

Abstract
Objective
Whilst there is robust evidence of elevated dopamine synthesis capacity once a psychotic disorder has developed, little is known about whether it is altered prior to the first episode of frank illness. We addressed this issue by measuring dopamine synthesis capacity in subjects at ultra high risk of psychosis, and then following them to determine their clinical outcome.
Method
This prospective study included thirty subjects who met standard criteria for being at ultra high risk of psychosis and twenty-nine healthy volunteers. Subjects were scanned using [18F]-DOPA positron emission tomography. The ultra high risk subjects were scanned at presentation and followed-up for at least three years to determine their clinical outcome. Six subjects had co-morbid schizotypal personality disorder and were excluded from the analysis (data are provided for comparison). Of the remaining subjects, nine developed a psychotic disorder subsequent to scanning (psychotic transition group), and 15 did not (non-transition group).
Results
There was a significant effect of group on striatal dopamine synthesis capacity (p=0.006). The psychotic transition group had greater dopamine synthesis capacity in the striatum (p=0.004, effect size=1.18) and its associative subdivision (p=0.015, effect size=1.24) than controls, and showed a positive correlation between dopamine synthesis capacity and symptom severity. Dopamine synthesis capacity was also significantly greater in the psychotic transition than the non-transition group (p=0.036).
Conclusions
These findings provide evidence that the onset of frank psychosis is preceded by presynaptic dopaminergic dysfunction. Further work is required to determine the specificity of elevated dopamine synthesis capacity to particular psychotic disorders.
Keywords: psychosis, schizophrenia, prodrome, at risk, vulnerability, dopamine, imaging, cause, pathophysiology, hypothesis, bipolar affective disorder, schizotypal personality disorder, psychosis continuum, psychosis spectrum
INTRODUCTION
A substantial body of evidence implicates dopaminergic dysfunction in the pathoetiology of psychotic disorders (1-4). In particular, converging in vivo evidence from positron emission tomography (PET) and other molecular imaging studies indicates that psychotic disorders are associated with dysregulated presynaptic striatal dopaminergic function (see reviews (1;5;6)). It has thus been proposed that striatal hyperdopaminergia underlies the onset of psychosis-preceding illness onset and increasing further to lead to the development of frank psychosis (4;7).
The onset of psychotic disorders is typically preceded by a prodromal phase, characterized by functional decline and attenuated psychotic symptoms - perceptual disturbances and paranoid ideas that are less severe than in frank psychosis (8;9). Individuals with these symptoms present to services seeking assessment and treatment (8;9). Structured assessments and operationalized criteria have been developed for identifying clinical features, termed an at risk mental state, that are associated with an ultra high risk of developing psychosis. Those meeting these criteria are said to be at ultra high risk for psychosis, as about one third will develop a psychotic illness within two years (9;10).
We have previously reported that, on average, dopamine synthesis capacity is higher in ultra high risk subjects than in healthy controls (11). However, at the time of that study the clinical outcome of the subjects was yet to be determined. As only a proportion of those at ultra high risk will subsequently develop a psychotic disorder, the relationship between dopamine dysfunction and the subsequent onset of illness is thus still unclear. In the present prospective study we therefore tested the hypothesis that dopamine synthesis capacity would be elevated compared to controls in the group of ultra high risk subjects that went on to develop psychosis after scanning. A related prediction was that within this group, the severity of presenting symptoms would be directly related to dopamine synthesis capacity. Because schizotypal personality disorder is independently associated with presynaptic dopaminergic dysfunction and psychotic-like symptoms (12-15), we excluded ultra high risk subjects with schizotypal personality disorder from the main analysis but have reported their data for comparison. As altered dopamine synthesis capacity may be localised to the associative subdivision of the striatum in psychotic disorders (11), we examined striatal subdivisions in addition to examining the striatum as a whole.
METHOD
Subjects
The study was approved by the hospitals’ research ethics committees. All subjects gave written informed consent to participate. Subjects meeting the Comprehensive Assessment of At Risk Mental States criteria for ultra high risk of psychosis on the basis of attenuated psychotic symptoms (8) (n=30, mean age=25.0 years [SD=4.1], 17 [57%] male) were studied using 6-[18F]fluoro-L-DOPA (18F-DOPA) positron emission tomography at clinical presentation. We have previously reported baseline data in a subgroup (n=24) of this ultra high risk sample (11), but at that stage they had not received follow-up so their subsequent clinical outcome was unknown. The present study includes follow-up data on all ultra high risk subjects included in our original baseline study (11) plus follow-up data on six additional ultra high risk subjects recruited after this study was published. A minimum of 3 years follow-up was used as the great majority of transitions to psychosis in ultra high risk subjects occur within this period (9;10). Six of the ultra high risk subjects met DSM-IV criteria for co-morbid schizotypal personality disorder. They were excluded from the analysis because the latter has been independently associated with elevated striatal dopamine release (13). However their data are shown separately for comparison (schizotypal personality disorder group). The analysis thus includes the remaining 24 ultra high risk subjects (15 [63%] male), and matched controls. All of these subjects were antipsychotic naive.
