingham MC, Lyles DS. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J Virol. 2001;75:12169–12181. doi: 10.1128/JVI.75.24.12169-12181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koyama AH. Induction of apoptotic DNA fragmentation by the infection of vesicular stomatitis virus. Virus Res. 1995;37:285–290. doi: 10.1016/0168-1702(95)00026-m. [DOI] [PubMed] [Google Scholar]
- 20.Bell JC, Lichty B, Stojdl DF. Getting oncolytic virus therapies off the ground. Cancer Cell. 2003;4:7–11. doi: 10.1016/s1535-6108(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of M protein of vesicular stomatitis virus to suppress interferon beta gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed M, Brzoza KL, Hiltbold EM. Matrix protein mutant of vesicular stomatitis virus stimulates maturation of myeloid dendritic cells. J Virol. 2006;80:2194–2205. doi: 10.1128/JVI.80.5.2194-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed M, Mitchell LM, Puckett S, Brzoza-Lewis KL, Lyles DS, Hiltbold EM. M protein mutant vesicular stomatitis virus stimulates maturation of TLR7-positive dendritic cells through TLR-dependent and -independent mechanisms. J Virol. 2009;83:2962–2975. doi: 10.1128/JVI.02030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer. 2008;8:228. doi: 10.1186/1471-2407-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahvi DM, Henry MB, Albertini MR, Weber SM, Meredith K, Schalch H, et al. Intratumoral injection of IL-12 plasmid DNA–results of a phase I/IB clinical trial. Cancer Gene Ther. 2007;14:717–723. doi: 10.1038/sj.cgt.7701064. [DOI] [PubMed] [Google Scholar]
- 28.Shan D, Chen L, Njardarson JT, Gaul C, Ma X, Danishefsky SJ, et al. Synthetic analogues of migrastatin that inhibit mammary tumor metastases in mice. Proc Natl Acad Sci USA. 2005;102:3772–3776. doi: 10.1073/pnas.0500658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H, Weiguo Z, Rong L, Abraham S, Kittrell FS, Medina D, et al. Blocking tumor growth, invasion, and metastasis by maspin in a syngeneic breast cancer model. Cancer Res. 2001;61:6945–6951. [PubMed] [Google Scholar]
- 30.Shi F, Rakhmilevich AL, Heise CP, Oshikawa K, Sondel PM, Yang N-S, et al. Intratumoral injection of interleukin-12 plasmid DNA, either naked or in complex with cationic lipid, results in similar tumor regression in a murine model. Mol Cancer Ther. 2002;1:949–957. [PubMed] [Google Scholar]
- 31.Weber SM, Qi C, Neal Z, Sondel PM, Mahvi DM. IL-12 cDNA direct injection: antimetastatic effect from a single injection in a murine hepatic metastases model. J Surg Res. 2004;122:210–217. doi: 10.1016/j.jss.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Rakhmilevich AL, Janssen K, Hao Z, Sondel PM, Yang N-S. Interleukin-12 gene therapy of a weakly immunogenic mouse mammary carcinoma results in reduction of spontaneous lung metastases via a T-cell-independent mechanism. Cancer Gene Ther. 2000;7:826–838. doi: 10.1038/sj.cgt.7700176. [DOI] [PubMed] [Google Scholar]
- 33.Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. Immune response in the absence of neurovirulence in mice infected with M protein mutant vesicular stomatitis virus. J Virol. 2008;82:9273–9277. doi: 10.1128/JVI.00915-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008;10:38–45. [PubMed] [Google Scholar]
- 35.Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of cancer by IL-12-based cytokine combinations. Exp Opin Biol Ther. 2007;7:1705–1721. doi: 10.1517/14712598.7.11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 37.Sung CK, Choi B, Wanna G, Genden EM, Woo S, Shin EJ. Combined VSV oncolytic virus and chemotherapy for squamous cell carcinoma. Laryngoscope. 2008;118:237–242. doi: 10.1097/MLG.0b013e3181581977. [DOI] [PubMed] [Google Scholar]
- 38.Kelly BJ, Fleeton MN, Atkins GJ. Potential of alphavirus vectors in the treatment of advanced solid tumors. Recent Patents Anticancer Drug Discov. 2007;2:159–166. doi: 10.2174/157489207780832432. [DOI] [PubMed] [Google Scholar]
- 39.Varghese S, Rabkin SD, Nielsen GP, MacGarvey U, Liu R, Martuza R. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007;67:9371–9379. doi: 10.1158/0008-5472.CAN-07-0674. [DOI] [PubMed] [Google Scholar]
- 40.Pulaski B, Clements VK, Pipeling MR, Ostrand-Rosenberg S. Immunotherapy with vaccines combining MHC class II/ CD80+ tumor cells with interleukin-12 reduces established metastatic disease and stimulates immune effectors and monokine induced by interferon gamma. Cancer Immunol Immunother. 2000;49:34–45. doi: 10.1007/s002620050024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen L, Hang T-G, Meseck M, Mandeli J, Fallon J, Woo S. Rejection of metastatic 4T1 breast cancer by attenuation of Treg cells in combination with immune stimulation. Mo Ther. 2007;15:2194–2202. doi: 10.1038/sj.mt.6300310. [DOI] [PubMed] [Google Scholar]
- 42.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 43.Elzaouk L, Moelling K, Pavlovic J. Anti-tumor activity of mesenchymal stem cells producing IL-12 in a mouse melanoma model. Exp Dermatol. 2006;15:865–874. doi: 10.1111/j.1600-0625.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- 44.Takeda K, Hayakawa Y, Atsuta M, Hong S, Van Kaer L, Kobayashi K, et al. Relative contribution of NK and NKT cells to the anti-metastatic activities of IL-12. Int Immunol. 2000;12:909–914. doi: 10.1093/intimm/12.6.909. [DOI] [PubMed] [Google Scholar]
- 45.Chiesa MD, Romagnani C, Thiel A, Moretta L, Moretta A. Multidirectional interactions are bridging human NK cells with plasmacytoid and monocyte-derived dendritic cells during innate immune responses. Blood. 2006;108:3851–3858. doi: 10.1182/blood-2006-02-004028. [DOI] [PubMed] [Google Scholar]
- 46.Schattner A, Rager-Zisman B. Lysis by natural killer cells requires viral replication and glycoprotein expression. Immunol Lett. 1986;13:261–268. doi: 10.1016/0165-2478(86)90111-2. [DOI] [PubMed] [Google Scholar]
- 47.Carey BL, Ahmed M, Puckett S, Lyles DS. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol. 2008;82:12104–12115. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]