

Abstract
Opiate addiction is associated with many adverse health and social harms, fatal overdose, infectious disease transmission, elevated health care costs, public disorder, and crime. Although community-based addiction treatment programs continue to reduce the harms of opiate addiction with narcotic substitution therapy such as methadone maintenance, there remains a need to find a substance that not only blocks opiate-type receptors (mu, delta, etc.) but also provides agonistic activity; hence the impetus arose for the development of a combination of narcotic antagonism and mu receptor agonist therapy. After three decades of extensive research the federal Drug Abuse Treatment Act 2000 (DATA) opened a window of opportunity for patients with addiction disorders by providing increased access to options for treatment. DATA allows physicians who complete a brief specialty-training course to become certified to prescribe buprenorphine and buprenorphine/naloxone (Subutex, Suboxone) for treatment of patients with opioid dependence. Clinical studies indicate buprenorphine maintenance is as effective as methadone maintenance in retaining patients in substance abuse treatment and in reducing illicit opioid use. With that stated, we must consider the long-term benefits or potential toxicity attributed to Subutex or Suboxone. We describe a mechanism whereby chronic blockade of opiate receptors, in spite of only partial opiate agonist action, may ultimately block dopaminergic activity causing anti-reward and relapse potential. While the direct comparison is not as yet available, toxicity to buprenorphine can be found in the scientific literature. In considering our cautionary note in this commentary, we are cognizant that to date this is what we have available, and until such a time when the real magic bullet is discovered, we will have to endure. However, more than anything else this commentary should at least encourage the development of thoughtful new strategies to target the specific brain regions responsible for relapse prevention.
1. Introduction
Opiate addiction continues to be associated with many adverse health and social harms, fatal overdose, infectious disease transmission, elevated health care costs, public disorder, and crime (Wood et al. 2005). It is a problem of national concern, especially with dramatically increased rates of abuse and dependence of prescription opioids and with almost three million Americans having abused heroin. Currently, the most effective treatment for this growing epidemic is opioid replacement therapy. Acceptance of this therapy has been progressive and its effectiveness is no longer in question. The two main approved medications for opioid maintenance therapy are methadone hydrochloride and buprenorphine hydrochloride. Each has unique characteristics that determine its suitability for an individual patient (Schottenfeld 2005). Dating back to Dole and Nyswander (1966), community-based addiction treatment programs continue to reduce the harms of opiate addiction with narcotic substitution therapy such as methadone maintenance. Methadone, a synthetic opiate agonist, has been shown to be effective in reducing withdrawal symptoms and the impulse to continue injecting opiates (Goldstein 1991). In addition, patients using methadone maintenance treatment had an average of 14 fewer relapse-related events (hospitalizations, emergency room visits, or outpatient detoxifications) per 1,000 months of enrollment than buprenorphine patients. Buprenorphine patients averaged 10 fewer relapse events than patients who received outpatient treatments, and 86 fewer events per 1,000 months than patients who received no treatment. Methadone appears to be somewhat more effective in reducing hospitalization emergency room and detoxification use than buprenorphine. Both of these treatments are more effective than drug-free treatment for opioid addiction. All forms of treatment are significantly less costly and more effective than no treatment (Butler 2008).
A review of the literature regarding the use of buprenorphine and methadone in medication assisted therapy (MAT) programs for opiate addiction shows that such programs have proven effective when looking at the following primary and secondary outcome indicators (USHHS Report 2003):
-
Primary Outcome Measures
Abstinence from illicit opiate use
Reduction in illicit opiate use
Reduction in the severity of withdrawal from opiate use
Retention in treatment for persons enrolled in opiate withdrawal or opiate cessation programs.
-
Secondary Outcome Measures
Level of injecting
Employment status
Housing status
Educational status
Criminality
Quality of life
However, the need to find a substance that not only blocks opiate-type receptors (mu, delta, etc.), but also provides agonistic activity, afforded the impetus for the development of a combination of narcotic antagonism and mu receptor agonist therapy (SAMHSA, 2007).
It is noteworthy that until 2000, medications for opioid dependence were limited to two opioid agonists, methadone and LAAM (withdrawn from market in 2003), or the narcotic antagonist naltrexone used for both opiate (Judson and Goldstein, 1984) and alcohol dependence (Blum et al., 1977). At that time, and even today, prescribing methadone is restricted to hospitals and federal- and state-approved opioid replacement substance abuse treatment programs. Currently, physicians can prescribe naltrexone, but patients must be opioid-free for several days prior to starting its use. According to Arfken et al. (2010), prior to 2002, the only pharmacological options for physicians treating opiate addicts were the antagonist naltrexone or the agonist methadone, both under strict regulations.
The federal Drug Abuse Treatment Act 2000 (DATA) opened a window of opportunity for patients with addiction disorders by providing increased access to options for treatment. DATA allows physicians to become certified to prescribe buprenorphine, by taking a short specialty-training course. Certified physicians can prescribe buprenorphine and buprenorphine/naloxone (Subutex, Suboxone) in a traditional office setting when treating patients with opioid dependence. Clinical studies indicate buprenorphine maintenance is as effective as methadone maintenance in retaining patients in substance abuse treatment and reducing illicit opioid use. Sublingual buprenorphine is more effective than Clonidine or Clonidine/naltrexone in short-term opioid detoxification treatment. Buprenorphine provides an additional tool to treat opioid addiction and improve the quality of lives of these patients. When the FDA approved the monoformulation of buprenorphine and buprenorphine/naloxone for the treatment of opioid dependence and placed both formulations in Schedule III, it became possible for physicians to prescribe these medications in their offices for detoxification or long-term maintenance (McNicholas, 2004; Stock and Shum, 2004). Restrictions on group practices including certification were then replaced in 2006 with limitation of 30 patients per physician, regardless of type of practice.
The regulations were modified again in January 2007 to allow up to 100 patients per physician after the physician had a year of experience prescribing buprenorphine under the 30-patient limit, and registered intent to treat more than 30 patients. Certainly the U.S. experiment in expanding Schedule iii-V medication for opioid dependence to physicians outside of formal substance abuse treatment facilities have resulted in expanded capacity (Arfken et al., 2010). Specifically, the number of physicians on the federal Center for Substance Abuse Treatment (CSAT) Locator List in 2004 was 2,518, and in 2008 it was 9,069. Of this combined total of 15,662 physicians, 13,095 (84%) were limited to 30 patients, whereas 2,567 were authorized to treat up to 100 patients. The total patient capacity nationwide at the end of 2008 was 649,550. The total number of patients treated from 2008 to 2010 is unknown.
2. Buprenorphine/Naloxone Is Clinically Effective for Reducing Withdrawal and Craving Behavior but Not Relapse
In the past 20 years, the development of new research technology has greatly advanced our understanding of the neurobiological mechanisms and therapeutic strategies for drug addiction. Many chemical medications have been produced, some of which have been widely used in clinical treatment of drug addiction (Kreek et al., 2002), such as methadone and buprenorphine for heroin replacement therapy (O’Brien, 2005), and naltrexone for heroin and alcohol anticraving treatment (Dackis and O’Brien, 2005; O’Brien, 2005). These medications, however, have been unable to prevent drug relapse following detoxification on a long-term basis (Heidbreder and Hagan, 2005; O’Brien, 2005). For example, naltrexone suppresses euphoria from heroin via its antagonistic effect on opiate receptors, but most heroin addicts would not accept it for long-term therapy (O’Brien, 2005). Methadone and buprenorphine, as opiate agonists having reward enhancement properties on an acute basis, are clinically effective in reducing withdrawal and craving for heroin during detoxification (Gold, 1993; Bruijnzeel et al. 2007), but it is difficult to use them to reduce the likelihood of relapse after detoxification (O’Brien, 2005). Recently our laboratory proposed that relapse from drugs of abuse including alcohol and opiates, were due in part to dopaminergic receptor supersensitivity, and we proposed that relapse prevention involved the activation instead of blocking dopaminergic activity we termed “deprivation-amplification relapse therapy” (DART). It has been shown by Doehring et al (2009) that compared with the control group, drug users carried more frequently the minor allele of DRD2 SNP rs1076560G>T SNP (P=0.022, odds ratio 2.343) or the ATCT haplotype of DRD2 rs1799978A>G, rs1076560G>T, rs6277C>T, ANKK1 rs1800497C>T (P=0.048, odds ratio 2.23), with similar tendencies for ANKK1 rs1800497C>T (P=0.056, odds ratio 2.12) and the TCCTCTT haplotype of DRD2 rs12364283T>C, rs1799732C del, rs4648317C>T, rs1076560G>T, rs6275C>T, rs6277C>T, and ANKK1 rs1800497C>T (P=0.059, odds ratio 2.31). The average and maximum daily methadone doses were significantly associated with the DRD2 rs6275C>T SNP (P=0.016 and 0.005 for average and maximum dose, respectively). Carriers of the variant rs6275T allele needed higher methadone doses than non-carriers. In addition, this variant was associated with a longer time to reach the maximum methadone dose (P=0.025).
