

Abstract
New treatments need to be developed for the significant human diseases of toxoplasmosis and malaria to circumvent problems with current treatments and drug resistance. Apicomplexan parasites causing these lethal diseases are deficient in pyrimidine salvage suggesting that selective inhibition of de novo pyrimidine biosynthesis can lead to a severe loss of UMP and dTMP pools thereby inhibiting parasite RNA and DNA synthesis. Disruption of Toxoplasma gondii carbamoyl phosphate synthetase II (CPSII) induces a severe uracil auxotrophy with no detectable parasite replication in vitro and complete attenuation of virulence in mice. Here we show that a CPSII cDNA minigene efficiently complements the uracil auxotrophy of CPSII deficient mutants restoring parasite growth and virulence. Our complementation assays reveal that engineered mutations within or proximal to the catalytic triad of the N-terminal glutamine amidotransferase (GATase) domain inactivate the complementation activity of T. gondii CPSII and demonstrate a critical dependence on the apicomplexan CPSII GATase domain in vivo. Surprisingly, indels present within the T. gondii CPSII GATase domain as well as the C-terminal allosteric regulatory domain are found to be essential. In addition several mutations directed at residues implicated in allosteric regulation in Escherichia coli CPS either abolish or markedly suppress complementation and further define the functional importance of the allosteric regulatory region. Collectively, these findings identify novel features of T. gondii CPSII as potential parasite-selective targets for drug development.
Keywords: Toxoplasma gondii, carbamoyl phosphate synthetase II, complementation, essential indels, mutations
1. Introduction
Current chemotherapeutic treatments of human infections with apicomplexan parasites selectively target distinguishing aspects of essential parasite enzymes or essential biological functions. While current therapies are often effective, challenges are presented by the development of drug resistance as well as problems associated with long-term treatment regimes. Consequently, development of new drug treatments exhibiting good efficacy, low toxicity, selectivity to parasite but not host, and delayed development of widespread drug resistance is a high piority. Apicomplexan parasites such as Plasmodium falciparum and Toxoplasma gondii rely on a de novo pyrimidine synthesis pathway that is an excellent target for chemotherapy to inhibit both RNA and DNA synthesis (Fox and Bzik, 2002; Chaudhary, 2007; Fox, 2007; Painter et al., 2007). Genetic disruption of carbamoyl phosphate synthetase II (CPSII) in T. gondii induces a severe uracil auxotrophy as demonstrated by the complete absence of parasite replication in vitro and extreme attenuation of virulence in murine infection, even in severely immune deficient IFN −/− mice (Fox and Bzik, 2002). CPSII knock out strains such as cps1-1 invade host cells but do not replicate in vivo (Fox and Bzik, 2002). Strain cps1-1 possesses a unique and potent ability to elicit strong and long-lasting host protective immune responses (Fox and Bzik, 2002; Ling et al., 2006; Shaw et al., 2006; Dzierszinski et al., 2007; Zhao et al., 2007; Dzierszinski and Hunter, 2008; Wilson et al., 2008).
Carbamoyl phosphate synthetase (CPS) catalyzes the production of carbamoyl phosphate from 2 ATP molecules, glutamine, bicarbonate, and water. In most organisms carbamoyl phosphate is used as the precursor molecule for biosynthesis of uridine monophosphate (UMP) and arginine (Beckwith et al., 1962; Jones, 1980; Davis, 1986). Surprisingly, parasites from the phylum Apicomplexa (Plasmodium sp. and T. gondii) are natural arginine auxotrophs and the parasite CPSII is the first committed step in pyrimidine biosynthesis (Fox et al., 2004).
Escherichia coli CPS consists of a large subunit (CPS domains) with binding sites for substrates and allosteric effectors, and an associated small subunit glutamine amidotransferase (GATase) belonging to the Class I amidotransferase superfamily that catalyzes the hydrolysis of glutamine to glutamate and ammonia (Beckwith et al., 1962; Foley et al., 1971; Raushel et al., 1978; Thoden et al., 1999b). In contrast, the eukaryotic CPSII has an N-terminal GATase that is fused to the CPS polypeptide, as well as possessing fused C-terminal domains that catalyze subsequent steps in pyrimidine biosynthesis (Jones, 1980; Davis, 1986; Davidson et al., 1993). Protozoa that include medically important parasites (Plasmodium, Toxoplasma, Babesia, Trypanosoma, Leishmaina) possess a single atypical and simplified eukaryotic CPSII comprising an N-terminal GATase fused to CPS via an atypically short linker but do not possess fused C-terminal domains that catalyze subsequent steps in pyrimidine biosynthesis (Aoki et al., 1994; Flores et al., 1994; Chansiri and Bagnara, 1995; Gao et al., 1998; Nara et al., 1998; Gao et al., 1999; Fox and Bzik, 2003). Because these subsequent enzyme activities of the de novo pyrimidine pathway are not directly fused to the C-terminus of protozoan forms of CPSII, we could easily engineer a functional CPSII minigene to evaluate genetic complementation of T. gondii CPSII function in vivo.
While the structure, function, and mechanisms of CPS catalysis and regulation have been well characterized for E. coli CPS (Thoden et al., 1997; Mora et al., 1999; Thoden et al., 1999b; Fresquet et al., 2000; Huang and Raushel, 2000a, 2000b, 2000c; Miles and Raushel, 2000), the larger CPSII polypeptides of eukaryotes are not yet well characterized at both the functional and structural level (Davidson et al., 1993; Graves et al., 2000; Fox and Bzik, 2003; Simmons et al., 2004; Kothe et al., 2005). Here, we report functional complementation of uracil auxotrophy in T. gondii based on CPSII minigenes. The development of an efficient system for complementation of uracil auxotrophy provides a new genetic model for positive selection and enables a direct genetic dissection of functional domains in a eukaryotic CPSII enzyme in vivo. Our results identify unique indels present in apicomplexan CPSII as distinct functionally important domains that may potentially serve as novel parasite-selective drug targets.