Healthy controls (n=29; 20 [69%] male) were recruited contemporaneously from the same geographical area and socio-demographic background by local advertisement and by approaching social contacts of ultra high risk subjects (after first receiving written permission to do so), and were matched to the ultra high risk group on the basis of age (within five years). Parental socio-economic status (SES) and ethnicity were determined using UK standard criteria (as described: http://www.ons.gov.uk/census/index.html). Exclusion criteria for all subjects were: history of neurological or medical disorder (other than past minor self-limiting illnesses) or head injury, illicit drug or alcohol abuse/dependency, pregnancy, or contraindication to scanning. All subjects were instructed to fast, and abstain from psychoactive substances including caffeine, tobacco and alcohol for at least 12 hours prior to scanning. All subjects received a urine drug screen one hour prior to scanning and clinical assessments to confirm that they had not recently taken any illicit substances. Additional exclusion criteria for control subjects were: family history of psychotic disorder, or personal history of any axis I or axis II psychiatric disorder (assessed using the Structured Clinical Interview for Diagnosis (16)).
Clinical outcomes at follow up were determined using the Structured Clinical Interview for Diagnosis (16). This assessment was conducted blind to the imaging data by an experienced psychiatrist independent of the study and trained in the use of the interview. All subjects recruited into the study completed follow-up. Nine of the ultra high risk subjects (38%) developed a psychotic disorder (the psychotic transition group) within the follow-up period. These disorders comprised: schizophrenia (n=7), schizophreniform disorder (n=1) and bipolar I affective disorder with a psychotic manic episode (n=1). The remaining ultra high risk subjects (n=15) have not developed a psychotic disorder (the non-transition group). None of the subjects in the schizotypal personality disorder group have developed a psychotic disorder to date.
Clinical measures
The following instruments were used to assess subjects at the time of the scan: Comprehensive Assessment of At Risk Mental States (CAARMS) (8), Positive And Negative Symptom Scale (PANSS) (17), Global Assessment of Function (GAF) (16) scales, and exposure to cigarettes and alcohol was assessed by structured questionnaire (adapted from (18), available on request).
Positron Emission Tomography (PET) scanning
Subjects received 6-[18F]fluoro-L-DOPA (18F-DOPA) PET imaging using an ECAT/EXACT3D PET scanner (Siemens/CTI, Knoxville, USA). 150mg carbidopa and 400mg entacapone were administered orally one hour prior to scanning to reduce the formation of radiolabeled metabolites. Subjects were positioned with the orbitomeatal line parallel to the transaxial plane of the tomograph and head position was marked and monitored throughout the scan via laser crosshairs and a camera. The 95 minute emission scan was preceded by a short transmission scan using a 150-MBq cesium-137 rotating point source to correct for attenuation and scatter. Thirty seconds after starting the emission scan a bolus intravenous injection of 150 MBq of 18F-DOPA was administered. Structural magnetic resonance imaging was performed to exclude intracranial abnormalities.
Image analysis
Region of interest analysis
The region of interest analysis was carried out blind to group status by ODH and comprised the whole striatum and the limbic, associative and sensorimotor subdivisions of the striatum (illustrated in supplementary figure 1). The whole striatum and its subdivisions were defined using previously described criteria and combine left and right sides (11;19). The striatal subdivisions reflect the topographical arrangement of corticostriatal projections, and the functional organisation of the striatum (19;20)- see supplementary information.
Regions of interest were automatically placed on individual 18F-DOPA PET dynamic images without observer bias by using statistical parametric mapping software (SPM5; Wellcome Department of Cognitive Neurology, London, England) to normalize the region of interest map to each individual PET space (using the PET summation image). A Patlak graphical analysis was used to calculate influx constants (kicer values) for the regions of interest relative to uptake in the reference region (21). The reference region was the cerebellum, as defined using the HamNet probabilistic brain atlas (22). Striatal 18F-DOPA kicer values (denoted as Ki in some previous publications (11)) reflect the presynaptic synthesis and storage of striatal dopamine for release, respond to experimental manipulation of brain dopaminergic systems, and correlate with the striatal release of dopamine (see review (23) and discussion in (11)).
Voxel-based analysis
The region of interest analysis was complemented by an independent voxel-based analysis. Voxel-based kicer parametric images of the brain were constructed from movement corrected images using a wavelet-based Patlak approach that provides a higher signal-to-noise ratio than the original graphical procedure (24). The parametric image for each subject was then normalised into standard space using the subject’s PET summation image and the 18F-DOPA template. Statistical parametric mapping was conducted using SPM5 (Wellcome Department of Imaging Neuroscience, London, UK) and a striatal mask defined according to previously described criteria (19), to compare groups. Results are presented corrected for multiple comparisons using random field theory as applied in SPM5 (p<0.05 corrected at the family wise error rate).