As these results are well known and seem very robust, it is a crucial research question to understand why methadone or buprenorphine fails to reduce the probability of relapse. Such understanding would help in revealing the neurobiological mechanisms of relapse and designing better therapeutic strategies. In this commentary, we provide the underlying basis for treatment approaches that could ultimately lead to reversal of hypodopaminergic function and possible relapse prevention.
3. Pharmacological Mechanisms of Action (MOA) of Buprenorphine and Naloxone
Buprenorphine is an opioid analgesic, derived from thebaine. Buprenorphine was initially classified as a “mixed agonist-antagonist analgesic” or a narcotic antagonist analgesic. The work of Martin et al. in 1976 (reviewed by Kreek, 2000) on the animal model of the chronic spinal dog substantiated the substance’s action as partial agonist at the mu-opioid receptor. These findings were underscored by the substance’s general pharmacological profile. Further, buprenorphine was one of the first narcotic analgesics to be assessed for its abuse liability in humans. Buprenorphine was eventually turned it into a widely used therapeutic agent in patients with opioid dependence because of this perceived lower abuse liability. Interest in buprenorphine spanning more than 30 years has been attributed to its unique pharmacological characteristics, including moderate intrinsic activity, high affinity to and slow dissociation from mu-opioid receptors or what is known as biophase distribution (Yassen et al., 2006).
Early pharmacological studies demonstrated buprenorphine has strong binding to opioid receptors, and an inverted U-shaped dose-response curve in rodents. In the rat paw formalin test, although buprenorphine demonstrated a bell-shaped dose-response curve against an acute noxious stimulus, it showed a classic sigmoidal curve in the later phase of the assay. In most preclinical antinociceptive tests, buprenorphine was shown to be fully efficacious, with an antinociceptive potency 25 to 40 times higher than morphine. A ceiling effect for respiratory depression (but not for analgesia) has been demonstrated in humans (Hans, 2007). Current studies are focusing on norbuprenorphine, an N-dealkylated metabolite of buprenorphine. Norbuprenorphine is a likely contributor to the overall pharmacology of buprenorphine; in the mouse writhing test, norbuprenorphine provides antinociceptive efficacy similar to buprenorphine, with analgesic activity shown to be dose-dependent (Cowan, 2003; Chang et al., 2006).
In terms mechanism of action of naloxone it is well established that the drug binds to delta, mu, and kappa opioid receptors. By doing so, it blocks the action of opiate-like drugs such as heroin and morphine. The chronic effects of another potent narcotic antagonist, naltrexone, was studied by Lesscher et al. (2003). Chronic treatment with the opioid antagonist naltrexone induced functional supersensitivity to opioid agonists, which may be explained by receptor up-regulation induced by opioid receptor blockade. These findings suggested opioid receptor subtype-selective regulation by chronic naltrexone treatment in mice, but it was less likely to occur with naloxone because of a very short half-life. However (and as elaborated below), it is still quite possible that the blockade of mu and delta receptors even with naloxone could induce a supersensitivity and may be involved in relapse similar to the supersensitivity of hypodopaminergic receptor density especially in carriers of the dopamine (DA) D2 receptor gene A1 allele for dopaminergic pathways (Blum et al., 2009).
A variety of studies, both laboratory based and clinical, have revealed the mechanisms of action of long-acting opioid agonists in treatment, including prevention of disruption of molecular, cellular, and physiologic events and, in fact, allowing normalization of those functions disrupted by chronic heroin use. A number of molecular biological studies have revealed single nucleotide polymorphisms of the human mu opioid receptor gene; the mu opioid receptor is the site of action of heroin, the major opiate drug of abuse, analgesic agents such as morphine, and the major treatment agents for heroin addiction. These findings support the early hypotheses of Dole’s laboratory that addiction may be due to a combination of genetic, drug-induced, and environmental (including behavioral) factors and also, that atypical stress responsivity may contribute to the acquisition and persistence of, as well as relapse to, use of addictive drugs (Kreek, 2003; Gold et al., 1982; Blum, 2009).
4. Reward Addiction Commonality: Activation instead of Blocking Mesolimbic Dopamine in Reward Circuitry Provides Long-Term Treatment Benefits for Opiate Dependence
It is well known that brain reward circuitry is regulated by neurotransmitter interactions and net release of DA in the nucleus accumbens (NAc) (Blum & Kozlowski, 1990). The major loci for feelings of well being and reward occur in the mesolimbic system of the brain. The natural sequence of events of the “brain reward cascade” leading to reward, involves the interrelationship of at least four important neurochemical pathways: serotonergic (5-HT); enkephalinergic (Enk), GABAergic (GABA), and dopaminergic (DA). The synthesis, vesicle storage, metabolism, release, and function of these neurotransmitters are regulated by genes and their expression are regulated by messenger RNA (mRNA) directed proteins. It has been postulated that genome orientated research will provide genetic testing that will categorize individuals as to their specific neurochemical makeup and thus provide useful information to assist in appropriate development of the most correct treatment options for the patient requiring psychiatric care (Malhotra et al., 2007).
DA is a substance with many important neurochemical functions and has been credited with resultant behavioral effects such as “pleasure,” “stress reduction” and “wanting”. Simply stated, without the normal functionality of DA, an individual will be lacking hedonic response and an inability to cope with stress (Koob & Le Moal, 2008). Thus, genetic hypodopaminergic activity of the brain predisposes an individual to seek substances and/or behaviors that will overcome this anhedonic state by activating mesolimbic dopaminergic centers (Volkow et al., 2002). It turns out that these substances and behaviors include: alcohol, opiates, psychostimulants, nicotine, carbohydrates, cannabinoids, gambling, sex, and indulgence in any excessive pleasure or thrill seeking behaviors, like video gaming etc. (Radwan et al., 2007; Epstein et al., 2007; da Silva et al., 2007; Kirsch et al., 2006; Costa-Mallen et al., 2005; Shahmoradgoli et al., 2005; Swan et al., 2007; Xu et al., 2004; Sprangler et al., 2004; Poce et al., 2003; Spitz et al., 1998; Comings et al., 1996). Use of these substances and engaging in these aforementioned behaviors commonly induces the release of neuronal DA into the synapse at the NAc, the reward center of the brain (Koob & Le Moal, 2008). Acute indulgence in these behaviors can be classified as self-medicating and leads to a preferential release of DA, which overcomes the hypodopaminergic state for that individual. The resultant self-medication provides a temporary relief of discomfort and a “pseudo feeling” of well being (Xiao et al., 2007). Unfortunately, chronic abuse of these psychoactive substances and excessive indulgence in the aberrant behaviors leads to inactivation of the brain reward cascade (i.e., neurotransmitter synthesis inhibition, neurotransmitter storage depletion, toxic formation of pseudo neurotransmitters, and receptor dysfunction (structural and or density)). These behaviors can also lead to neurotransmitter dysfunction via depletion. Therefore, both substance seeking and pathological behaviors as ways of providing a feel good response (a “fix”) result in ever escalating and uncontrollable craving behavior. It has been well established that individuals possessing certain genetic polymorphisms (variations) are particularly prone to amplified polymorphic expressions with environmental or lifestyle insult and will be at increased risk for impulsive, compulsive, and addictive behaviors (Blum et al, 2000). Such common genetic antecedents influencing the natural brain reward cascade provide the understanding that impulsive, compulsive, and addictive behaviors are commonly linked and support the emerging concept of Reward Deficiency Syndrome (RDS) as an umbrella term to characterize and classify these commonly linked genetically induced behaviors (Comings & Blum, 2000; Bowirrat & Oscar-Berman, 2005; Green et al., 1999). In this scenario, any and all of these abusable psychoactive drugs or pathological behaviors are candidates for addiction (tolerance/dependence) and are chosen by the individual as a function of genetic and environmental factors (e.g., availability, peer pressure, etc) (Blum et al., 2000).
While DA is critical to maintain normalization of natural rewards, the neuronal release of DA into NAc synaptic sites is somewhat complex. In 1989 our laboratory proposed an interactive cascade of events of mesolimbic function that lead to net DA release (Blum and Kozlowski, 1990). It was termed the “brain reward cascade’ (see Figure 1).
Fig 1. Brain Reward Cascade.