2. Materials and methods
2.1. Plasmid Construction
A functional CPSII minigene encoding the authentic 1687 amino acids of carbamoyl phosphate synthetase was constructed by sequential coupling of defined cDNA segments generated by reverse transcriptase/PCR. First, a 1829 bp cDNA for the N-terminal GATase domain of CPSII was amplified from polyA+ mRNA from the RH strain (5′-ACTAGTGGTGATGACGACGACAAGATGCCTCACAGTGGAGGGC-3′ and 5′-GATATCCACGTGTCGCGGCCGCGCTCTC-3′). The 1829 bp cDNA was introduced (SpeI/EcoRV) into pET41b (SpeI/XhoI-blunted). Next an N-terminal section of the CPS domain cDNA comprising bp 1829 to bp 3532 was generated (5′-GAGAGCGCGGCCGCGAC-3′ and 5′-CACGTGGAGGCGAGACGTCGTCGTC-3′) and fused to the GATase domain (NotI/PmlI). The remainder of the CPS domain was constructed by amplifying two cDNA segments, bp 3003-4097 and bp 4097-5064 (5′-AGTACTTGATGAATTCACCG-3′ and 5′-TTTCTGCGAGATCTTCTTCACG-3′, 5′-GCGTGAAGAAGATCTCGCAG-3′ and 5′-ATCGATCACGTGATTTTTGAGGCCAGTATTCATCC-3′, respectively), and then the two C-terminal segments were fused in PCR4TOPO (EcoRI/BglII). Finally the C-terminal section of CPS was fused with the N-terminal section in PET41b (EcoRI/PmlI) and the complete 5064 bp CPSII minigene coding sequence was determined to verify authenticity.
5′ UTR and 3′ UTR were amplified from RH genomic DNA. 5′ UTR to bp −516 was amplified (5′-GCTAGCGTGGACCCCCATTATCCTTCGC-3′ and 5′-ACTAGTCACTCGTCGAATGGTTGCGTCTG-3′), and 5′ UTR to bp −2057 was amplified (5′-GCTAGCGTGGACCCCCATTATCCTTCGC-3′ and 5′-ACTAGTGAAATCGCGATCAACGCGACAG-3′). The 3′ UTR (920 bp) was amplified (5′-AGTACTTGCACCACCACCACCACCACTAATTTCCAATACTTTCGCCAAAAACGTTCC-3′ and 5′-GCGCACGTGGTTGAGAGCTTGACCCGCATGCA-3′). Finally 5′ UTR segments (ScaI/SpeI) were fused into the CPSII minigene (SpeI), and subsequently the 3′ UTR (ScaI/PmlI) was fused into the above plasmid(s) (ScaI/PmlI).
2.2. Site-directed mutagenesis
Mutations were first introduced into the either the GATase or CPS domains using Stratagene’s PCR based QuikChange® II XL Site-Directed Mutagenesis Kit. Products were Dpn I digested, transformed into XL-10 Gold Ultracomp cells, and subsequently transferred into the full CPS II complementation vector. Forward and reverse complementary primers containing the desired mutations were used to create the desired mutations. Plasmids with correct coding region and engineered CPSII minigene mutation(s) were verified by sequencing prior to transfection experiments.
2.3. Parasite culture and transfection
Tachyzoites of strain cps1-1 were maintained in human foreskin fibroblasts with or without uracil supplementation (300 M) (Fox and Bzik, 2002). Wild type or CPSII minigene plasmids containing defined mutations were transfected (20 g) into the cps1-1 background and selections were performed without drug addition in the absence of uracil using previously described methods (Fox and Bzik, 2002). Briefly 1 × 107 freshly isolated tachyzoites of strain cps1-1 were transfected in 0.4 ml electroporation buffer (Donald and Roos, 1993). Growth and complementation in the absence of uracil supplementation was scored as described below. Transfected cps1-1 parasites growing in the absence of uracil were cloned by limiting dilution.
2.4. Determination of parasite growth rate in cps1-1 complementation assays
Following transfection of strain cps1-1 with wild-type or mutant CPSII minigenes fresh monolayers of HFF cells were infected in 5 ml of infection medium with 25% of the transfected parasites. At 2h post-transfection monolayers were washed 2 times with PBS to remove parasites that had not invaded and fresh infection medium returned to cultures. At 36 h post-transfection the growth rate was measured by scoring tachyzoites per vacuole by examination of randomly selected vacuoles in light microscopy. Vacuoles containing one parasite per vacuole were excluded from counting. A total of 50 vacuoles with 2 or more parasites were scored to determine a mean of parasites per vacuole in each transient transfection assay. The growth rate was then converted to a relative doubling time based on a 36 h growth period. Experiments were independently repeated at least three times. A student t-test was used to calculate the standard error of the mean.
2.5. Determination of transient complementation efficiency
Following transfection of strain cps1-1 with wild-type or mutant CPSII minigenes fresh monolayers of HFF cells were infected in 5 ml of infection medium with 25% of the transfected parasites. At 2h post-transfection monolayers were washed 2 times with PBS to remove parasites that had not invaded and fresh infection medium was returned to cultures. At 36 h post-transfection the transient transfection efficiency was measured at the same time as growth rate by scoring the number of vacuoles containing two or more parasites per light microscope “field” at a fixed objective during the scoring of 50 vacuoles as described above in section 2.4. Vacuoles with only one parasite per vacuole were excluded from counting. The transient complementation efficiency is reported as % of control vacuoles observed using the wild-type Pc 4 CPSII minigene. Experiments were repeated a minimum of three independent times and a student t-test was used to calculate the standard error of the mean.
2.6. Determination of stable complementation efficiency
Strain cps1-1 was transfected with wild-type or mutant CPSII minigenes. Immediately following transfection the contents of the transfection curvette (and the first wash of the curvette with infection medium) was transferred into 20 ml of infection medium. Serial dilutions of the transfected tachyzoites were prepared in infection medium and fresh HFF monolayers in duplicate 25 cm2 flasks were infected with 2%, 0.5%, or 0.1% of total transfected parasites and plaque forming units (PFU) were scored. The infected HFF flasks were left undisturbed for 7 days and then monolayers were fixed and stained with Coumassie Blue to score the number of PFU. The stable complementation efficiency is reported as % of control PFU observed using the wild-type Pc 4 CPSII minigene. Experiments were repeated a minimum of three independent times and a student t-test was used to calculate the standard error of the mean. Control experiments using the pET41b plasmid without the CPSII minigene were conducted a minimum of 4 times and no PFU were observed in the absence of uracil supplementation.