Statistical analysis
The data were normally distributed as assessed using the Kolmogorov-Smirnov test apart from the substance use data. After confirming homogeneity of variance with Levene’s test, analysis of variance was used to determine if there was an effect of group (psychotic transition, non-transition and control groups) on striatal kicer value, demographic, and clinical variables for parametric variables, and the Kruskal-Wallis test was used for non-parametric variables. Where there were significant group effects, independent t-tests using Bonferroni correction for multiple comparisons (four striatal regions across three groups) were used to determine if mean kicer values were significantly elevated in the psychotic transition group compared to the controls in line with the main hypothesis, and to determine if there were significant differences in mean kicer values between the non-transition group and control, and psychotic transition and non-transition groups. The relationship between kicer values and symptom scores was explored using Pearson’s correlation coefficient and the influence of individual data points assessed using Cook’s distance-centred leverage plots. Mean kicer values in the schizotypal personality disorder group were compared with those in the psychotic transition and non-transition groups using independent t-tests, but this analysis should be considered exploratory as we had no a priori hypotheses for this comparison. A two-tailed significance level of p=0.05 was used throughout.
RESULTS
Demographic and clinical characteristics (table 1)
Table 1.
Demographic and clinical data by group at presentation. There were no significant differences between groups (all p-values>0.1).
| Control (N=29) |
Non-transition (N=15) |
Psychotic transition (N=9) |
||||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | |
| Age/yrs | 25.6 | 4.0 | 23.8 | 3.7 | 24.9 | 3.1 |
| Parental SESa | 3.0 | 0.9 | 3.0 | 0.9 | 2.9 | 0.9 |
| Cigarettes/day | 2.8 | 5.2 | 5.2 | 5.5 | 5.2 | 5.8 |
| Alcohol: units/week |
10.0 | 10.5 | 4.8 | 4.6 | 6.5 | 6.8 |
| CAARMS totalb | - | - | 35.1 | 20.3 | 40.3 | 22.1 |
| CAARMS Positiveb |
- | - | 7.4 | 4.1 | 8.1 | 3.0 |
| PANSS total | - | - | 44.5 | 12.7 | 50.2 | 21.9 |
| PANSS Positive |
- | - | 11.4 | 3.3 | 13.8 | 5.7 |
| GAF | - | - | 59.6 | 14.2 | 56.0 | 11.3 |
SES=socio-economic status. Higher scores indicate lower socio-economic status (range from 1-5, where 1=highest and 5=lowest)
CAARMS=Comprehensive Assessment of At Risk Mental State (higher scores indicate more severe high risk symptoms).
There was no significant effect of group on mean age (F=1.12, df=2,50, p=0.33), parental SES (F=0.04, df=2,44, p=0.96), cigarette (H=3.0; df=2; p=0.23), or alcohol use (H=3.4; df=2; p=0.18), or on radioactivity administered (mean [SD] for psychotic transition=148.2 [5.2], non-transition=148.8 [3.4], controls=142.9 [14.0] MBq; F=1.82 df=2,49; p=0.17). The ethnic composition of the groups was white British/white other psychotic transition: n=5 (56%), non-transition: n=8 (53%), control: n=15 (52%) groups; black British/black other: psychotic transition: n=3 (33%), non-transition: n=5 (33%), control: n=12 (41%) groups; other ethnicity: psychotic transition: n=1 (11%), non-transition: n=3 (20%), control: n=2 (7%) groups.
There was no significant difference in the clinical characteristics between the psychotic transition and non-transition groups at the time of imaging (table 1; CAARMS total: t=0.60 df=22, p=0.56; CAARMS positive: t=0.45, df=22, p=0.66; PANSS total: t=0.81, df=22, p=0.43; PANSS positive: t=1.31, df=22, p=0.20; GAF score: t=0.65, df=22, p=0.53).
Striatal dopamine synthesis capacity (Figure 1)
Figure 1.