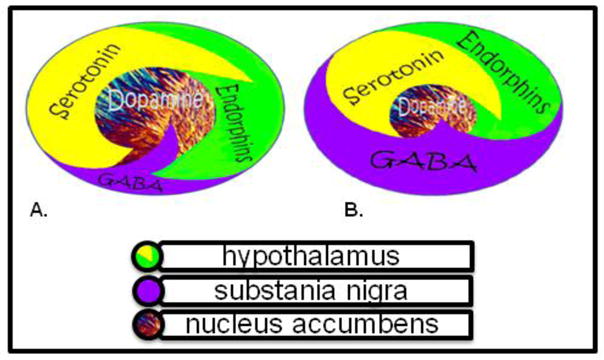
Fig 1a. Schematic represents the normal physiologic state of the neurotransmitter interaction at the mesolimbic region of the brain. Briefly in terms of the “Brain Reward Cascade” first coined by Blum and Kozlowski [90]: serotonin in the hypothalamus stimulates neuronal projections of methionine enkephalin in the hypothalamus which in turn inhibits the release of GABA in the substania nigra thereby allowing for the normal amount of DA to be released at the NAc (reward site of Brain). Fig 1b Represents hypodopaminergic function of the mesolimbic region of the brain. It is possible that the hypodopaminergic state is due to gene polymorphisms as well as environmental elements including both stress and neurotoxicity from aberrant abuse of psychoactive drugs (i.e. alcohol, heroin, cocaine etc). Genetic variables could include serotonergic genes (serotonergic receptors [5HT2a]; serotonin transporter 5HTlPR); endorphinergic genes (mu OPRM1 gene; proenkephalin (PENK) [PENK polymorphic 3′ UTR dinucleotide (CA) repeats}; GABergic gene (GABRB3) and dopaminergic genes (ANKKI Taq A; DRD2 C957T, DRD4 7R, COMT Val/met substation, MAO-A uVNTR, and SLC6A3 9 or 10R). Any of these genetic and or environmental impairments could result in reduced release of DA and or reduced number of dopaminergic receptors. (Brain reward cascade -modified with permission from IIOAB Journal, Blum et al. IIOAB, 2010, 11(2) 1–14.)
The interactions of activities in the separate subsystems mentioned above merge together into the much larger global system. These activities take place simultaneously and in a specific sequence, merging like a cascade. The end result is a sense of peace, pleasure, and well being when these systems work normally. Other research has confirmed that the reward sensation is related to complex cascade reactions involving several neurotransmitters and structures in the limbic system (Volkow et al. 2002). The ultimate result of the process is the activation of the mesolimbic DA pathway, which starts in the tegmental ventral area and ends at the DA D2 receptors on the cell membranes of neurons located in the NAc and the hippocampus (Volkow et al. 2007).
The process, as described by Blum and Kozlowski (1990), starts in the hypothalamus with the excitatory activity of serotonin-releasing neurons. This causes the release of the opioid peptide met-enkephalin in the ventral tegmental area, which inhibits the activity of neurons that release the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). The disinhibition of DA-containing neurons in the ventral tegmental area allows them to release DA in the NAc and (via amygdala) in certain parts of the hippocampus, permitting the completion of the cascade and the development of the reward sensation (Carelli, 2002). Usually, if the cascade is working properly, the reward or feeling of well-being is obtained provided certain basic conditions are fulfilled (Blum & Kozlowski, 1990)
5. Traditional Long-Term Blockade Leads to Mood Changes and Suicide Ideation
Most recent examples of pharmaceuticals that block DA release and or receptor activation include Acomplia (Rimonabant), the cannabinoid (CB1) receptor blocker, and possibly Gabapentin. While there are numerous studies supporting the therapeutic benefits of Acomplia as an anti-craving drug, the long-term adverse effects resulted in a recent rejection by the United States Federal Drug Administration (FDA). A recent PUBMED search revealed 1007 papers on Acomplia. Since the prevalence of obesity continues to increase, there is a demand for effective and safe anti-obesity agents that can produce and maintain weight loss and improve co morbidity. Christensen et al. (2007) conducted a meta-analysis of all published randomized controlled trials to assess the efficacy and safety of the newly approved anti-obesity agent Rimonabant. They searched the Cochrane database and Controlled Trials Register, Medline via Pubmed, Embase via WebSpirs, Web of Science, Scopus, and reference lists up to July 2007. They collected data from four double-blind, randomized controlled trials (including 4105 participants) that compared 20 mg per day Rimonabant with placebo. Patients given Rimonabant had a 4.7 kg (95% CI 4.1–5.3 kg; p < 0.0001) greater weight reduction after 1 year than did those given placebo. Rimonabant caused significantly more adverse events than did placebo (Odds Ratio (OR) = 1.4; p = 0.0007; number needed to harm = 25 individuals [95% CI 17–58]), and 1.4 times more serious adverse events (OR = 1.4; p = 0.03; number needed to harm = 59 [27–830]). Patients given Rimonabant were 2.5 times more likely to discontinue the treatment because of depressive mood disorders than were those given placebo (OR = 2.5; p = 0.01; number needed to harm = 49). Furthermore, anxiety caused more patients to discontinue treatment in Rimonabant groups than in placebo groups (OR = 3.0; p = 0.03; number needed to harm = 166). Their findings suggest that 20 mg per day of Rimonabant increases the risk of adverse psychiatric events – i.e., depressed mood disorders and anxiety; despite depressed mood being an exclusion criterion in these trials. Taken together with the recent US Food and Drug Administration finding of increased risk of suicide during treatment with Rimonabant, these researchers recommended increased alertness by physicians to these potentially severe adverse psychiatric reactions. Concerning this report, we propose that the negative effects on mood are due to the continued blockade of naturally required DA release at the NAc.
Gabapentin is a gamma-aminobutyric acid (GABA) analogue, with GABAmimetic pharmacological properties. Gabapentin is used for the treatment of seizures, anxiety and neuropathic pain. It has been proposed that Gabapentin may be useful in the treatment of cocaine dependence. However, clinical trials with Gabapentin have shown conflicting results, while preclinical studies are sparse. In one study, Peng et al. (2008) investigated the effects of Gabapentin on intravenous cocaine self-administration and cocaine-triggered reinstatement of drug-seeking behavior, as well as on cocaine-enhanced DA in the NAc. They found that Gabapentin (25–200 mg/kg, i.p., 30 min or 2 h prior to cocaine) failed to inhibit intravenous cocaine (0.5 mg/kg/infusion) self-administration under a fixed-ratio reinforcement schedule or cocaine-triggered reinstatement of cocaine-seeking behavior. In vivo microdialysis showed that the same doses of Gabapentin produced a modest increase (approximately 50%, p < 0.05) in extracellular NAc GABA levels, but failed to alter either basal or cocaine-enhanced NAc DA. These data suggest that Gabapentin is a weak GABA-mimic drug. At the doses tested, it has no effect in the addiction-related animal behavioral models. This is in striking contrast to positive findings in the same animal models shown by another GABAmimetic – gamma-vinyl GABA – by Garner’s group (see Blum et al., 2000 for review). Based on our current theoretical model we are opposed to the use of Gabapentin to treat substance seeking behavior especially in long term care.
Other than a few scientific groups that suggest serotonergic/dopaminergic agonist therapy (Rothman et al., 2007), most strategies embrace dopaminergic receptor blockade/attenuation of DA release (Malhorta et al., 2007; Koob et al., 2008; Blum et al., 2000; Comings & Blum, 2000; Bowirrat & Oscar-Berman, 2005; Green et al., 1999; Suzuki et al., 2010). We propose that, in most circumstances, utilization of amino acid precursors affecting positive dopaminergic activation is a better alternative (Chen et al., 2011)
6. Proposed Relapse Mechanisms
It is well known that after prolonged abstinence, individuals who use their drug of choice experience a powerful euphoria that often precipitates relapse. While a biological explanation for this conundrum has remained elusive, we hypothesize that this clinically observed “supersensitivity” might be tied to genetic dopaminergic polymorphisms. Another therapeutic conundrum relates to the paradoxical finding that the dopaminergic agonist bromocriptine induces stronger activation of brain reward circuitry in individuals who carry the DRD2 A1 allele compared with DRD2 A2 allele carriers. Because carriers of the A1 allele relative to the A2 allele of the DRD2 gene have significantly lower D2 receptor density, a reduced sensitivity to DA agonist activity would be expected in the former (Kirsch et al., 2006). Thus, it is perplexing that with low D2 density there is an increase in reward sensitivity with the DA D2 agonist bromocriptine. Moreover, under chronic or long-term therapy with D2 agonists, such as bromocriptine, it has been shown in vitro that there is a proliferation of D2 receptors. One explanation for this relates to the demonstration that the A1 allele of the DRD2 gene is associated with increased striatal activity of L-amino acid decarboxylase, the final step in the biosynthesis of DA. This appears to be a protective mechanism against low receptor density and would favor the utilization of an amino acid neurotransmitter precursor like L-tyrosine for preferential synthesis of DA. This seems to lead to receptor proliferation to normal levels and results in significantly better treatment compliance only in A1 carriers (Blum et al., 2009).