2.7. Virulence assays
Adult 6-8 week old C57Bl/6 mice were obtained from Jackson Labs and mice were maintained in tecniplast Seal Safe mouse cages on vent racks at the Dartmouth-HitchcockMedical Center mouse facility. All mice were cared for and handled according to Animal Care and Use Program of Dartmouth College using NIH approved Institutional animal care and use committee guidelines. Tachyzoites were isolated from freshly lysed HFF monolayers and were purified by filtration through sterile 3 m nuclepore membranes. Tachyzoites were washed in PBS, counted in a hemocytometer to determine parasite number, and a PBS solution prepared with 1 × 104 or 1 × 107 tachyzoites per ml. Groups of 4 mice were injected intraperitoneally (i.p.) with 0.2 ml (2 × 106 tachyzoites for strain cps1-1, or 2 × 103 tachyzoites for cloned isolates obtained after transfections with Pc 4, N348A, and 873-910 plasmids. Mice were then monitored daily for degree of illness and survival. The virulence assays were performed twice.
3. Results and Discussion
3.1. Complementation of uracil auxotrophy and virulence
The cps1-1 mutant of T. gondii invades host cells but due to pyrimidine starvation exhibits no detectable growth rate in the absence of uracil supplementation (Fox and Bzik, 2002). Providing a functional CPSII gene to the cps1-1 mutant should restore production of carbamoyl phosphate required for biosynthesis of UMP. Due to the large size and intron/exon complexity of the genomic DNA locus of CPSII (Fox and Bzik, 2003), we constructed a cDNA minigene encoding the 1687 amino acid CPSII polypeptide under the control of authentic CPSII 5′ UTR and 3′ UTR regulatory regions (see Materials and Methods for details of plasmid construction). To examine complementation, plasmids representing a promoter-less minigene coding region construct as well as minigenes under the control of either 0.5 kb or 2.0 kb of 5′ UTR (Fig. 1) were transfected into the cps1-1 uracil auxotrophic T. gondii mutant and cultured in HFF cells in the absence or presence of uracil. Tachyzoites per parasite vacuole were scored 36h after transfection. The promoter-less construct Pc 0 as well as the 0.5 kb 5′ UTR construct Pc 2 failed to complement the cps1-1 mutant and did not restore any detectable growth rate in the absence of uracil. In contrast, the 2 kb 5′ UTR CPSII minigene Pc 4 efficiently complemented the cps1-1 mutant and restored a normal tachyzoite growth rate in the absence of uracil.
Figure 1.
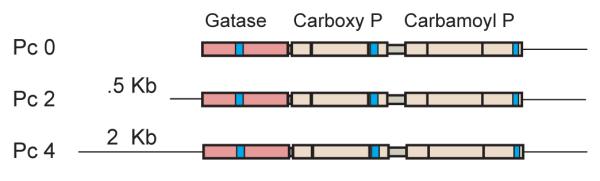
Domain structure of T. gondii CPSII is shown relative to GATase, Carboxy phosphate (P), and carbamoyl phosphate (P) domains (not to scale). Plasmids Pc 0, Pc 2, and Pc 4 possess 0 kb, 0.5 kb, and 2 kb 5′UTR, respectively, a coding cassette of CPSII cDNA, and 0.92 kb 3′ UTR element.
In quantitative experiments, plasmids Pc 0, Pc 2, and Pc 4 were transfected into strain cps1-1 and total plaque forming units (PFU) determined at 7 days after transfection in the absence of uracil supplementation. Plasmids Pc 0, Pc 2, and the vector pET41b produced no PFU (0%) following transfection of cps1-1. In contrast, during the course of this study 16 independent transfections of 1 × 107 cps1-1 tachyzoites were performed using plasmid Pc 4 and the mean of PFU was determined to be 1.8% ± 0.14 of the original 1 × 107 transfected tachyzoites.
PFU arising from transfection experiments with Pc 4 or mutant alleles of CPSII (sections 3.2 and 3.3) were cloned by limiting dilution and stable isolates were examined for virulence in mice. The cps1-1 strain is essentially completely avirulent (Fig. 2) (Fox and Bzik, 2002). In contrast, transfection of cps1-1 with plasmid Pc 4 produced stable clones that exhibit the high virulence phenotype of the parental RH strain in C57Bl/6 mice (Fig. 2).
Figure 2.
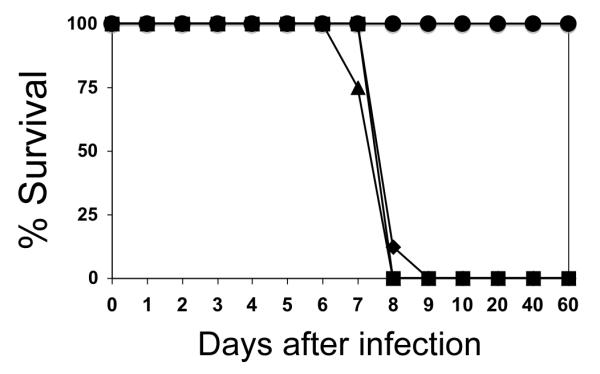
Virulence of complemented cps1-1 strains. Virulence of strains was determined by intraperitoneal injection of C57BL/6 mice with 2 × 106 tachyzoites of strain cps1-1 (circles), or 2 × 103 tachyzoites of a cloned isolate of cps-1 complemented with Pc 4 (tiangles), mutant A348 (squares), or mutant 873-910 (diamonds). Groups of 4 mice were infected intraperitoneally with freshly isolated tachyzoites. Data from two experiments is shown as % survival of animals compared to cps1-1 infection.
3.2. Functional analysis of the glutamine amidotransferase domain of CPSII
The requirement of the fused eukaryotic GATase domain to produce ammonia for CPS function in apicomplexan CPSII has not been previously examined in vivo. We constructed the mutation C345 to A345 in plasmid Pc 4 in an essential catalytic triad residue of the GATase domain equivalent to C269 that abolished GATase activity in E. coli (Rubino et al., 1987) (Table 1). The T. gondii C345 to A345 mutation completely abolished complementation activity indicating that CPSII is dependent on a functional GATase domain for the production of ammonia in vivo (Table 2). The dependence of T. gondii CPSII activity on the amidotransferase domain validates the analysis of unique sites within this domain as parasite specific drug targets. We next targeted the proximal N348 residue that is selectively present in most protozoan CPSII enzymes (Fox and Bzik, 2003) (Supplemental Figure). Mutation of N348 to R348 abolished complementation activity whereas mutation of N348 to A348 modestly reduced the initial growth rate (from 7.4 to 8.4 h) in the 36 h growth assay but did not significantly interfere with the efficiency of transient or stable complementation (Table 2). A stable clone isolated after transfection with the A348 mutant exhibited the high virulence phenotype of the parental RH strain in C57Bl/6 mice (Fig. 2).
TABLE 1.