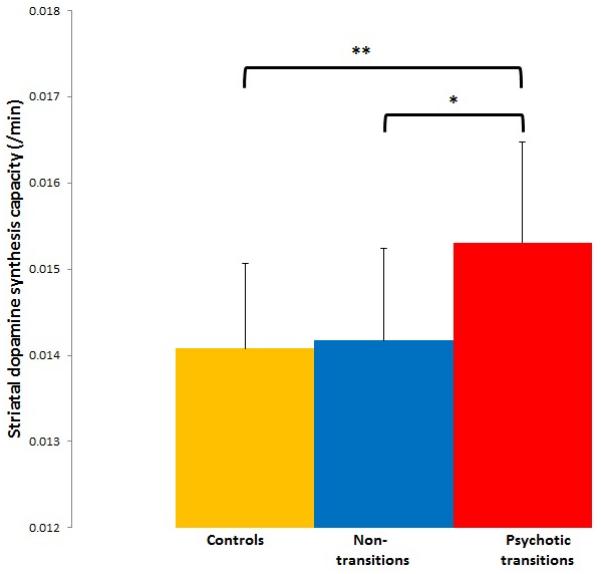
Showing mean (error bars=SD) dopamine synthesis capacity (kicer values) for the whole striatum in psychotic transition (N=9), non-transition (N=15) and control (N=29) groups. There were significant differences between psychotic transition and control groups (**p=0.004 corrected) and between the psychotic transition and non-transition groups (*p=0.015 corrected).
The region of interest analysis revealed that there was a significant effect of group on mean kicer value in the whole striatum (figure 1, F=5.8, df=2, 50, p=0.006), and in its associative subdivision (F=5.8, df=2,50, p=0.006), but not in the limbic (F=2.35, df=2,50, p=0.11) or sensorimotor subdivisions (F=2.5, df=2,50, p=0.09). The group effect remained significant after excluding the one subject within the psychotic transition group who had developed a non-schizophreniform psychosis (whole striatum: F=5.1, df=2, 49, p=0.01; associative striatum: F=4.8, df=2,49, p=0.013).
Comparison between psychotic transition and control groups
The comparison of the psychotic transition and control groups showed that, after adjustment for multiple comparisons, mean kicer values were significantly elevated in the psychotic transition group in the whole striatum (mean [SD]/min: psychotic transition=0.0153 [0.0012], control=0.0140 [0.0010]; p=0.004 corrected), and in its associative subdivision (mean [SD]/min: psychotic transition=0.0149 [0.0011], controls=0.0136 [0.0010]; p=0.015 corrected). The effect size of the elevation in kicer was d=1.18 in the whole striatum and d=1.24 in the associative striatum. There was no difference in the limbic (mean [SD]/min: psychotic transition=0.0153 [0.0013], controls=0.0140 [0.0019]; p=0.2) or sensorimotor (mean [SD]/min: psychotic transition=0.0164 [0.0015], controls=0.0152 [0.0015], p=0.095) striatal subdivisions. The voxel-based analysis also identified a greater kicer in the psychotic transition than the control group in a voxel cluster with its focus in the left head of caudate, which lies within the associative subdivision (figure 2). This difference was significant at p<0.05, corrected for multiple comparisons using the family wise error rate, and remained significant after excluding the psychotic transition subject with a non-schizophreniform psychosis. The control>psychotic transition group contrast revealed no significant difference, even at an uncorrected statistical threshold (p<0.05).
Figure 2.
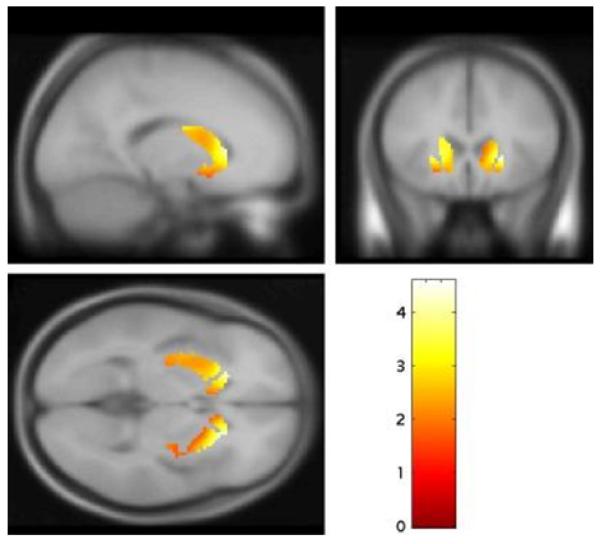
Increased dopamine synthesis capacity, relative to controls (N=29), in subjects who were scanned when they presented with prodromal symptoms and subsequently developed a psychotic disorder (psychotic transition group, N=9). The most significant increase was in the head of the left caudate nucleus (p=0.007 family wise error rate corrected).
Comparison between the psychotic transition and non-transition groups
After adjustment for multiple comparisons, kicer value was significantly elevated in the psychotic transition compared to the non-transition group in the whole striatum (figure 1, mean [SD]/min non-transition=0.0142 [0.0011]; p=0.036), and its associative subdivision (mean [SD]/min non-transition=0.0136 [0.0012]; p=0.015), but not in the limbic (mean [SD]/min non-transition=0.0149 [0.0015], p>0.99), or sensorimotor (mean [SD]/min non-transition=0.0153 [0.0013], p=0.222) striatal subdivisions. The voxel-based analysis also identified a greater striatal kicer in the psychotic transition than the non-transition group with a peak in the caudate (figure 3, p=0.036 corrected at the family wise error rate). The contrast non-transition>psychotic transition showed no significant differences, even at an uncorrected statistical threshold (p<0.05).
