

Abstract
Prenylation is an essential post-translational modification in all eukaryotes. Here we describe the synthesis of a 340-member library of peptides containing free C-termini on cellulose membranes. The resulting library was then used to probe the specificity of protein farnesyltransferase from S. cerevisiae.
Post-translational lipidation of a variety of proteins, including Ras, with isoprenoids is essential for normal cellular functions and has important roles in numerous diseases. Consequently there is considerable interest in understanding the enzymes involved in this process and in developing inhibitors that may serve as drugs.1 Protein prenylation involves the addition of C15 (farnesyl) or C20 (geranylgeranyl) isoprenoids near the C-termini of proteins and is catalyzed by protein prenyltransferases.2 Following prenylation, additional enzymatic processing including proteolysis3 and methylation4 occurrs. To study the substrate specificity of these enzymes, the primary strategy employed to date has involved the synthesis, purification and assaying of individual peptides.5,6 While that strategy has yielded important insights, it limits the extent of sequence space that can be studied due to the large number of possible combinations. Combinatorial approaches provide possible strategies for increasing the number of sequences that can be examined. However, combinatorial studies of protein prenylation present a number of challenges. First, prenylation is a post-translational modification that occurs at the C-terminus of a protein. Since solid phase peptide synthesis is typically performed from the C- to N-terminus, more complex synthetic procedures are required. Next, enzymes involved in prenylation are large (~80 kDa or larger) or membrane-bound limiting their penetration into resins typically employed for solid phase synthesis. Finally, isoprenoids are not intrinsically chromogenic and are sensitive to acidic conditions and electrophiles rendering their their addition to peptides on resin difficult to monitor and their subsequent cleavage and sequence analysis more complex.
Protein farnesyltransferase (PFTase) catalyzes the addition of a farnesyl group to the thiol of a specific cysteine residue present in a tetrapeptide Ca1a2X-box sequence2 as shown in Fig. 1. Given that considerable interest exists in developing prenylation inhibitors selective for fungi or protozoa, it is clear that additional studies that investigate prenyltransferase specificity are needed. In this report, we describe a methodology that can be used to prepare and screen peptides as substrates for PFTase.
Fig. 1.
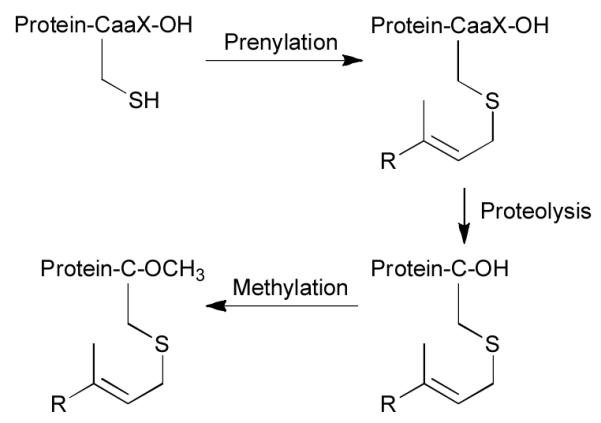
C-terminal prenylation and subsequent proteolysis and methylation. The “R” group can be a farnesyl or geranylgeranyl group.
Our approach for the preparation of peptide libraries with free C-termini (Fig. 2 and Scheme S2) is based on the pioneering work of Kania, Zuckermann and Marlowe7 and others.8,9 A four residue (BBLL) linker was initially added onto amine-functionalized solid surfaces followed by coupling to the γ-carboxyl group of Fmoc-Glu-ODmab; this resulted in the installation of an orthogonally (Dmab) protected acid that could be used for subsequent on-resin cyclization. Fmoc removal and coupling of an HMPA linker followed by ester bond formation produced a depsipeptide containing an acid-labile ester bond. Seven residues were then sequentially added ending with a linker to permit cleavage of the final product from the resin post-synthesis. After Dmab removal from Glu, the resulting free amino- and carboxyl-groups were coupled to yield a cyclic peptide on resin; treatment with modified Reagent K caused cleavage of the internal ester linkage and afforded the desired linear peptide with a free C-terminus.