We propose that low D2 receptor density and polymorphisms of the D2 gene are associated with risk for relapse of substance abuse, including alcohol dependence, heroin craving, cocaine dependence, methamphetamine abuse, nicotine sensitization, and glucose craving. With this in mind, we suggest a putative physiological mechanism that may help to explain the enhanced sensitivity following intense acute dopaminergic D2 receptor activation: “denervation supersensitivity.” Rats with unilateral depletions of neostriatal DA display increased sensitivity to DA agonists estimated to be 30 to 100 x in the 6-hydroxydopamine (6-OHDA) rotational model. However, it is difficult to explain the extent of behavioral supersensitivity by a simple increase in receptor density. Thus, the administration of DA D2 agonists would target D2 sensitization and attenuate relapse, especially in D2 receptor A1 allele carriers. This hypothesized mechanism is supported by clinical trials utilizing amino acid neurotransmitter precursors, enkephalinase, and catechol-O-methyltransferase (COMT) enzyme inhibition, which have resulted in attenuated relapse rates in reward deficiency syndrome (RDS) probands (Blum et al., 2007). If future translational research reveals that DA agonist therapy reduces relapse in RDS, it would support the proposed concept, which we term “deprivation-amplification relapse therapy” (DART). This term couples the mechanism for relapse (which is “deprivation-amplification,” especially in DRD2 A1 allele carriers) with natural D2 agonist therapy utilizing amino acid precursors, COMT, MOA and enkephalinase inhibition therapy (Blum et al., 2009).
7. Long-Term Therapy with Buprenorphine/Naloxone Combination May Induce Anti-Reward
In brief, legislation has enabled physicians to treat opioid-dependent patients with an office-based maintenance program using buprenorphine, a partial mu-opioid receptor agonist. Clinical studies have indicated that buprenorphine effectively manages opioid addiction. Buprenorphine is more effective than placebo for managing opioid addiction but may not be superior to methadone if high doses are needed. It is comparable to lower doses of methadone, however. treatment phases include induction, stabilization, and maintenance. Buprenorphine therapy should be initiated at the onset of withdrawal symptoms and adjusted to address withdrawal symptoms and cravings. Advantages of buprenorphine include low abuse potential and high availability for office use. Disadvantages include high cost and possible lack of effectiveness in patients who require high methadone doses. Most family physicians are required to complete eight hours of training before they can prescribe buprenorphine for opioid addiction. However, as a cautionary note we are proposing that while short-term therapy seems very appropriate this may not be the case for prolonged maintenance therapy with this potent combination of drugs. Over the past two decades, a number of neuroimaging studies have shed some light on this potential dilemma.
The combination of buprenorphine/naloxone while having acute benefits in the treatment of heroin addiction has been found unable to prevent drug relapse and may even increase the chance for relapse (Heidreder & Hogan, 2005; O’Brien, 2005). Naloxone suppresses euphoria from heroin via its antagonistic effect on opiate receptors, but most heroin addicts would not accept it for long-term therapy (O’Brien, 2005; Chen et al., 2004). Methadone and buprenorphine, as opiate agonists, are clinically effective in reducing withdrawal and craving for heroin during detoxification, but it is difficult to use them to reduce the likelihood of relapse after detoxification (O’Brien, 2005). As these results are well known and seem very robust, it is important to understand why methadone or buprenorphine fail to reduce the probability of relapse. Such understanding would help by revealing the neurobiological mechanisms of relapse and facilitate the design of better therapeutic strategies.
In a recent study, Mei et al. (2010) examined the acute effects of buprenorphine on brain responses to heroin-related cues to reveal the neurobiological and therapeutic mechanisms of addiction and relapse. Fifteen heroin addicts at a very early period of abstinence, were studied in two separate periods 10–15 min apart: an early period (5–45 min) and a later period (60–105 min) after sublingual buprenorphine, roughly covering the onset and peak of buprenorphineplasma level. During both periods, functional magnetic resonance imaging (fMRI) scanning with heroin-related visual stimuli was performed followed by questionnaires. Under the effect of buprenorphine, brain responses to heroin-related cues showed decrease in amygdala, hippocampus, ventral tegmental area, and thalamus but no changes in ventral striatum nor orbital-prefrontal-parietal cortices. As an uncontrolled trial, these preliminary results suggested that buprenorphine has specific brain targets in reducing withdrawal and craving during early abstinence, and that ventral striatum and orbital-prefrontal and parietal cortices may be the key targets in developing therapy for drug addiction and relapse.
We highlight this with the realization that the cingulate gyrus is a site responsible in part for drug relapse. Converging neuropsychological and functional neuroimaging evidence indicates that the dorsal anterior cingulate cortex (dACC) is dysfunctional in drug-addicted populations. Few studies, however, have investigated the biochemical and physiological properties of the dACC in such populations. Yücel et al. (2007) used proton magnetic resonance spectroscopy ((1)H-MRS) together with fMRI to probe dACC biochemistry and physiological activity during performance of a behavioral control task in 24 opiate-dependent individuals (maintained on a stable dose of methadone or buprenorphine at the time of study) and 24 age, gender, intelligence, and performance-matched healthy subjects. While both groups activated the dACC to comparable levels, the opiate-using group displayed relatively increased task-related activation of frontal, parietal, and cerebellar regions, as well as reduced concentrations of dACC N-acetylaspartate and glutamate/glutamine. In addition, the opiate-using group failed to show the expected correlations between dACC activation and behavioral measures of cognitive control. These findings suggested that the dACC is biochemically and physiologically abnormal in long-term opiate-dependent individuals. Furthermore, opiate addicts required increased, perhaps compensatory, involvement of the fronto-parietal and cerebellar behavioral regulation network to achieve normal levels of task performance/behavioral control. Moreover, in the Mei et al. (2010) study brain regions with an unchanged fMRI response to heroin-related cues from early to late periods included the ventral striatum, orbital and lateral prefrontal cortex, and parietal cortex, suggesting that their responses were not modulated by buprenorphine. Thus, these results showing lack of effect of Buprenorphine on these known key brain regions linked to relapse (De Ridder et al., 2011) may elucidate the limited therapeutic effects on relapse using this drug.
Accordingly, the striatum is a core region of the reward system in addiction (Everitt and Robbins, 2005; Koob and Volkow 2010). The pattern of unchanged activation in the striatum may reflect sustained expectation of the high reward level of addictive drugs (Volkow et al., 2003 a,b, 2006a). This would imply that, for more of a radical cure of addiction, the striatum should be targeted to degrade the already elevated reward level from drug abuse due to tolerance and sensitization (Nestler, 2009) especially dopaminergic receptor supersensitivity (Blum et al., 2009).
Recent animal studies on the incubation of cocaine craving have indicated such a possibility, that is to target the GluR2-lacking AMPA receptors in the ventral striatum (Conrad et al., 2008). Orbital and lateral regions of prefrontal cortex are known to be involved in motivation, drive, control, and inhibition (Wilson et al., 2004; Weiss, 2005; Volkow et al., 2009). The parietal cortex is known to be involved in impulsivity (Lee et al., 2005), attention (Lawrence et al., 2002), and craving (Garavan et al., 2000) in addicts. Frontal and parietal regions should be important therapeutic targets for addiction attenuation as they may function together with the striatum to result in the persistence and uncontrollability of compulsive drug seeking. These neurobiological findings may partly underpin key addiction-related phenomena, such as poor inhibitory control of drug-related behavior in the face of adverse consequences, and may be of relevance to the design of future treatment studies.
It is of some interest that a recent study from our laboratory as one example, found that a complex KB220Z overcame cingulate gyrus abnormalities in psychostimulant abusers undergoing protracted abstinence (Blum et al., 2010). Specifically, positive outcomes demonstrated by quantitative electroencephalographic (qEEG) imaging in a randomized, triple-blind, placebo-controlled, crossover study involving oral KB220Z™ showed an increase of alpha waves and low beta wave activity in the parietal brain region. Using t statistics, significant differences observed between placebo and KB220Z™ consistently occurred in the frontal regions after week 1 and then again after week 2 of analyses (p = 0.03). This is the first report to demonstrate involvement of the prefrontal cortex in the qEEG response to a natural putative D2 agonist (KB220Z™), especially evident in DA D2 A1 allele subjects.
Independently, we have further supported this finding with an additional study of three polydrug abusers undergoing protracted abstinence who carried the DRD2 A1 allele. Significant qEEG differences were found between those who received one dose of placebo compared with those who were administered KB220Z™. KB220Z™ induced positive regulation of the dysregulated electrical activity of the brain in these addicts. The results are indicative of a phase change from low amplitude or low power in the brain to a more regulated state by increasing an average of 6.169 mV(2) across the prefrontal cortical and cingulate gyrus region (see Figure 2).
Fig 2. KB220Z compared to Placebo in Psychostimulant Abusers.
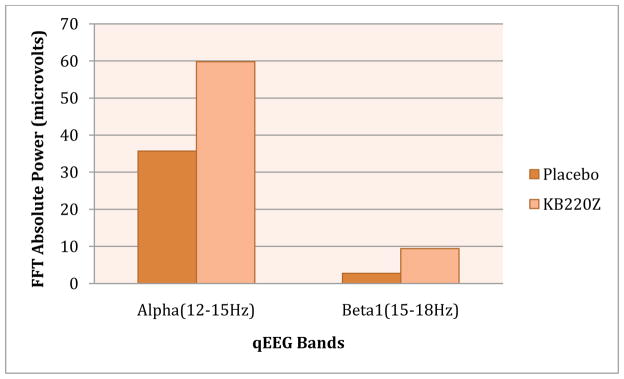
Illustrates positive response of KB220Z compared to placebo in triple blind randomized placebo -controlled study in psychostimulant abusers undergoing protracted abstinence. Modified from data presented by Blum et al (2010).