Effects of mutations in E. coli CPS or Hamster CPSII
| Mutation | Location in Tg | Effect on CPS or CPSII |
|---|---|---|
| Ec C269A | Tg C345 | Abolishes GATase activity |
| Ec G359F | Tg G435 | Ammonia leaks, uncoupling GATase & CPS |
| Ha T456A | Tg T533 | Abolishes MAPK activation |
| Ec E761A | Tg E1316 | Abolishes ornithine activation and UMP repression |
| Ec H781K | Tg H1336 | Reduces CPS activity, ornithine activation, and UMP repression |
| Ec T974A | Tg T1530 | Reduces ornithine activation, IMP activation, and UMP repression |
| Ha S1345A | Tg T1530 | Reduces PRPP activation |
| Ec T1042A | Tg T1649 | Reduces ornithine binding |
Escherichia coli (Ec); Hamster (Ha); T. gondii (Tg)
See text for references and Supplemental Figure for alignments. Table 1 identifies the location of residues in T. gondii CPSII that correspond to residues previously examined in mutagenesis studies in either E. coli or Hamster CPS enzymes.
TABLE 2.
Effect of GATase mutations on T. gondii CPSII complementation activity
| GATase Mutation | growth rate (h) | transient efficiency1 (% of control vacuoles) |
stable efficiency2 (% of control PFU) |
|---|---|---|---|
| Tg Pc 4 (w.t.) | 7.4 ± 0.08 | 100 | 100 |
| Tg Δ172-229 | nd | 0 | 0 |
| Tg C345A | nd | 0 | 0 |
| Tg N348R | nd | 0 | 0 |
| Tg N348A | 8.2 ± 0.27 | 96 ± 6.6 | 91 ± 6.8 |
| Tg P385R | 11.5 ± 0.95 | 18 ± 4.0 | 1.3 ± 0.51 |
| Tg G435F | 12.4 ± 0.45 | 8.4 ± 2.3 | 0.6 ± 0.27 |
| Tg Δ455-457 | 7.5 ± 0.15 | 98 ± 9.8 | 103 ± 13 |
| Tg Δ454-470 | 8.9 ± 0.51 | 64 ± 19 | 11 ± 4.2 |
no growth detected (nd)
Table 2 shows the effect of engineered mutations in the GATase domain of T. gondii CPSII on parasite growth rate, transient complementation efficiency, and stable complementation efficiency in cps1-1 complementation assays.
Transient complementation is scored as the % of control vacuoles of Pc 4 transfection.
Stable complementation is scored as % of control PFU of Pc 4 transfection.
Amino acid 385 adjacent to residues comprising the catalytic triad is uniquely a proline residue in T. gondii CPSII (Fox and Bzik, 2003) (Supplemental Figure). Changing amino acid P385 to R385 had a dramatic effect on reducing the initial parasite growth rate (from 7.4 to 11.5 h), and reduced transient complementation efficiency to 18% and stable complementation efficiency to 1.3% of the control (Table 2). Clones obtained from transfection with the R385 mutant indicated that 3 to 7 copies of the CPSII minigene were stably integrated in complemented parasites (data not shown).
The naturally occurring CPS enzyme consists of an , -heterodimeric protein with an “ammonia tunnel” which channels ammonia produced by the small subunit GATase to the binding site for the first molecule of MgATP located within the N-terminal half of CPS in the carboxy phosphate domain (Miles et al., 1993; Thoden et al., 1997; Miles et al., 1998; Huang and Raushel, 1999; Thoden et al., 1999b; Huang and Raushel, 2000b, 2000a, 2000c; Miles and Raushel, 2000; Miles et al., 2002). The carboxy phosphate domain first phosphorylates (MgATP) bicarbonate to carboxy phosphate which activates GATase activity ~1000-fold (Miles and Raushel, 2000). Carboxy phosphate then combines rapidly with ammonia delivered in a highly coordinated fashion from the GATase domain to form carbamate (Thoden et al., 1999a). Perforation of the ammonia tunnel in E. coli CPS via mutation of G359 to F359 results in ammonia leakage from the tunnel and loss of CPS activity (Table 1). The Gly residue corresponding to E. coli G359 is universally conserved in all GATases that are coupled with CPS activity (Fox and Bzik, 2003) (Supplemental Figure). Mutation of T. gondii G435 to F435, corresponding to the E. coli G359 to F359 mutation, caused a marked reduction in the initial parasite growth rate (from 7.4 to 12.4 h), and reduced transient complementation efficiency to 8.4% and stable complementation efficiency to 0.6% of the control. These results suggest that disruption of the T. gondii CPSII ammonia tunnel markedly decreased CPSII complementation activity in vivo (Table 2), again revealing the strict dependence of the parasite CPS on ammonia produced by the fused GATase activity of CPSII.
We also examined the domain in T. gondii CPSII (amino acids 454-470) that links GATase with CPS domains. While deletion of amino acids 455 to 457 had no detectable effect on complementation activity, deletion of amino acids 454 to 470 caused a significant reduction in the initial parasite growth rate from 7.4 to 8.9 h, as well as a reduction in stable complementation efficiency to 11% of the control (Table 2).
3.3. Deletion of indels
Apicomplexan CPSII enzymes contain locations where novel insertions of amino acids (indels) occur at several locations within the GATase and CPS domains (Fig. 3 and Supplemental Figure). While ribozyme targeting of a P. falciparum CPSII indel at the RNA level was previously shown to inhibit parasite proliferation (Flores et al., 1997), previous studies have not directly addressed the functional importance of indels in parasite proteins. The unusually frequent occurrence of novel insertions of low or high complexity within protozoan parasite proteins, particularly in Plasmodium sp. and T. gondii (Cherkasov et al., 2006; DePristo et al., 2006), would provide parasite selective drug targets in instances where the indel provides a necessary function. Functional complementation of CPSII in T. gondii enabled a genetic test of essential indels. In the GATase domain we targeted the T. gondii CPSII indel location where other apicomplexan CPSII also exhibit a large amino acid insertion that other protozoans, fungi, mammals, and prokaryotes do not share (Fig. 3 and Supplemental Figure). Deletion of the GATase indel (E171-A229) completely abolished CPSII function as demonstrated by the inability of this mutant to complement the uracil auxotrophy of cps1-1 (Table 2) These results further establish this novel parasite-specific indel as a possible parasite-selective drug target within the essential GATase domain.
Figure 3.