Figure 3.
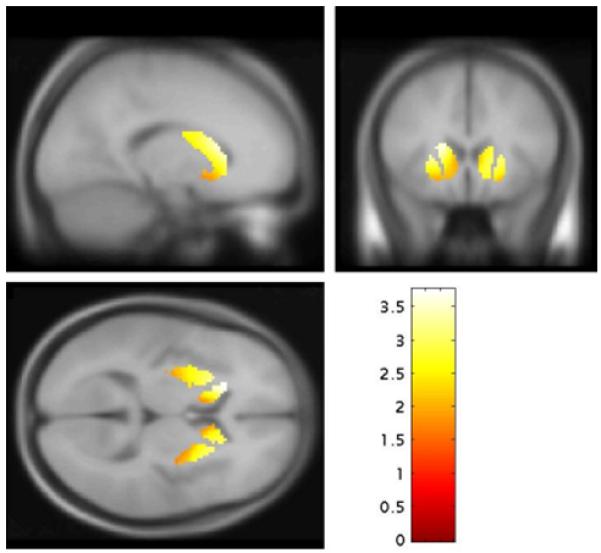
Increased dopamine synthesis capacity in subjects who were scanned when they presented with prodromal symptoms and subsequently developed a psychotic disorder (psychotic transition group, N=9) relative to subjects who presented with similar symptoms but did not develop a psychotic disorder (non-transition group, N=15). The most significant increase was in the left caudate (p=0.036 family wise error rate corrected).
Comparison between the non-transition and control groups
The region of interest analysis indicated that there was no significant difference in mean kicer value in the non-transition group compared to controls in the whole striatum (p>0.9), or its subdivisions (all p>0.3) and there were no significant differences in the corresponding voxel-based analysis for the contrast non-transition>controls, even at an uncorrected threshold (p<0.05).
The relationship between striatal dopamine synthesis capacity and symptoms
Within the psychotic transition group, there was a significant positive relationship between whole striatal kicer values and both the total CAARMS (figure 4; r=0.67, p=0.049) and total PANSS scores (r=0.71, p=0.032). These findings were not driven by outlying or high influence data points, as assessed using Cooks distance-centered leverage plots. These correlations were not evident in the non-transition group (r=0.14, p=0.619 and r=0.02, p=0.940 respectively).
Figure 4.
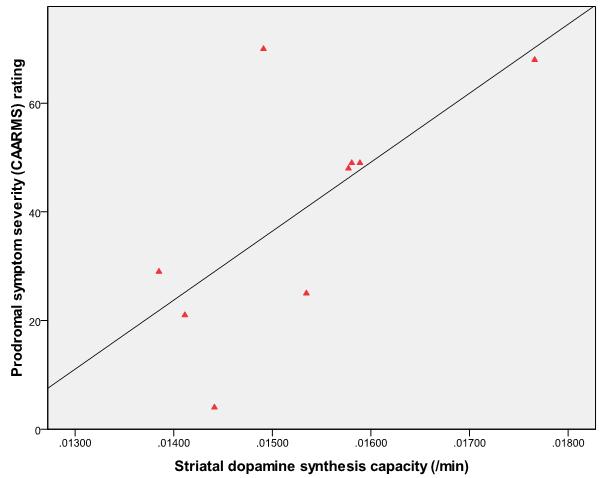
Showing greater severity of prodromal symptoms (total CAARMS score) is associated with greater dopamine synthesis capacity in the whole striatum (kicer value/min) at presentation in subjects who subsequently developed psychosis (N=9, r=0.67, p=0.049).
Striatal dopamine synthesis capacity in the schizotypal personality disorder group
The kicer values for the schizotypal personality disorder group and the statistical comparisons are shown in the supplementary information. There was no significant difference in the kicer values between the schizotypal personality disorder and psychotic transition groups (p-values>0.3). However, there was a significant elevation in kicer values in the schizotypal personality disorder group compared to controls in the whole striatum (p=0.006) and its associative (p=0.003) and sensorimotor (p=0.047) but not limbic (p>0.5) subdivisions, and compared to the non-transition group in the associative (p=0.023) subdivision, but this was only at a trend level in the whole striatum (p=0.053) and not significant in the sensorimotor (p=0.095) or limbic (p>0.5) subdivisions.