8. Brain Glucose Metabolism and Buprenorphine and Naloxone Effects
Buprenorphine
Over the past decade, functional neuroimaging has contributed greatly to our knowledge about the neuropharmacolgy of substance misuse in man (Volkow et al., 2003a). Techniques such as functional magnetic resonance imaging, positron emission tomography (PET) or single photon emission tomography (SPET) can measure changes in regional cerebral activity, whereas changes in neuropharmacological parameters (e.g., receptor number and neurotransmitter levels) can be directly measured only with PET and SPET. Recently, a series of studies have shown common effects of substances of misuse on the brain, such as an acute increase in DA release (followed by hypo function after chronic use) and cue exposure-induced activation of the frontal cortex (Lingford-Hughes, 2005).
In terms of brain glucose metabolism, Walsh et al. (1994) found that buprenorphine compared to placebo significantly reduced cerebral glucose metabolism (CMRglc) and the regional cerebral metabolic rate for glucose (rCMRglc) by up to 32% in all but three of 22 bilateral and in four midline regions (p < .05). No region showed an increase in rCMRglc. Buprenorphine also produced miosis, respiratory depression, and subjective ratings of euphoria and sedation in comparison to placebo (p < .05). These observations extend previous findings of reduced CMRglc following acute treatment with morphine and other non-opioid euphorogenic drugs.
The above result has similarly been observed with cocaine as well. Cocaine produced euphoria and reduced glucose utilization globally (mean reduction, 14%). Twenty-six of 29 brain regions (all neocortical areas, basal ganglia, portions of the hippocampal formation, thalamus, and midbrain) showed significant decrements (5% to 26%) in the regional cerebral metabolic rate for glucose. The findings demonstrated that reduced cerebral metabolism is associated with cocaine-induced euphoria (London et al., 1990a). it is noteworthy that buprenorphine only partially reversed this cocaine-induced abnormality (Holman et al., 1993). While similar effects were seen with morphine, the reduced CMRglc was not correlated with morphine induced euphoria (London et al., 1990b)
Naloxone
Experiments were conducted by Dow-Edwards et al. (1989) to determine some of the metabolic correlates of tonic opioid activity in the central nervous system under conditions previously examined for changes in monoamine levels. The glucose metabolic rates in seven brain regions were determined by autoradiographic visualization of 14C-deoxyglucose incorporation in female rats after eight days of chronic exposure to naltrexone pellets and 10 days after pellet removal. Chronic administration of naltrexone resulted in a significant decrease in the metabolic activity of neurons in the striatum. This could have impact on relapse induction. Other brain areas examined under this condition were not significantly affected. These results indicated that tonic opioid input is an important determinant of metabolic activity in the striatum. In addition, these results indicated that conditions previously shown to alter regional content of monoamines do not necessarily produce concomitant changes in regional glucose utilization.
The interaction of naloxone and brain activity of this substance may be tied to genetic antecedents such as the DA D2 receptor Taq A1 allele. Ritchie and Noble (1996) measured the [3H]naloxone binding in frontal gray cortex, caudate nucleus, amygdala, hippocampus, and cerebellar cortex obtained post mortem from human alcoholic and nonalcoholic subjects. Binding was found to be higher in alcoholics than in nonalcoholics for all of the brain regions examined, with a significant difference in the frontal cortex. When subjects were grouped by the presence or absence of the A1 (minor) allele of the D2 DA receptor gene, [3H]naloxone binding was lower in all brain regions examined of subjects with the A1 allele than in those without this allele, with a significant difference in the caudate nucleus. These findings suggested that one of the consequences of chronic alcohol exposure in humans is an enhancement of the brain opiate receptor system. However, the decreased [3H]naloxone binding observed in subjects with the A1 allele may be a compensatory response to their decreased dopaminergic modulation of opiate receptor activity having relapse consequences due to D2 receptor supersensitivity (Ritchie & Noble, 2003; Seeman, 2010; Blum et al., 2009).
Additionally, neostriatal GABAergic neurons projecting to the globus pallidus synthesize the opioid peptide enkephalin, while those innervating the substantia nigra pars reticulata and the entopeduncular nucleus synthesize dynorphin. The differential control exerted by DA on the activity of these two efferent projections concerns also the biosynthesis of these opioid peptides. It was found that a strong reduction of glutamate decarboxylase messenger RNA expression was detected over pallidal neurons following either naloxone or haloperidol treatment. The amplitude of the variations of mu opioid receptor density and of preproenkephalin and preprodynorphin messenger RNA levels suggested that the regulation of neostriatal and pallidal micro opioid receptors is more susceptible to a direct opioid antagonism (Naltrexone) while the biosynthesis of opioid peptides in the neostriatum is more dependent on the dopaminergic transmission. The down-regulation of mu opioid receptors following haloperidol probably represents an adaptive change to increased enkephalin biosynthesis and release. The haloperidol-induced increase in neostriatal preprodynorphin messenger RNA expression might result from an indirect, intermittent stimulation of neostriatal D1 receptors. The haloperidol-induced decrease of pallidal glutamate decarboxylase messenger RNA expression suggests, in keeping with the current functional model of the basal ganglia, that the activation of the striatopallidal projection produced by the interruption of neostriatal dopaminergic transmission reduces the GABAergic output of the globus pallidus.
The reduction of pallidal glutamate decarboxylase messenger RNA expression following opioid receptor blockade indicates an indirect, excitatory influence of enkephalin upon globus pallidus neurons and, consequently, a functional antagonism between the two neuroactive substances (GABA and enkephalin) of the striatopallidal projection in the control of globus pallidus output. Through this antagonism enkephalin could partly attenuate the GABA-mediated effects of a dopaminergic denervation on pallidal neuronal activity leading to dopaminergic “denervation” supersensitivity and relapse potential (Mavridis and Besson, 1999).
Further work by Chen et al. (1994) suggested that persistent inhibition of D2 DA receptors differentially regulates the expression of D1 and D2 DA receptor mRNA in striatum, and that the magnitude, duration and interval of inhibiting dopaminergic transmission may be important factors in regulating DA receptor mRNA expression. These results also suggested that D2 DA antagonists indirectly down-regulate opioid receptors by increasing the expression of proenkephalin mRNA, thereby increasing enkephalin which, in turn, decreases opioid receptors in striatum an important neuro-substrate for heroin relapse.
The notion of supersensitivity to even opiate agonists has been further investigated. Albeit not with naloxone, chronic treatment with the opioid antagonist naltrexone induces functional supersensitivity to opioid agonists, which may be explained by receptor up-regulation induced by opioid receptor blockade. The levels of opioid receptor subtypes through the brain of mice were determined after chronic naltrexone treatment using quantitative in vitro autoradiography. Treatment with naltrexone clearly induced up-regulation of mu- (mean 80%) and, to a lesser extent, delta-opioid receptors (mean 39%). The up-regulation of mu- and delta-opioid receptors was evident throughout the brain, although there was variation in the percentage change across brain regions. In contrast, consistent up-regulation of kappa-opioid receptors was observed in cortical structures only and was not so marked as for mu- and delta-opioid receptors. In noncortical regions kappa-opioid receptor expression was unchanged. Taken together, the findings suggested opioid receptor subtype-selective regulation by chronic naltrexone treatment in mice (Yoburn et al., 1995). It could be argued that this effect does not occur with the dosage levels used with the combination of buprenorphine and naloxone but must be considered especially in sensitive genetic patients.
9. A Caution about Toxicity
Drug interactions are a leading cause of morbidity and mortality. Methadone and buprenorphine are frequently prescribed for the treatment of opioid addiction. Patients needing treatment with these medications often have co-occurring medical and mental illnesses that require medication treatment. The abuse of illicit substances is also common in opioid-addicted individuals. These clinical realities place patients being treated with methadone and buprenorphine at risk for potentially toxic drug interactions. A substantial literature has accumulated on drug interactions between either methadone or buprenorphine with other medications when ingested concomitantly by humans. One such interaction involves certain benzodiazepines (e.g., Flunitrazepam; FLZ) and the concomitant abuse of buprenorphine. FLZ that is widely abused and augments buprenorphine toxicity appeared the most potent to increase mu-cell surface receptor density at the lowest dose of 0.2 mg/kg. Among people using buprenorphine and benzodiazepines, the effects described here are likely to influence addictive behaviors and induce toxic effects that could be quantitatively different due to the quality of the benzodiazepines (Poisnel et al., 2009).