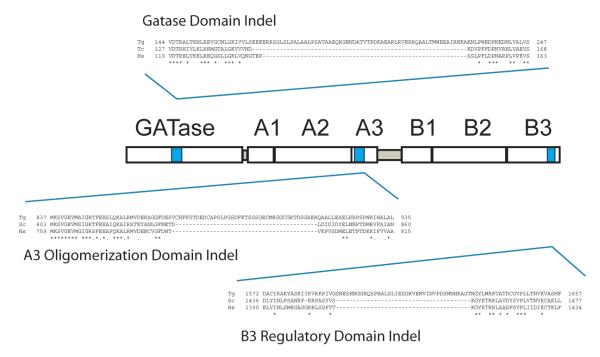
The T. gondii indels location in the GATase, the CPS.A3 and CPS.B3 domains is illustrated. The CPS domain consists of two halves, A & B, functional synthetase domains which can be subdivided into sections 1, 2, and 3 (A.1, A.2, & A.3 and B.1, B2, & B.3) corresponding to functional subdomains described by the structure of E. coli CPS (Thoden et al., 1999a; Thoden et al., 1999b). A.2 and B.2 correspond to the catalytic subdomains. The binding for MgATP in both domains is wedged between the A.2 and A.3 and B.2 and B.3 domains.
CPS is controlled via allosteric mechanisms acting through specific allosteric effectors and their binding interactions with the C-terminal domain of CPS.B (Braxton et al., 1999; Thoden et al., 1999c; Fresquet et al., 2000; Pierrat et al., 2002). Due to the complex assembly of 5 substrates via 4 chemical reactions at three active sites separated by ~100 Å (Thoden et al., 1997; Thoden et al., 1999b), the CPS enzyme activities are highly synchronized with one another and activity is tightly regulated by allosteric end-product repression as well as allosteric activation. The C-terminal ~150 amino acids of the large CPS subunit contain the regulatory domain controlling allosteric regulation of CPS activity where binding pockets exist that mediate direct physical interaction with allosteric effector molecules (Mora et al., 1999; Thoden et al., 1999a; Fresquet et al., 2000). Interaction of allosteric effectors with the C-terminal regulatory domain directly trigger conformational changes in CPS affecting activity and/or synchronization of active sites (Thoden et al., 1999b). T. gondii and B. bovis CPSII share an indel location within the C-terminal regulatory domain (Fig. 3 and Supplemental Figure). To examine whether this novel indel was essential to CPSII function we constructed a deletion of the T. gondii C-terminal indel (G1592 to R1628). This deletion completely abolished CPSII complementation activity (Table 3).
TABLE 3.
Effect of CPS mutations on T. gondii CPSII complementation activity
| CPS Mutation | growth rate (h) | transient efficiency (% of control vacuoles) |
stable efficiency (% of control PFU) |
|---|---|---|---|
| Tg Pc 4 (w.t.) | 7.4 ± 0.08 | 100 | 100 |
| Tg T533A | 7.4 ± 0.18 | 105 ± 7.2 | 98 ± 6.9 |
| Tg S581A | 7.3 ± 0.14 | 109 ± 11 | 104 ± 8.5 |
| Tg Δ873-910 | 8.2 ± 0.27 | 113 ± 12 | 65 ± 11 |
| Tg E1316A | nd | 0 | 0 |
| Tg E1318A | nd | 0 | 0 |
| Tg H1336K | nd | 0 | 0 |
| Tg T1430A | 7.7 ± 0.21 | 86 ± 5.7 | 82 ± 8.4 |
| Tg T1530A | 8.6 ± 0.25 | 51 ± 13 | 10 ± 3.2 |
| Tg T1530 fs | nd | 0 | 0 |
| Tg Δ1592-1628 | nd | 0 | 0 |
| Tg T1649A | 8.5 ± 0.21 | 65 ± 5.2 | 37 ± 7.0 |
no growth rate detected (nd). Table 3 shows the effect of engineered mutations in the CPS domains of T. gondii CPSII on parasite growth rate, transient and stable complementation efficiency in cps1-1 complementation assays. See Table 2 for descriptions of complementation.
The carboxy terminal region of the CPSII.A domain (domain A3, Fig. 3) contains the oligomerization domain known to coordinate the formation of tetramers of E. coli CPS (Thoden et al., 1997; Kim and Raushel, 2001). On the N-terminal side of the putative CPSII oligomerization domain a novel indel of ~34 amino acids is present in T. gondii CPSII (Supplemental Figure) (Fox and Bzik, 2003). Deletion of this indel (C873-G910) caused a minor, but detectable, disruption in complementation activity based on a reduced initial growth rate (from 7.4 to 8.2 h), similar transient complementation efficiency (113%), and slightly reduced stable complementation efficiency to 65% of the control (Table 3). Interestingly, the more subtle effect of this indel deletion in the T. gondii CPSII oligomerization domain is potentially similar to the relatively minor effect on E. coli CPS activity previously observed in mutants blocked in oligomerization contact regions that prevent tetramer but not dimer formation (Kim and Raushel, 2001). A stable clone obtained from transfection with the 873-910 mutant exhibited the high virulence phenotype of the parental RH strain in C57Bl/6 mice (Fig. 2).