DISCUSSION
We found that dopamine synthesis capacity was elevated with a large effect size (>1) in the subjects who presented with prodromal signs of psychosis and went on to a first episode of a psychotic disorder. This elevation was evident relative to healthy controls and to subjects who presented with similar clinical features but who did not go on to develop psychosis. In the psychotic transition group, there was a direct relationship between the magnitude of dopaminergic dysfunction and the severity of prodromal symptoms at presentation. Dopamine synthesis capacity in the ultra high risk subjects who did not subsequently develop psychosis was not significantly different from that in controls, and was not correlated with symptoms at presentation.
These findings support the dopamine hypothesis (4;7) by providing, to our knowledge, the first evidence that dopaminergic dysfunction predates the subsequent onset of frank psychotic illness in people with symptoms that are truly prodromal to a psychotic disorder, and extend our finding of a longitudinal increase in dopamine synthesis capacity in this group (25). This does not preclude the involvement of other neurotransmitter systems which may act upstream to alter dopaminergic function (26;27). For example, animal studies show that cortical damage can alter striatal dopaminergic function (28;29) and human neuroimaging data indicate that cortical dysfunction is related to striatal dopaminergic function in both ultra high risk (30) and schizophrenic subjects (31;32). Furthermore, we have recently found an altered relationship between striatal dopaminergic and cortical glutamatergic indices in psychotic transition subjects (33), suggesting that both may be involved in the development of psychosis.
Findings in the schizotypal personality disorder group
The finding that dopamine synthesis capacity was elevated in the schizotypal personality disorder group compared to controls is consistent with evidence that individuals with schizotypal personality disorder show elevated dopamine release, similar to that in remitted schizophrenia and at a level intermediate between that in acute schizophrenia and controls (13). As schizotypal personality disorder is independently associated with an increased lifetime risk of schizophrenia relative to the general population (see review (14)), the subjects in the schizotypal personality disorder group probably continue to have an elevated risk of developing schizophrenia despite not developing it during the follow-up period. Our data along with previous findings in schizotypal personality disorder, thus, suggest that dopaminergic dysfunction may be related to the vulnerability to schizophrenia spectrum disorders, rather than frank psychosis per se.
However, as a proportion of ultra high risk subjects in any given sample are likely to also have co-morbid schizotypal personality disorder (8), the lack of differences between the psychotic transition and schizotypal personality disorder groups limits the utility of this PET measure as a marker for impending psychosis, although greater sensitivity may be achieved using novel image analytic approaches (34).
Methodological considerations
A critical consideration in all longitudinal studies in ultra high risk samples is that some subjects who have not developed psychosis might still do so subsequent to the end of the follow up period. However, follow up of ultra high risk subjects indicates that the great majority of transitions occur within the first 24 months, after which time the transition rate sharply decelerates (9;10). In the present study, all the subjects were followed up for at least 36 months but, occasional transitions may still occur several years after presentation. The development of psychosis has also been associated with reductions in grey matter volume, but in the cerebral cortex rather than in the striatum (35). Moreover, the PET normalisation procedure we employed is accurate even when there is structural change (36), and striatal volume loss would, if anything, reduce, rather than increase, kicer values (37). It, thus, seems unlikely that our neurochemical findings were secondary to structural alterations in the striata of the psychotic transition group. However, given the evidence of altered cortico-striatal interactions in ultra high risk subjects, future work should investigate the relationship between cortical changes and striatal dopaminergic function. Whilst we could not detect a significant difference between dopamine synthesis capacity in the non-transition group and healthy controls, we cannot exclude the possibility that this reflected a lack of statistical power. Studies in larger samples are needed to clarify if dopamine synthesis capacity in the non-transition group is intermediate between that in psychotic transition subjects and controls.
Specificity of the findings
Our key clinical outcome was the development of a first episode of a psychotic disorder, and as such the findings support a link between dopamine dysfunction and psychosis in general rather than as a manifestation of a particular psychotic disorder. Nevertheless, all but one of the cases that developed psychosis met diagnostic criteria for a schizophreniform disorder, and exclusion of the one subject who met criteria for bipolar disorder did not alter the results. It is possible that the findings may vary according to the type of psychotic disorder they predate, and this issue may be addressed in future studies.
Our finding that the dopaminergic abnormality in the psychotic transition group was localised to the associative striatum extends previous findings in schizophrenia that elevated dopamine synthesis capacity (11) and synaptic dopamine levels (38) are localised to the associative striatum, by indicating for the first time that this is also the case in the prodrome that precedes the first episode of psychosis. The associative striatum is functionally linked to the dorso-lateral prefrontal cortex (see supplementary figure 1), a cortical area that independent research has shown to be the site of functional impairments in ultra high risk subjects (39). Our finding in the associative striatum thus suggests that frontal-striatal interactions may be important in the development of psychosis. Whilst [18F]-DOPA PET measurements in the striatum show high reliability (40), reliability is lower in smaller structures. However, the voxel-based analysis identified the peak difference to the left head of caudate, which is within the associative striatum, and so supports the associative localisation of dopaminergic abnormalities, and also suggests there may be a lateralisation effect which warrants further investigation. Finally, our findings are unlikely to be an effect of antipsychotic medication, as all the ultra high risk subjects were antipsychotic naïve.