Moreover, there is some evidence that buprenorphine can induce liver toxicity. Herve et al. (2004) reported on several cases of acute hepatitis with buprenorphine. According to Peyrière et al. (2009), most cases of hepatotoxicity related to buprenorphine have occurred in hepatitis C-infected patients. The main mechanism for buprenorphine-induced hepatitis is a mitochondrial defect, exacerbated by cofactors with additional potential to induce mitochondria dysfunction (e.g., HCV, alcohol, concomitant medications). The diagnosis of intravenous buprenorphine-induced hepatitis was classified as probable in two cases. While it is true that buprenorphine was morphine-like but was 25 to 50 times more potent than morphine and was longer-acting and is acceptable to drug addicted individuals (Jasinski et al., 1976), there are cases of fatal overdose and suicide ideation (Gaulier et al., 2000; Tracqui et al., 1998; Kintz, 2001).
10. Pharmacokinetics of Mixed Agonist/Antagonists of Opiate Receptors
Detoxification from opiate addiction has been a medical problem for as long as opiate drugs have been available. Treatment before the discovery of Clonidine involved giving another opioid drug with less dangerous consequences of chronic use, such as the long-acting and orally administered once-a-day methadone, for another opioid mu agonist like heroin, which must be taken intravenously many times a day, thus making rehabilitation, work, and avoidance of hepatitis, HIV, and other illnesses difficult (Gold, 1993)
Although methadone has proved to be very beneficial, it still has significant abuse potential. Naloxone, because it blocks the effects of all opiates, has facilitated the transformation from addiction to a drug-free state for many recovering addicts but blocks DA release in the reward site of the brain (Blum et al., 1976; Blum et al., 2009; Gold et al., 1982). By alleviating withdrawal symptoms and by lessening the detoxification period, Clonidine similarly has improved the prospect of recovery from opiate addiction. Relapse, whether withdrawal is treated with Clonidine or other new agents or not, occurs with great regularity because repeated opiate use can induce a new acquired drive state, i.e., the drive for opiates. In addition, with powerful withdrawal symptoms during abstinence, opiate relapse is difficult to prevent without an adequate treatment program (Gold, 1993; Blum et al., 2009; Blum & Gold, 2010).
Medical magic bullets like buprenorphine/naloxone and Clonidine are needed to be given for opiate withdrawal distress as part of a recovery program which not only allows the brain to re-establish normal homeostatic changes in the drug-free state but also provides sufficient motivation for new approaches to achieving and sustaining pleasurable existence. However, an important cautionary note regarding for example the indiscriminate use of Clonidine for detoxification from all psychoactive drugs is the finding that Clonidine may exacerbate ethanol withdrawal in mice (Blum et al., 1983).
Similarly, the combination of coupling a mixed opiate agonist/antagonist may have properties which also must be cautiously considered before the widespread adoption of such an approach. Hume et al. (2007) reported on preclinical studies investigating opioid receptor occupancy with oxycodone (mu- and kappa-receptor agonist), morphine (mu-receptor agonist), and buprenorphine (partial agonist at the mu-receptor and antagonist at the delta- and kappa-receptors), each given at antinociceptive doses. In vivo binding of [(11)C] diprenorphine was not significantly reduced after treatment with the full agonists but was reduced by approximately 90% by buprenorphine. In addition, given that [(11)C] diprenorphine is a non-subtype-specific PET tracer, there was no regional variation that might feasibly be interpreted as due to differences in opioid subtype distribution. This experiment showed that mu-receptor binding of agonist like morphine or potentially heroin was significantly reduced with buprenorphine. Although a positive outcome for short-term therapy, in the long term, administration of buprenorphine will result in a attenuation of mu receptor occupancy of the natural opioid enkephalin and will ultimately result in reduced release of DA at the accumbens brain region. This in turn may lead to generalized drug seeking behavior.
Moreover, morphine has been universally assumed to act solely on opiate receptors, and predominantly on mu receptors. In consonance with this, several studies have demonstrated that opiate mu agonists and dopaminergic agonists and antagonists are incapable of binding each other’s receptors, except at extremely high concentrations (nor, for that matter, is acetylcholine, serotonin, gamma-hydroxybutyrate, norepinephrine, or histamine able to bind opiate receptors). Yet, while other neurotransmitter antagonists (e.g. alpha- and beta-adrenoceptor-blocking agents) are for the most part limited in their effect on opiate-induced responses, many of the central effects elicited by morphine and other opioids have been found to be markedly potentiated by DA antagonists and reversed by direct and indirect DA agonists. Even more significantly, DA antagonists (especially those appreciably inhibiting DA release selectively) can also mimic many of these effects in low to moderate doses (Feigenbaum and Yanai, 1984).
In fact, the amplitude of the variations of mu opioid receptor density in the neostriatum is more dependent on the dopaminergic transmission. The down-regulation of mu opioid receptors following haloperidol represents probably an adaptive change to increased enkephalin biosynthesis and release (Mavridis and Besson, 1999). In essence then, we have increased enkephalin and the reduced availability of mu-receptors, thereby preventing enkephalin attenuation of the GABA-mediated effects of a dopaminergic denervation on pallidal neuronal activity leading to a hypodopaminergic function in the long-term (Theile et al., 2011).
Finally, at one level it makes pharmacological sense to combine a partial agonist-antagonist like buprenorphine and the opiate receptor antagonist naloxone together for the treatment of opiate addiction; however, to some degree the interactive pharmacokinetics argues against this combination especially in the long-term. Galynker et al. (1996) studied the pharmacokinetics of naloxone. Naloxone, an opiate receptor antagonist, administered 30–40 min after tracer injection at a dose of 1.0 mg/kg I.V., reduced [11C] buprenorphine binding in thalamus, striatum, cingulate gyrus, and frontal cortex to values 0.25 to 0.60 of that with no intervention. There were minimal (< 15%) effects on cerebellum. Naloxone treatment significantly reduced the slope of the Patlak plot in receptor-containing regions. These results demonstrated that [11C] buprenorphine can be displaced by naloxone in vivo. We are therefore suggesting that If naloxone can displace buprenorphine (even at doses used in the combination), which may have something to do with alternate days of treatment regimen, the combination rational may undergo rethinking at least for long-term maintenance therapy. It is noteworthy, however, that the combination of naltrexone (naloxone not studied) does shorten opioid detoxification with buprenorphine in the short-term (Umbricht et al., 1999).
In terms of actual concentrations of naloxone it has been determined for area under the concentration-time curves (AUC0)-24 hours (0.421 vs. 0.374 ng ×hr/mL) and peak concentration [Cmax] (0.186 vs. 0.186 ng/mL) [Bruce et al. 2010].
To reiterate, buprenorphine/naloxone (Suboxone) comprises the partial mu-opioid receptor agonist buprenorphine in combination with the opioid antagonist naloxone in a 4: 1 ratio. When buprenorphine/naloxone is taken sublingually as prescribed, the naloxone exerts no clinically significant effect, leaving the opioid agonist effects of buprenorphine to predominate. However, when buprenorphine/naloxone is parenterally administered in patients physically dependent on full agonist opioids, the opioid antagonism of naloxone causes withdrawal effects, thus reducing the abuse potential of the drug combination (Orman and Keating 2009). Moreover, buprenorphine and naloxone sublingual (S.L.) dose formulations may decrease parenteral buprenorphine abuse (Harris et al. 2000).
Ciraulo et al. (2006) investigated, the pharmacokinetic and pharmacodynamic properties of multiple doses of sublingual tablets containing either buprenorphine alone or buprenorphine and naloxone. Subjects were experienced opiate users who received escalating doses (4–24 mg) of buprenorphine either alone or in combination with naloxone. Cmax and AUCs increased for both buprenorphine and naloxone with escalating doses. Significant differences were found across the range of doses administered for dose-adjusted Cmax for both tablet formulations and for the dose-adjusted AUCs for the buprenorphine-naloxone tablets.
It is noteworthy that buprenorphine undergoes extensive first-pass metabolism and therefore has very low oral bioavailability; however its bioavailability sublingually is extensive enough to make this a feasible route of administration for the treatment of opioid dependence. The mean time to maximum plasma concentration following sublingual administration is variable, ranging from 40 minutes to 3.5 hours. Buprenorphine has a large volume of distribution and is highly protein bound (96%). It is extensively metabolised by N-dealkylation to norbuprenorphine primarily through cytochrome P450 (CYP) 3A4. The terminal elimination half-life of buprenorphine is long and there is considerable variation in reported values (mean values ranging from 3 to 44 hours). Most of a dose of buprenorphine is eliminated in the feces, with approximately 10–30% excreted in urine. The presence of naloxone does not appear to influence the pharmacokinetics of buprenorphine (Elkader and Sproule, 2005).