3.4. CPSII regulatory domains
Suppression of mammalian CPSII activity is highly dependent on the presence or absence of regulated phosphorylation at a distinct MAP kinase site at (T456) in the carboxy phosphate CPSII.A domain (Graves et al., 2000). T. gondii and other lower eukaryotic forms of CPSII are distinct from mammalian CPSII in lacking this critical MAPK site (Fox and Bzik, 2003). T. gondii CPSII shares the Threonine residue corresponding to the mammalian T456 position and this residue was mutated to exclude the possibility that a novel parasite kinase may control CPSII activation. Mutation of T533 to A533 in T. gondii CPSII had no significant effect on complementation activity (Table 3). We also examined whether the nearby putative MAPK core SP site present in T. gondii but absent in mammalian CPSII was necessary for activity. Mutation of S581 to A581 also had no detectable effect on complementation activity (Table 3). E. coli CPS activity is repressed by UMP, strongly activated by ornithine and weakly activated by IMP, whereas eukaryotic CPSII is typically activated by PRPP and is repressed by UTP (or UDP in kinetiplastids) (Jones, 1980; Nara et al., 1998). Strikingly, T. gondii CPSII is insensitive to allosteric activation by PRPP, and relatively high levels of UTP were required for suppression (Asai et al., 1983). To gain further insight into the importance of allosteric regulatory regions and the type of regulation occurring in T. gondii CPSII we constructed mutations in several amino acid residues that were conserved between the T. gondii and E. coli C-terminal regulatory domains that were also known to mediate allosteric control in E. coli CPS (Table 1). While IMP induces only modest allosteric effects on E. coli CPS activity, ornithine potently activates the glutamine dependent ATPase and ATP synthesis reactions and thereby markedly upregulates activity by increasing the affinity of CPS for its nucleotide substrates while overriding the strong effect of UMP to suppress the catalytic activity of CPS (Braxton et al., 1999). We targeted conserved ornithine binding sites identified in T. gondii CPSII analogous to those previously identified in E. coli (Table 1). The K loop coordinates the binding of a potassium ion and includes the conserved residue H781 that also coordinates the transmission of the allosteric regulatory signals from the C-terminal regulatory domain (Thoden et al., 1999a; Pierrat et al., 2002). Mutation of H781 to K781 in E. coli CPS reduced the magnitude of the allosteric effects of both ornithine and UMP, decreased the allosteric response to IMP, and also diminished the catalytic activity of CPS by one to two orders of magnitude in the absence of allosteric effectors (Pierrat et al., 2002). In T. gondii we found that a CPSII minigene with the analogous mutation (H1336 to K1336) failed to detectably complement the uracil auxotrophy of the cps1-1 mutant (Table 3). A second mutation (E761 to A761) also within the K loop of E. coli CPS was previously found to be crucial to the transmission of the allosteric activation signal by ornithine, but did not affect catalytic turnover in the absence of effectors. This mutation also eliminated feedback repression by UMP and decreased activation by IMP (Pierrat et al., 2002). In T. gondii CPSII we found the analogous mutation of E1316 to A1316 resulted in a complete loss of complementation activity by the mutant CPSII minigene (Table 3). Interestingly, a second mutation (E1318A to A1318) at a nonconserved residue two amino acids downstream of E1316 also resulted in a complete loss of complementation activity by the mutant CPSII minigene (Table 3).
To further define the extent of the impact of mutations within the allosteric regulatory region we also targeted a residue conserved between T. gondii and the E. coli CPS.B3 allosteric regulatory region (T974) that strongly influenced the allosteric response to ornithine in E. coli CPS (Table 1). The mutation T974 to A974 disrupted the IMP/UMP binding pocket in E. coli CPS and not only abolished UMP inhibition and IMP activation, but also decreased activation by ornithine (Fresquet et al., 2000). Interestingly, this site also plays a role in allosteric control in mammalian CPSII as well since mutation of the corresponding hamster residue S1355 to A1355 nearly abolished activation by PRPP and lowered overall CPSII activity ~5-fold (Simmons et al., 2004). While T. gondii CPSII is nonresponsive to PRPP in vitro, mutation of the analogous residue in T. gondii CPSII (T1530 to A1530) significantly reduced CPSII complementation activity based on a reduced initial parasite growth rate (from 7.4 to 8.6 h), and reduced transient complementation efficiency to 51% and stable complementation efficiency to 10% of the control suggesting several copies of this mutant allele are necessary to fully support parasite growth (Table 3). A second conserved residue (T1042) in the E. coli regulatory domain plays a direct role in ornithine binding (Pierrat et al., 2002). Mutation of T1042 to A1042 in E. coli CPS reduces the magnitude of the allosteric response to ornithine. The analogous mutation T1649 to A1649 in T. gondii had the more modest effect of slightly lowering the initial growth rate (from 7.4 to 8.5 h), and reduced transient complementation efficiency to 65% and stable complementation efficiency to 37% of the control. The overall importance of the relatively nonconserved T. gondii CPSII C-terminal regulatory region as a putative drug target could readily be seen through a frameshift mutation that was incorporated into T1530 that effectively deleted much of the CPS.B3 regulatory region and abolished complementation (Table 3).
Our results suggest that residues that are known to mediate allosteric interaction in E. coli CPS and mammalian CPSII are likely to play a role in allosteric regulation in T. gondii CPSII. The major negative impact on the ability to complement by mutating residues that selectively affect up-regulation by ornithine in E. coli CPS, regardless of whether the allosteric effects of UMP or IMP are maintained, may indicate allosteric regulation by ornithine or a related effector in Toxoplasma gondii and could indicate points at which to intervene biochemically. It is notable that T. gondii is metabolically distinct from animals and many eukaryotes in being naturally auxotrophic for both ornithine and purines (Chaudhary, 2007; Fox, 2007). These natural auxotrophic requirements for parasite growth may be linked to novel strategies for allosteric regulation of CPSII, control of pyrimidine biosynthesis and balancing of purine and pyrimidine pools in protozoan parasites (Asai et al., 1983; Nara et al., 1998; Fox and Bzik, 2003). Further work is necessary to determine the specific allosteric effectors controlling T. gondii CPSII. Previous studies show that indels occur more frequently within genes from T. gondii and Plasmodium sp. and may represent parasite-selective drug targets (Cherkasov et al., 2006; DePristo et al., 2006). Functional data supporting this latter hypothesis has not been previously reported. Here we show that deletion of the GATase indel as well as the C-terminal regulatory indel completely abolished complementation activity of CPSII minigenes. Our results suggest a functional and essential role for a number of C-terminal amino acids known to transmit the allosteric regulatory signals in mammalian CPSII or E. coli CPS. T. gondii CPSII is dependent on GATase for production of ammonia in vivo, and an ammonia tunnel potentially analogous to the ammonia tunnel described in E. coli CPS appears to be present. Additional approaches are necessary to determine the specific mechanism(s) responsible for mutations in CPSII that abrogate complementation activity. In summary our results reveal unique features of T. gondii CPSII that may provide novel parasite-selective drug targets for the inhibition of CPSII function, thereby starving the parasite of essential pyrimidines required for RNA and DNA synthesis.
Supplementary Material
Supplemental Figure. Sequence alignment of CPSII genes from Escherichia coli (EC), Toxoplasma gondii (Tg), Plasmodium falciparum (Pf), Babesia bovis (Bb), Trypanosoma cruzi (Tc), Leishmania major (Lm), Saccharomyces cerevisiae (Sc), Homo sapiens (Hs). Locations of indels in Toxoplasma gondii and locations where point mutations were constructed are shown in red type.
Acknowledgements
We gratefully thank Elmer R. Pfefferkorn for discussion. This work was funded in part by National Institutes of Health Grant AI41930 (to D.J.B.)
Footnotes
Note: Supplementary data associated with this article.