Fig. 2.

Synthesis of C-terminal peptides exemplified by a RAGCVIX library. Reagents and conditions: (i) DIC coupling of Fmoc-Aa (2×), then capping, then 20% piperidine, then BPB staining; (ii) DIC coupling of HMPA (2×); (iii) 0.5 M Fmoc-Aa and 0.5 M 6-Cl-HOBt in DMF/CH2Cl2, 0.5 M DIC in CH2Cl2, 0.2 M DMAP in CH2Cl2 (6-8×), then capping, then 20% piperidine, then BPB staining; (iv) DIC coupling of Fmoc-Aa (2×), then capping, then 20% piperidine, then BPB staining; (v) 0.5 M photolysis linker, 0.5 M Et3N in DMF (3-4×); (vi) 2% hydrazine; (vii) 0.05 M BOP, 0.05 M 6-Cl-HOBt and 0.1 M DIEA in DMF (2×); (viii) modified reagent K. PL represents a cleavable linker so that peptides can be cleaved from resin and analyzed by MS. B represents β-alanine.
In order to verify the synthesized peptides, a cleavable linker should be installed so that peptides can be released from the solid surface and checked by MS. After considering the orthogonality of the conditions between peptide release and peptides synthesis and the quality of MS spectra of the released peptides, a new photocleavablelinker, shown in bottom left of Fig. 2 was designed and prepared (see Scheme S1). The resulting residue was incorporated into peptides as “PL” using the strategy outlined in Fig. 2. It should be noted that while the linker introduces a branch point and generates an epimeric mixture due to its stereogenic center, it is sufficiently upstream from the CaaX box that it does not contact the enzyme.10
Next, we began to examine the enzymatic prenylation of these molecules. Unfortunately, treatment of TentaGel beads terminating in the sequence CVIA with PFTase and FPP for prolonged reaction time did not result in the formation of any detectable farnesylated product by MS after photocleavage; experiments employing a more hydrophilic PEGA resin as the solid support for synthesis gave similar negative results. Those results suggest that the large size of PFTase, a heterodimer of 80-90 kDa, limits the ability of the enzyme to penetrate into the interior of many conventional supports used for SPPS.11 Thus we elected to pursue a parallel synthesis strategy where peptides were synthesized on amine-functionalized cellulose filters (SPOT synthesis). Gratifyingly, synthesis of C-terminal peptides ending in CVIA using the synthetic procedure described above proceeded smoothly and analysis after enzymatic prenylation with FPP showed essentially quantitative conversion of the peptide to the corresponding farnesylated product.
While MS analysis of peptides released from the resin after photocleavage worked well for the detection of prenylated peptides in the development of this methodology, such an approach is not ammenable to the screening of large libraries of peptides. Hence, we explored several approaches including the transfer of fluorescent isoprenoids12 and a number of two step procedures involving the transfer of alkyne- or aldehyde-containing isoprenoid analogues, followed by click reaction with biotin or a myc epitope-based peptide and subsequent streptavidin- or antibody-based detection. In evaluating those methods, we sought to minimize background labeling and eliminate any reaction with peptides in which the requisite cysteine residue present in the CaaX box had been changed to serine as a control. Of those various methods, the best results were obtained when an alkyne-containing analogue of FPP13,14 was used for PFTase-catalyzed reactions followed by click reaction with biotin-azide and subsequent detection with streptavidin-alkaline phosphatase (SP-AP) using a chromogenic substrate. Hence, that procedure was used for all library screening described here. It should be noted that the alkyne-containing analogue employed here has a Vmax value comparable to that of FPP13 and that screening was performed under saturating substrate conditions.