There is the question as to the real need for the addition of naloxone along with buprenorphine to treat opiate dependence. Accordingly, the addition of naloxone does not affect the efficacy of buprenorphine for two reasons: (1) naloxone is poorly absorbed sublingually relative to buprenorphine and (2) the half-life for buprenorphine is much longer than for naloxone (32 vs. 1 h for naloxone). The sublingual absorption of buprenorphine is rapid and the peak plasma concentration occurs 1 h after dosing. The plasma levels for naloxone are much lower and decline much more rapidly than those for buprenorphine (Chiang and Hawks, 2003). However in 2009, a highly sensitive method was developed to measure naloxone and its metabolite nornaloxone in human plasma, urine, and human liver microsomes (HLM). The mean recoveries were 69.2% for naloxone and 32.0% for nornaloxone. Specifically, in human subjects receiving 16 mg buprenorphine and 4 mg naloxone, naloxone was detected for up to 2 h in all three subjects and up to 4 h in one subject. Mean AUC(0–24) was 0.303 +/− 0.145 ng/mL.h; mean C(max) was 0.139 +/− 0.062 ng/mL; and T(max) was 0.5 h. In 24-h urine samples, about 55% of the daily dose was excreted in either conjugated or unconjugated forms of naloxone and nornaloxone in urine via the p450 system (Fang et al. 2009).
In considering the concept as set forth herein that long term effects of naloxone (even at sublingual low doses) may have any prolonged action to affect dopaminergic activity by blocking mu receptors is unlikely. However, we must consider the role of polymorphisms of the P450 gene. Polymorphisms in the cytochrome P450 (CYP) family may have had the most impact on the fate of pharmaceutical drugs. CYP2D6, CYP2C19 and CYP2C9 gene polymorphisms and gene duplications account for the most frequent variations in phase I metabolism of drugs since nearly 80% of drugs in use today are metabolised by these enzymes. Approximately 5% of Europeans, 1% of Asians, 5–14% of Caucasians, 5% Africans lack CYP2D6 activity, and these individuals are known as poor metabolizers. CYP2C9 is another clinically significant drug metabolizing enzyme that demonstrates genetic variants (Kosarac et al. 2009). Thus we must be cognizant of these facts and as such the long term effects of the combination of buprenorphine and naloxone must be monitored accordingly (Zhou et al. 2009).
One important question to answer would be what the long-term effects of buprenorphine are alone without the combination of naloxone on dopaminergic reward pathways. Research is encouraged in order to determine the best approach to combat the deleterious effects of opioid dependence. It seems parsimonious to also consider the potential of combining buprenorphine with a natural D2 agonist such as KB220Z for relapse prevention.
11. Intelligent Rationale for Using Naloxone not Naltrexone
There are two commercially available PO (sublingual) buprenorphine preparations. One has naloxone (Suboxone) and the other doesn’t (Subutex). Clinically they can be considered to have identical effects since the naloxone is not absorbed via the PO route; the added naloxone in Suboxone has no clinical effect unless administered parenterally (IV or IM) and is added only to decrease street value by making Suboxone undesirable for intravenous use.
The possibility of precipitating withdrawal in chronic opioid users and the opioid receptor blockade effect (from other opioids) has nothing to do with the added naloxone and occurs whether one is administering Suboxone or Subutex. It is purely a result of the buprenorphine. The withdrawal precipitating potential results from the fact that buprenorphine is a partial opioid receptor agonist.
Thus, if a patient is taking opioids like Oxycontin or heroin chronically and is given buprenorphine, the other opioid is displaced and replaced abruptly reducing the receptor activation from 100% to around 60–70%. This manifests as withdrawal. If Suboxone is crushed and given IV to someone chronically taking IV drugs like heroin, the naloxone would decrease the opioid activation even more than the buprenorphine, i.e., to zero, causing severe withdrawal. It also would block the immediate “buzz” in people not using opioids chronically. Thus, buprenorphine acts like “superglue” in that it displaces other opioids from the opioid receptor, yet only partially activates it.
All of the opioid receptor antagonism/blocking from SL buprenorphine comes from its’ tremendous receptor affinity which can displace or prevent the binding of other opioids (except those with strong receptor affinities themselves such as Fentanyl, Sufentanyl, and possibly Dilaudid). There are several important differences that account for why Bekett-Benckiser chose naloxone over naltrexone. Although both are opioid receptor antagonists, there are big differences:
Naloxone works IV or IM. Naltrexone is a PO drug
Naloxone lasts only around 30–45 minutes. Naltrexone can last for days.
12. From Bench to Bedside: A Clinical Perspective
We find it very interesting that our clinical skills and observations are now being validated by the neurobiology. For example, when Suboxone became clinically available, one of us (JF) designed a program to specialize in treating patients who were interested in abstinence and not maintenance.
“We had to specifically describe the program and refuse thousands of patients who were interested in only in getting the drug and not working the cognitive and emotional aspects of a recovery plan. We designed groups that combined an educational approach focusing of both the neurobiology of addiction but also recovery. We stressed the importance of the DA and opioids systems to hedonic tone in order to seek and maintain sobriety, rather than to maintain an active drug using lifestyles. We had to create a new language, entitled “Recovery Dose Equivalency”, to counter the “insulin dependence” model of maintenance.
Patients had to be re-introduced to the idea of modulation of mood and cognitive state as an active recovery process that had to include dose reduction of buprenorphine over time. We stressed that the rate of dose lowering had to parallel the rate of acquisition of recovery skills. We created workbooks and asked patients to monitor rewarding activities and attempt to grade them. Patients were instructed that they could not lower their dose of buprenorphine until there was a positive effect from such recovery based activities – good diet, exercise, talking therapy, self help, etc.. They learned to measure what worked for them so that they could be confident that dose reduction would not lead to automatic relapse.
In this process, we surely see lots of patients who probably have genetic polymorphisms that set them up for a low hedonic tone that becomes obvious over the time of dose reduction. Their effort to increase DA and endorphins to achieve happiness (Blum et al 2009) and good feelings is either inefficient endogenously, or they are too lazy to work a good recovery program to facilitate such an internal response. We do not push our patients too fast – we have treated hundreds of patients who have remained on buprenorphine for years – but their current dose is 70–80% lower than their initial dose. Those patients who appear to make an effort but are unsuccessful are probably genetically unable to – but we still try and go slower.
We also observed that relapse had little to do with cravings, but more with the ability to enhance frontal lobe and emotional circuitry. In fact, neural adaptation in the medial prefrontal cortex (mPFC) is thought to play a crucial role in vulnerability to relapse to drug seeking (Van den Oever, 2010). Measuring the quality of recovery is not easy to do, but we the authors do not believe it is possible in current methadone programs because of the costs, the untested genetic makeup of the population, and the fact that most counseling consists of medication management and not recovery skill development.”
With this said, we are cognizant that until we have the ability to offer opiate addicts a safe, cost effective substitute, a global scientific challenge indeed, we do applaud the current treatment community and the government for providing the treatment tool of buprenorphine and naloxone. This has led to the saving of thousands from unavoidable premature death from narcotic overdose.
12. Conclusions
There is much support for the short-term benefits of the combination of buprenorphine/naloxone (Walley et al., 2008). The benefits of narcotic antagonism have been reviewed by many investigators (Blum et al., 2004; O’Brien, 2005; Kreek, 2000; Gold et al., 1982). In addition, Jasinski et al. (1976) provided evidence that buprenorphine was morphine-like but was 25 to 50 times more potent than morphine and was longer-acting. They suggested that little if any physical dependence of clinical significance was produced by buprenorphine. The effects of morphine to 120-mg doses were blocked by buprenorphine, a blockade that persisted for 29 1/2 hours. In humans, buprenorphine has less intrinsic activity than morphine, and as such, has a low abuse potential. Moreover, the drug has potential for treating narcotic addiction since it is acceptable to addicts, is long acting, produces a low level of physical dependence such that patients may be easily detoxified, is less toxic than drugs used for maintenance therapy, and blocks the effects of narcotics. However, the addiction treatment community is faced with potential problems associated with long-term anti-relapse treatment with the combination of these two drugs.
The work of Mei et al. (2010) showing that at least for buprenorphine by itself there was no activation at the ventral striatum and orbital-prefrontal-parietal cortices suggests that this lack of activation as measured by fMRI may provide in part an explanation as to why this treatment does not affect relapse. The authors correctly suggested that future therapeutic approaches to prevent relapse should target the ventral striatum and orbital-prefrontal-parietal cortices. Thus, with this in mind Blum and Gold (2011) proposed the possibility that consideration should be given to the utilization of natural D2 agonist therapy following needed confirmation of preliminary work (Blum et al., 2010; Miller at al., 2010) specifically with those with genetically induced DA deficiency.
Based upon initial results with large populations receiving D2 agonist therapy with KB220/KB220Z (Blum et al., 2010; Miller et al., 2010; Blum et al., 2011), we propose that offering a safe, nonaddicting, natural dopaminergic receptor agonist that potentially up-regulates instead of down-regulates dopaminergic receptors, could be at least one option to utilize in the long term to prevent relapse rather than the combination of buprenorphine/naloxone alone. In fact, earlier research has already shown that the combination of the KB220 variant in combination with narcotic antagonism increased compliance in serious long-term methadone by 7.16 fold (Chen et al., 2004).