References
- Aoki T, Shimogawara R, Ochiai K, Yamasaki H, Shimada J. Molecular characterization of a carbamoyl-phosphate synthetase II (CPS II) gene from Trypanosoma cruzi. Adv Exp Med Biol. 1994;370:513–516. doi: 10.1007/978-1-4615-2584-4_108. [DOI] [PubMed] [Google Scholar]
- Asai T, O’Sullivan WJ, Kobayashi M, Gero AM, Yokogawa M, Tatibana M. Enzymes of the de novo pyrimidine biosynthetic pathway in Toxoplasma gondii. Mol Biochem Parasitol. 1983;7:89–100. doi: 10.1016/0166-6851(83)90037-3. [DOI] [PubMed] [Google Scholar]
- Beckwith JR, Pardee AB, Austrian R, Jacob F. Coordination of the synthesis of the enzymes in the pyrimidine pathway of Escherichia coli. J. Mol. Biol. 1962;5:618–635. doi: 10.1016/s0022-2836(62)80090-4. [DOI] [PubMed] [Google Scholar]
- Braxton BL, Mullins LS, Raushel FM, Reinhart GD. Allosteric dominance in carbamoyl phosphate synthetase. Biochemistry. 1999;38:1394–1401. doi: 10.1021/bi982097w. [DOI] [PubMed] [Google Scholar]
- Chansiri K, Bagnara AS. The structural gene for carbamoyl phosphate synthetase from the protozoan parasite Babesia bovis. Mol Biochem Parasitol. 1995;74:239–243. doi: 10.1016/0166-6851(95)02499-9. [DOI] [PubMed] [Google Scholar]
- Chaudhary K, Fox BA, Bzik DJ. Toxoplasma gondii: The Model Apicomplexan Parasite: Perspectives and Methods. Elsevier; London: 2007. [Google Scholar]
- Cherkasov A, Lee SJ, Nandan D, Reiner NE. Large-scale survey for potentially targetable indels in bacterial and protozoan proteins. Proteins. 2006;62:371–380. doi: 10.1002/prot.20631. [DOI] [PubMed] [Google Scholar]
- Davidson JN, Chen KC, Jamison RS, Musmanno LA, Kern CB. The evolutionary history of the first three enzymes in pyrimidine biosynthesis. Bioessays. 1993;15:157–164. doi: 10.1002/bies.950150303. [DOI] [PubMed] [Google Scholar]
- Davis RH. Compartmental and regulatory mechanisms in arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol. Rev. 1986;50:280–313. doi: 10.1128/mr.50.3.280-313.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Zilversmit MM, Hartl DL. On the abundance, amino acid composition, and evolutionary dynamics of low-complexity regions in proteins. Gene. 2006;378:19–30. doi: 10.1016/j.gene.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Dzierszinski F, Pepper M, Stumhofer JS, LaRosa DF, Wilson EH, Turka LA, Halonen SK, Hunter CA, Roos DS. Presentation of Toxoplasma gondii antigens via the endogenous major histocompatibility complex class I pathway in nonprofessional and professional antigen-presenting cells. Infect Immun. 2007;75:5200–5209. doi: 10.1128/IAI.00954-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierszinski FS, Hunter CA. Advances in the use of genetically engineered parasites to study immunity to Toxoplasma gondii. Parasite Immunol. 2008;30:235–244. doi: 10.1111/j.1365-3024.2007.01016.x. [DOI] [PubMed] [Google Scholar]
- Flores MV, O’Sullivan WJ, Stewart TS. Characterisation of the carbamoyl phosphate synthetase gene from Plasmodium falciparum. Mol Biochem Parasitol. 1994;68:315–318. doi: 10.1016/0166-6851(94)90176-7. [DOI] [PubMed] [Google Scholar]
- Flores MV, Atkins D, Wade D, O’Sullivan WJ, Stewart TS. Inhibition of Plasmodium falciparum proliferation in vitro by ribozymes. J Biol Chem. 1997;272:16940–16945. doi: 10.1074/jbc.272.27.16940. [DOI] [PubMed] [Google Scholar]
- Foley R, Poon J, Anderson PM. Characterization of the reactive sulfhydryl groups in carbamyl phosphate synthetase of Escherichia coli. Biochemistry. 1971;10:4562–4569. doi: 10.1021/bi00800a034. [DOI] [PubMed] [Google Scholar]
- Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature. 2002;415:926–929. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
- Fox BA, Bzik DJ. Organisation and sequence determination of glutamine-dependent carbamoyl phosphate synthetase II in Toxoplasma gondii. Int J Parasitol. 2003;33:89–96. doi: 10.1016/s0020-7519(02)00214-x. [DOI] [PubMed] [Google Scholar]
- Fox BA, Gigley JP, Bzik DJ. Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol. 2004;34:323–331. doi: 10.1016/j.ijpara.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Fox BA, Chaudhary K, Bzik DJ. Toxoplasma: Molecular and Cellular Biology. Horizon Bioscience; Norwich: 2007. [Google Scholar]
- Fresquet V, Mora P, Rochera L, Ramon-Maiques S, Rubio V, Cervera J. Site-directed mutagenesis of the regulatory domain of Escherichia coli carbamoyl phosphate synthetase identifies crucial residues for allosteric regulation and for transduction of the regulatory signals. J Mol Biol. 2000;299:979–991. doi: 10.1006/jmbi.2000.3794. [DOI] [PubMed] [Google Scholar]
- Gao G, Nara T, Nakajima-Shimada J, Aoki T. Molecular characterization of a carbamoyl-phosphate synthetase II (CPS II) gene from Leishmania mexicana. Adv Exp Med Biol. 1998;431:237–240. doi: 10.1007/978-1-4615-5381-6_46. [DOI] [PubMed] [Google Scholar]
- Gao G, Nara T, Nakajima-Shimada J, Aoki T. Novel organization and sequences of five genes encoding all six enzymes for de novo pyrimidine biosynthesis in Trypanosoma cruzi. J Mol Biol. 1999;285:149–161. doi: 10.1006/jmbi.1998.2293. [DOI] [PubMed] [Google Scholar]
- Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS, 3rd, Evans DR. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403:328–332. doi: 10.1038/35002111. [DOI] [PubMed] [Google Scholar]
- Huang X, Raushel FM. Deconstruction of the catalytic array within the amidotransferase subunit of carbamoyl phosphate synthetase. Biochemistry. 1999;38:15909–15914. doi: 10.1021/bi991805q. [DOI] [PubMed] [Google Scholar]
- Huang X, Raushel FM. Restricted passage of reaction intermediates through the ammonia tunnel of carbamoyl phosphate synthetase. J Biol Chem. 2000a;275:26233–26240. doi: 10.1074/jbc.275.34.26233. [DOI] [PubMed] [Google Scholar]