First, we prepared a 20-member library via manual SPOT synthesis15 in which the C-terminal residue of the CVIX sequence was varied to contain any of the 20 proteogenic amino acids. In the first experiment, upon completion of the synthesis, individual spots were excised from the membranes and cut in half. In each case, one half-spot was subjected to enzymatic prenylation with OPP-Far-alkyne followed by click reaction with biotin-azide and visualization with SP-AP. The remaining half-spots were used to confirm that the desired peptides were present on the cellulose filters (Fig. S1). In a secondary experiment for evaluating whether the peptides were prenylated, individual spots were cut in half after completion of the enzymatic prenylation. One half-spot was subjected to screening as described above. Peptides from the other half-spots were photocleaved followed by analysis via MALDI-MS. (Fig. S2). For analysis of the spots subjected to prenylation and visualization with SP-AP, the intensity of the color was normalized to that obtained with CVIQ (Fig. S4 and Fig. S5). Based on that analysis, eight residues at the X position gave signals comparable to, or greater than A, including E, Q, D, N, G, M, S, and T. Of those, D and E did not show levels of prenylation (ratio of prenylated peptide to unprenylated peptide < 0.5, Fig S5) in the secondary MALDI-MS-based screen. Ds-GCVID also showed low reaction rate in a continuous spectrofluorimetric assay (Table S1). If streptavidin-horseradish peroxidase (SP-HRP) was used in lieu of SP-AP for screening, peptides containing D and E at the X position manifested much less signal suggesting that those sequences are not efficient substrates. However, the overall level of background staining obtained with SP-HRP was significantly greater than that observed with SP-AP limiting the dynamic range of the the screen. Hence, our preferred procedure is to employ SP-AP as the primary screen and then use SP-HRP in subsequent screening when necessary.
To extend these results, we next employed automated SOPT synthesis to build a 17×20 CVa2X library with the X being one of the 20 proteogenic amino acids except Q, P, Y and a2 being one of the 20 proteogenic amino acids. The procedure for peptide synthesis was similar except at step (iii) where 1,1′-carbonyldiimidazole in DMF was used for coupling.8 Mass spectrometric analysis of selected peptides from the library showed the presence of the desired peptides with the exception of peptides containing W at the X position where some side products were again observed (similar to the results from the manual synthesis noted above). Synthesis, enzymatic prenylation and screening were performed in duplicate using the method described above. The intensity of the color was normalized to that obtained with CVIN and color coded with the most intense signals shown in red (Fig. 3 and Fig. S6).
Fig. 3.
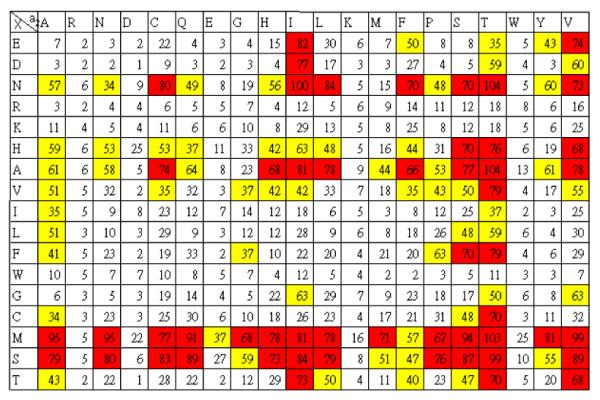
Evaluation of the extent of farnesylation of a RAGCVa2X library of peptides. The library subjected to enzymatic prenylation with an alkyne-containing analogue of FPP followed by click reaction with biotin-azide and subsequent reaction with SP-AP using a chromogenic substrate. Color intensity was quantified by Image J software. For comparison, the intensity was normalized relative to that observed with Asn at the X position. The library was synthesized and screened two times and the average color intensities were color coded. Key: below 33% (white); between 34 and 65% (yellow); above 65% (red).
Two general trends emerge from inspection of this data. First, analysis clearly indicates that PFTase from S. cerevisiae prefers M or S residues at the X position. This can be concluded from simple inspection of the number of high intensity (red) signals in their respective rows (Fig. 3) or more quantitatively by comparison of the summation of all intensities in a given row (Fig. S7). Such an analysis reveals that N, and A, and to a lesser extent, H V, F and T are also tolerated at the X position. A similar analysis indicates a preference for I, S, T and V residues at the a2 position with a number of others occuring less frequently. In addition, at the level of individual sequence analysis, a survey of all of the peptide sequences giving rise to the the strongest signals (53 total in red), 17 have previously been reported to be substrates for the mammalian PFTase;6 only two sequences, CVID and CVIE, that are not good substrates appeared as strong signals (as noted above). Additionally, 38 out of the 53 sequences are predicted to be substrates for the related human PFTase using the computer algorithm PrenPS16 (Fig. S8). Moreover, of the 15 predicted by PrenPS to not be substrates, two have been evaluated in vitro; both of those have been shown to be effective substrates. Hence, we conclude that the strategy described here using C-terminal peptide libraries generated by robotic spot synthesis can be used to identify bona fide substrate sequences.