Conclusions
Elevated striatal dopamine synthesis capacity predates the onset of psychosis in those who go on to develop psychosis, and is linked to the severity of prodromal symptoms. Further work is required to determine whether dopaminergic dysfunction is specific to psychotic disorders, or increases in a graded fashion across the putative psychosis spectrum.
Supplementary Material
Acknowledgments
This work was funded by grant U.1200.04.007.00001.01 from the Medical Research Council (MRC) and the BRC. We are grateful to the volunteers, staff of OASIS and LEO teams (South London and Maudsley NHS Foundation Trust) and GE Imanet.
Footnotes
Disclosures
The authors have no conflicts of interest.
Reference List
- 1.Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol. 2004;7:S1–S5. doi: 10.1017/S1461145704004110. [DOI] [PubMed] [Google Scholar]
- 2.Heinz A, Schlagenhauf F. Dopaminergic dysfunction in schizophrenia: salience attribution revisited. Schizophr Bull. 2010;36:472–485. doi: 10.1093/schbul/sbq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry. 2007;(Suppl 51):s13–s18. doi: 10.1192/bjp.191.51.s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 5.Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol. 1999;13:358–371. doi: 10.1177/026988119901300405. [DOI] [PubMed] [Google Scholar]
- 6.Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des. 2009;15:2550–2559. doi: 10.2174/138161209788957528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howes OD, Kapur S. The Dopamine Hypothesis of Schizophrenia: Version III--The Final Common Pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yung AR, Yuen HP, McGorry PD, Phillips LJ, Kelly D, Dell’Olio M, Francey SM, Cosgrave EM, Killackey E, Stanford C, Godfrey K, Buckby J. Mapping the onset of psychosis: the Comprehensive Assessment of At-Risk Mental States. Aust N Z J Psychiatry. 2005;39:964–971. doi: 10.1080/j.1440-1614.2005.01714.x. [DOI] [PubMed] [Google Scholar]
- 9.Cannon TD, Cadenhead K, Cornblatt B, Woods SW, Addington J, Walker E, Seidman LJ, Perkins D, Tsuang M, McGlashan T, Heinssen R. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch.Gen.Psychiatry. 2008;65:28–37. doi: 10.1001/archgenpsychiatry.2007.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yung AR, Nelson B, Stanford C, Simmons MB, Cosgrave EM, Killackey E, Phillips LJ, Bechdolf A, Buckby J, McGorry PD. Validation of “prodromal” criteria to detect individuals at ultra high risk of psychosis: 2 year follow-up. Schizophr Res. 2008;105:10–17. doi: 10.1016/j.schres.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, Bramon-Bosch E, Valmaggia L, Johns L, Broome M, McGuire PK, Grasby PM. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch.Gen.Psychiatry. 2009;66:13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- 12.Siever LJ, Amin F, Coccaro EF, Bernstein D, Kavoussi RJ, Kalus O, Horvath TB, Warne P, Davidson M, Davis KL. Plasma homovanillic acid in schizotypal personality disorder. Am J Psychiatry. 1991;148:1246–1248. doi: 10.1176/ajp.148.9.1246. [DOI] [PubMed] [Google Scholar]
- 13.Abi-Dargham A, Kegeles LS, Zea-Ponce Y, Mawlawi O, Martinez D, Mitropoulou V, O’Flynn K, Koenigsberg HW, Van Heertum R, Cooper T, Laruelle M, Siever LJ. Striatal amphetamine-induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I]iodobenzamide. Biol Psychiatry. 2004;55:1001–1006. doi: 10.1016/j.biopsych.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Siever LJ, Davis KL. The pathophysiology of schizophrenia disorders: perspectives from the spectrum. Am J Psychiatry. 2004;161:398–413. doi: 10.1176/appi.ajp.161.3.398. [DOI] [PubMed] [Google Scholar]
- 15.Siever LJ, Amin F, Coccaro EF, Trestman R, Silverman J, Horvath TB, Mahon TR, Knott P, Altstiel L, Davidson M. CSF homovanillic acid in schizotypal personality disorder. Am J Psychiatry. 1993;150:149–151. doi: 10.1176/ajp.150.1.149. [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. Fourth edition American Psychiatric Association Press; Washington DC: 1994. [Google Scholar]
- 17.Kay SR, Fiszbein A. Opler LA: The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 18.Barkus EJ, Stirling J, Hopkins RS, Lewis S. Cannabis-induced psychosis-like experiences are associated with high schizotypy. Psychopathology. 2006;39:175–178. doi: 10.1159/000092678. [DOI] [PubMed] [Google Scholar]
- 19.Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300. doi: 10.1097/01.WCB.0000048520.34839.1A. [DOI] [PubMed] [Google Scholar]