- Huang X, Raushel FM. An engineered blockage within the ammonia tunnel of carbamoyl phosphate synthetase prevents the use of glutamine as a substrate but not ammonia. Biochemistry. 2000b;39:3240–3247. doi: 10.1021/bi9926173. [DOI] [PubMed] [Google Scholar]
- Huang X, Raushel FM. Role of the hinge loop linking the N- and C-terminal domains of the amidotransferase subunit of carbamoyl phosphate synthetase. Arch Biochem Biophys. 2000c;380:174–180. doi: 10.1006/abbi.2000.1913. [DOI] [PubMed] [Google Scholar]
- Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP synthesis. Ann Rev Biochem. 1980;49:253–279. doi: 10.1146/annurev.bi.49.070180.001345. [DOI] [PubMed] [Google Scholar]
- Kim J, Raushel FM. Allosteric control of the oligomerization of carbamoyl phosphate synthetase from Escherichia coli. Biochemistry. 2001;40:11030–11036. doi: 10.1021/bi011121u. [DOI] [PubMed] [Google Scholar]
- Kothe M, Purcarea C, Guy HI, Evans DR, Powers-Lee SG. Direct demonstration of carbamoyl phosphate formation on the C-terminal domain of carbamoyl phosphate synthetase. Protein Sci. 2005;14:37–44. doi: 10.1110/ps.041041305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles BW, Mareya SM, Post LE, Post DJ, Chang SH, Raushel FM. Differential roles for three conserved histidine residues within the large subunit of carbamoyl phosphate synthetase. Biochemistry. 1993;32:232–240. doi: 10.1021/bi00052a030. [DOI] [PubMed] [Google Scholar]
- Miles BW, Banzon JA, Raushel FM. Regulatory control of the amidotransferase domain of carbamoyl phosphate synthetase. Biochemistry. 1998;37:16773–16779. doi: 10.1021/bi982018g. [DOI] [PubMed] [Google Scholar]
- Miles BW, Raushel FM. Synchronization of the three reaction centers within carbamoyl phosphate synthetase. Biochemistry. 2000;39:5051–5056. doi: 10.1021/bi992772h. [DOI] [PubMed] [Google Scholar]
- Miles BW, Thoden JB, Holden HM, Raushel FM. Inactivation of the amidotransferase activity of carbamoyl phosphate synthetase by the antibiotic acivicin. J Biol Chem. 2002;277:4368–4373. doi: 10.1074/jbc.M108582200. [DOI] [PubMed] [Google Scholar]
- Mora P, Rubio V, Fresquet V, Cervera J. Localization of the site for the nucleotide effectors of Escherichia coli carbamoyl phosphate synthetase using site-directed mutagenesis. FEBS Lett. 1999;446:133–136. doi: 10.1016/s0014-5793(99)00197-0. [DOI] [PubMed] [Google Scholar]
- Nara T, Gao G, Yamasaki H, Nakajima-Shimada J, Aoki T. Carbamoyl-phosphate synthetase II in kinetoplastids. Biochim Biophys Acta. 1998;1387:462–468. doi: 10.1016/s0167-4838(98)00127-7. [DOI] [PubMed] [Google Scholar]
- Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- Pierrat OA, Javid-Majd F, Raushel FM. Dissection of the conduit for allosteric control of carbamoyl phosphate synthetase by ornithine. Arch Biochem Biophys. 2002;400:26–33. doi: 10.1006/abbi.2002.2768. [DOI] [PubMed] [Google Scholar]
- Raushel FM, Anderson PM, Villafranca JJ. Kinetic mechanism of Escherichia coli carbamoyl-phosphate synthetase. Biochemistry. 1978;17:5587–5591. doi: 10.1021/bi00619a001. [DOI] [PubMed] [Google Scholar]
- Rubino SD, Nyunoya H, Lusty CJ. In vivo synthesis of carbamyl phosphate from NH3 by the large subunit of Escherichia coli carbamyl phosphate synthetase. J Biol Chem. 1987;262:4382–4386. [PubMed] [Google Scholar]
- Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, Yap GS. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J Immunol. 2006;176:7263–7271. doi: 10.4049/jimmunol.176.12.7263. [DOI] [PubMed] [Google Scholar]
- Simmons CQ, Simmons AJ, Haubner A, Ream A, Davidson JN. Substitutions in hamster CAD carbamoyl-phosphate synthetase alter allosteric response to 5-phosphoribosyl-alpha-pyrophosphate (PRPP) and UTP. Biochem J. 2004;378:991–998. doi: 10.1042/BJ20031228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I. Structure of carbamoyl phosphate synthetase: a journey of 96 A from substrate to product. Biochemistry. 1997;36:6305–6316. doi: 10.1021/bi970503q. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Raushal FM, Benning MM, Rayment I, Holden HM. The structure of carbamoyl phosphate synthetase determined to 2.1 angstrom resolution. Acta Crystallogr D. 1999a:8–24. doi: 10.1107/S0907444998006234. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Raushel FM, Benning MM, Rayment I, Holden HM. The structure of carbamoyl phosphate synthetase determined to 2.1 A resolution. Acta Crystallogr D Biol Crystallogr. 1999b;55:8–24. doi: 10.1107/S0907444998006234. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Raushel FM, Wesenberg G, Holden HM. The binding of inosine monophosphate to Escherichia coli carbamoyl phosphate synthetase. J Biol Chem. 1999c;274:22502–22507. doi: 10.1074/jbc.274.32.22502. [DOI] [PubMed] [Google Scholar]
- Wilson DC, Matthews S, Yap GS. IL-12 signaling drives CD8+ T cell IFN-gamma production and differentiation of KLRG1+ effector subpopulations during Toxoplasma gondii Infection. J Immunol. 2008;180:5935–5945. doi: 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wilson D, Matthews S, Yap GS. Rapid elimination of Toxoplasma gondii by gamma interferon-primed mouse macrophages is independent of CD40 signaling. Infect Immun. 2007;75:4799–4803. doi: 10.1128/IAI.00738-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure. Sequence alignment of CPSII genes from Escherichia coli (EC), Toxoplasma gondii (Tg), Plasmodium falciparum (Pf), Babesia bovis (Bb), Trypanosoma cruzi (Tc), Leishmania major (Lm), Saccharomyces cerevisiae (Sc), Homo sapiens (Hs). Locations of indels in Toxoplasma gondii and locations where point mutations were constructed are shown in red type.