From a biological perspective, it should be noted that in previous work, CaaX-box sequences where X is A, M, Q or S have been shown to be prenylated by PFTase from S. cerevisiae;17 however, N and T have not been reported as substrates for the yeast enzyme. These results highlight the utility this library approach for exploring the sequence specificity of PFTase and other enzymes involved in the subsequent processing of prenylated proteins; MS data suggests that the membrane-bound protease Rce1 can also act on these immobilized peptides (Fig. S10). Efforts to employ such arrays to probe the specificity of the prenyltransferases and other prenylated protein processing enzymes are underway.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: Abbreviations, experimental details and additional data. See DOI: 10.1039/b000000x/
Notes and references
- 1.Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, Waldmann H. Nat. Chem. Biol. 2006;2:518–528. doi: 10.1038/nchembio818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berndt N, Hamilton AD, Sebti SM. Nat. Rev. Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyartchuk VL, Ashby MN, Rine J. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 4.Clarke S, Vogel JP, Deschenes RJ, Stock J. Proc. Natl. Acad. Sci. U. S. A. 1988;85:4643–4647. doi: 10.1073/pnas.85.13.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krzysiak AJ, Scott SA, Hicks KA, Fierke CA, Gibbs RA. Bioorg. Med. Chem. Lett. 2007;17:5548–5551. doi: 10.1016/j.bmcl.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hougland JL, Hicks KA, Hartman HL, Kelly RA, Watt TJ, Fierke CA. J. Mol. Biol. 2010;395:176–190. doi: 10.1016/j.jmb.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kania RS, Zuckermann RN, Marlowe CK. J. Am. Chem. Soc. 1994;116:8835–8836. [Google Scholar]
- 8.Boisguerin P, Ay B, Radziwill G, Fritz RD, Moelling K, Volkmer R. ChemBioChem. 2007;8:2302–2307. doi: 10.1002/cbic.200700518. [DOI] [PubMed] [Google Scholar]
- 9.Hard RL, Liu J, Shen J, Zhou P, Pei D. Biochemistry. 2010;49:10737–10746. doi: 10.1021/bi101014s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 11.Kress J, Zanaletti R, Amour A, Ladlow M, Frey JG, Bradley M. Chem.–Eur. J. 2002;8:3769–3772. doi: 10.1002/1521-3765(20020816)8:16<3769::AID-CHEM3769>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 12.Dursina B, Reents R, Delon C, Wu Y, Kulharia M, Thutewohl M, Veligodsky A, Kalinin A, Evstifeev V, Ciobanu D, Szedlacsek SE, Waldmann H, Goody RS, Alexandrov K. J. Am. Chem. Soc. 2006;128:2822–2835. doi: 10.1021/ja052196e. [DOI] [PubMed] [Google Scholar]
- 13.Hosokawa A, Wollack J, Zhang Z, Chen L, Barany G, Distefano MD. Int. J. Pept. Res. Ther. 2007;13:345–354. [Google Scholar]
- 14.DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M, Distefano MD. Chem. Biol. Drug Des. 2010;76:460–471. doi: 10.1111/j.1747-0285.2010.01037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilpert K, Winkler DF, Hancock RE. Nat. Protoc. 2007;2:1333–1349. doi: 10.1038/nprot.2007.160. [DOI] [PubMed] [Google Scholar]
- 16.Maurer-Stroh S, Eisenhaber F. PLoS Comput. Biol. 2007;3:R55–1. [Google Scholar]
- 17.Caplin BE, Hettich LA, Marshall MS. Biochim. Biophys. Acta. 1994;1205:39–48. doi: 10.1016/0167-4838(94)90089-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.