- 20.Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 22.Hammers A, Allom R, Koepp MJ, Free SL, Myers R, Lemieux L, Mitchell TN, Brooks DJ, Duncan JS. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:224–247. doi: 10.1002/hbm.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumakura Y, Cumming P. PET studies of cerebral levodopa metabolism: a review of clinical findings and modeling approaches. Neuroscientist. 2009;15:635–650. doi: 10.1177/1073858409338217. [DOI] [PubMed] [Google Scholar]
- 24.Turkheimer FE, Aston JA, Asselin MC, Hinz R. Multi-resolution Bayesian regression in PET dynamic studies using wavelets. Neuroimage. 2006;32:111–121. doi: 10.1016/j.neuroimage.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, Valmaggia L, Allen P, Murray R, McGuire P. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.20. (Epub ahead of print, March 01): PMID: 21358709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinz A, Romero B, Gallinat J, Juckel G, Weinberger DR. Molecular brain imaging and the neurobiology and genetics of schizophrenia. Pharmacopsychiatry. 2003;36:S152–S157. doi: 10.1055/s-2003-45123. [DOI] [PubMed] [Google Scholar]
- 27.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chrapusta SJ, Egan MF, Wyatt RJ, Weinberger DR, Lipska BK. Neonatal ventral hippocampal damage modifies serum corticosterone and dopamine release responses to acute footshock in adult Sprague-Dawley rats. Synapse. 2003;47:270–277. doi: 10.1002/syn.10179. [DOI] [PubMed] [Google Scholar]
- 29.Pycock CJ, Kerwin RW, Carter CJ. Effect of lesion of cortical dopamine terminals on subcortical dopamine receptors in rats. Nature. 1980;286:74–76. doi: 10.1038/286074a0. [DOI] [PubMed] [Google Scholar]
- 30.Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, Montgomery AJ, Grasby PM, McGuire P. Abnormal prefrontal activation directly related to pre-synaptic striatal dopamine dysfunction in people at clinical high risk for psychosis. Mol Psychiatry. 2011;16:67–75. doi: 10.1038/mp.2009.108. [DOI] [PubMed] [Google Scholar]
- 31.Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, Weinberger DR, Berman KF. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- 32.Bertolino A, Breier A, Callicott JH, Adler C, Mattay VS, Shapiro M, Frank JA, Pickar D, Weinberger DR. The relationship between dorsolateral prefrontal neuronal N-acetylaspartate and evoked release of striatal dopamine in schizophrenia. Neuropsychopharmacology. 2000;22:125–132. doi: 10.1016/S0893-133X(99)00096-2. [DOI] [PubMed] [Google Scholar]
- 33.Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, O’Gorman RL, McLean MA, Barker GJ, McGuire P. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry. 2010;68:599–602. doi: 10.1016/j.biopsych.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 34.Bose SK, Turkheimer FE, Howes OD, Mehta MA, Cunliffe R, Stokes PR, Grasby PM. Classification of schizophrenic patients and healthy controls using [18F] fluorodopa PET imaging. Schizophr Res. 2008;106:148–155. doi: 10.1016/j.schres.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Phillips L, Velakoulis D, Yung A, McGorry PD, Wood SJ, van Erp TG, Thompson PM, Toga AW, Cannon TD, Pantelis C. Progressive brain structural changes mapped as psychosis develops in ‘at risk’ individuals. Schizophr Res. 2009;108:85–92. doi: 10.1016/j.schres.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson’s disease A 3D [(18)F]dopa-PET study. Brain. 1999;122:1637–1650. doi: 10.1093/brain/122.9.1637. [DOI] [PubMed] [Google Scholar]
- 37.Binkofski F, Reetz K, Gaser C, Hilker R, Hagenah J, Hedrich K, van ET, Thiel A, Buchel C, Pramstaller PP, Siebner HR, Klein C. Morphometric fingerprint of asymptomatic Parkin and PINK1 mutation carriers in the basal ganglia. Neurology. 2007;69:842–850. doi: 10.1212/01.wnl.0000267844.72421.6c. [DOI] [PubMed] [Google Scholar]
- 38.Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- 39.Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, Grasby PM, McGuire PK. Abnormal frontostriatal interactions in people with prodromal signs of psychosis: a multimodal imaging study. Arch Gen Psychiatry. 2010;67:683–91. doi: 10.1001/archgenpsychiatry.2010.77. [DOI] [PubMed] [Google Scholar]
- 40.Egerton A, Demjaha A, McGuire P, Mehta MA, Howes OD. The test-retest reliability of 18F-DOPA PET in assessing striatal and extrastriatal presynaptic dopaminergic function. Neuroimage. 2010;50:524–31. doi: 10.1016/j.neuroimage.2009.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.