In considering our cautionary note in this commentary we must realize that to date this is what we have available and until such a time when the real magic bullet is discovered we will have to endure. However, this commentary more than anything else, should at least encourage the development of thoughtful new strategies to target the brain regions responsible for relapse prevention. Thus, we are encouraging more research on the withdrawal symptoms and other chronic effects of buprenorphine/naloxone. Moreover, research directed to the potential combination of buprenorphine/KB220Z, to specifically assess their potential for maintenance and the prevention of relapse in the treatment of opioid dependence.
Fig 3.

Represents a thumb nail schematic of the salient points expressed in this commentary to assist in the understanding opioid addiction, opioid substitution therapy and an alternative modality.
The figure shows the Opioid addiction adverse effects (fatal overdose, infectious disease transmission, elevated health care costs, public disorder, and crime) and the available treatments. The traditional narcotic substitution therapy (e.g. methadone maintenance), does not target or block delta or mu receptors but provides agonistic activity. However, the new combination treatment of narcotic antagonism and mu receptor agonist therapy (even at very low doses of Naloxone) seems parsimonious. Clinical studies indicate that buprenorphine maintenance is as effective as methadone maintenance in retaining patients in substance abuse treatment and in reducing illicit opioid use. The figure delineates the negative effect on reward circuitry whereby chronic blockade of opioid receptors, even with partial opioid agonist action, may ultimately block dopaminergic activity, causing anti-reward effects and increasing relapse potential. Based upon initial results with large populations receiving D2 agonist therapy withKB220/KB220Z, we propose that offering a safe, nonaddicting, natural dopaminergic receptor agonist that potentially up-regulates instead of down-regulates dopaminergic receptors, could be at least one option to utilize in the long term to prevent relapse rather than the combination of buprenorphine/naloxone alone.
Acknowledgments
We thank the staff of PATH Foundation NY. The writing of this article was funded in part by LifeGen, Inc., San Diego, CA. Other support came from NIAAA grants R01-AA07112 and K05-AA00219 and the Medical Research Service of the US Department of Veterans Affairs (to MO-B).
Footnotes
Authors’ Contributions
KB wrote the first draft of the paper. The figures were developed by Margret Madigan, an AB. The entire final article was edited by Margaret Madigan. JF, JG, JB, SS, TS, MO-B, UD, MK, SM, FF, BWD, THJC, ALCC,DB and ERB reviewed and edited the manuscript. UD was responsible for literature search and appropriate reference style. All the authors read and approved the final manuscript.
Competing Interests:
The patented KB220Z therapy has been exclusively licensed to LifeGen, Inc., whereby KB and BWD owns stock. JG, and FF are partners with LifeGen, Inc. All other authors declare that they have no competing interest.
References
- Blum K, Braverman ER, Holder JM, Lubar JF, Monastra VJ, Miller D, Lubar JO, Chen TJ, Comings DE. Reward deficiency syndrome: a biogenetic model for the diagnosis and treatment of impulsive, addictive, and compulsive behaviors. J Psychoactive Drugs. 2000;32:i–iv. doi: 10.1080/02791072.2000.10736099. [DOI] [PubMed] [Google Scholar]
- Blum K, Briggs AH, DeLallo L. Clonidine enhancement of ethanol withdrawal in mice. Subst Alcohol Actions Misuse. 1983;4:59–63. [PubMed] [Google Scholar]
- Blum K, Chen ALC, Bowirrat A, Downs BW, Waite RL, Reinking J, et al. Genes and Happiness. Gene Ther Mol Biol. 2009;13:91–129. [Google Scholar]
- Blum K, Chen TJ, Downs BW, Bowirrat A, Waite RL, Braverman ER, Madigan M, Oscar-Berman M, DiNubile N, Stice E, Giordano J, Morse S, Gold M. Neurogenetics of dopaminergic receptor supersensitivity in activation of brain reward circuitry and relapse: proposing “deprivation-amplification relapse therapy” (DART) Postgrad Med. 2009;121:176–96. doi: 10.3810/pgm.2009.11.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum K, Chen TJ, Meshkin B, Waite RL, Downs BW, Blum SH, Mengucci JF, Arcuri V, Braverman ER, Palomo T. Manipulation of catechol-O-methyl-transferase (COMT) activity to influence the attenuation of substance seeking behavior, a subtype of Reward Deficiency Syndrome (RDS), is dependent upon gene polymorphisms: a hypothesis. Med Hypotheses. 2007;69:1054–60. doi: 10.1016/j.mehy.2006.12.062. [DOI] [PubMed] [Google Scholar]
- Blum K, Futterman S, Wallace JE, Schwertner HA. Naloxone-induced inhibition of ethanol dependence in mice. Nature. 1977;265:49–51. doi: 10.1038/265049a0. [DOI] [PubMed] [Google Scholar]
- Blum K, Mark S, Gold MS. Neuro-chemical activation of brain reward meso-limbic circuitry is associated with relapse prevention and drug hunger: A commentary. Med Hypotheses. 2011 doi: 10.1016/j.mehy.2011.01.005. (in press) [DOI] [PubMed] [Google Scholar]
- Blum K, Kozlowski GP. Ethanol and neuromodulator interactions: a cascade model of reward. In: Ollat H, Parvez S, Parvez H, editors. Alcohol and Behavior. Utrecht, Netherlands: VSP Press; 1990. pp. 131–149. [Google Scholar]
- Blum K, Wallace JE, Schwerter HA, Eubanks JD. Morphine suppression of ethanol withdrawal in mice. Experientia. 1976;32:79–82. doi: 10.1007/BF01932634. [DOI] [PubMed] [Google Scholar]
- Bowirrat A, Oscar-Berman M. Relationship between dopaminergic neurotransmission, alcoholism, and Reward Deficiency Syndrome. Am J Med Genet B Neuropsychiatr Genet. 2005;132B:29–37. doi: 10.1002/ajmg.b.30080. [DOI] [PubMed] [Google Scholar]
- Bruce RD, Altice FL, Moody DE, Morse GD, Andrews L, Lin SN, Fang WB, Ma Q, Friedland GH. Pharmacokinetic interactions between buprenorphine/naloxone and once-daily lopinavir/ritonavir. J Acquir Immune Defic Syndr. 2010;54(5):511–4. doi: 10.1097/qai.0b013e3181d3cad3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Marcinkiewcz C, Isaac S, Booth MM, Dennis DM, Gold MS. The effects of buprenorphine on fentanyl withdrawal in rats. Psychopharmacology (Berl) 2007;191:931–41. doi: 10.1007/s00213-006-0670-2. [DOI] [PubMed] [Google Scholar]
- Butler M, Kane RL, McAlpine D, Kathol RG, Fu SS, Hagedorn H, Wilt TJ. AHRQ Publication No 09-E003. Rockville, MD: Agency for Healthcare Research and Quality; Oct, 2008. Integration of Mental Health/Substance Abuse and Primary Care No. 173. (Prepared by the Minnesota Evidence-based Practice Center under Contract No. 290-02-0009.) [Google Scholar]
- Carelli RM. The nucleus accumbens and reward: neurophysiological investigations in behaving animals. Behav Cogn Neurosci Rev. 2002;1:281–296. doi: 10.1177/1534582302238338. [DOI] [PubMed] [Google Scholar]
- Chang Y, Moody DE, McCance-Katz EF. Novel metabolites of buprenorphine detected in human liver microsomes and human urine. Drug Metab Dispos. 2006;34:440–448. doi: 10.1124/dmd.105.006148. [DOI] [PubMed] [Google Scholar]
- Chen JF, Aloyo VJ, Qin ZH, Weiss B. Irreversible blockade of D2 dopamine receptors by fluphenazine-N-mustard increases D2 dopamine receptor mRNA and proenkephalin mRNA and decreases D1 dopamine receptor mRNA and mu and delta opioid receptors in rat striatum. Neurochem Int. 1994;25:355–66. doi: 10.1016/0197-0186(94)90143-0. [DOI] [PubMed] [Google Scholar]
- Chen TJH, Blum K, Chen LCH, Bowirrat A, Downs BW, Madigan MA, Waite RL, Bailey JA, Kerner M, Yelandi S, Giordano J, Morse S, Miller D, Braverman ER. Neurogenetics and clinical evidence for the putative activation of the brain reward circuitry by a neuroadaptagen: proposing an addiction candidate gene panel map. J Psychoactive Drugs. 2011;43(2):108–27. doi: 10.1080/02791072.2011.587393. [DOI] [PubMed] [Google Scholar]
- Chen TJ, Blum K, Payte JT, Schoolfield J, Hopper D, Stanford M, Braverman ER. Narcotic antagonists in drug dependence: pilot study showing enhancement of compliance with SYN-10, amino-acid precursors and enkephalinase inhibition therapy. Med Hypotheses. 2004;63:538–48. doi: 10.1016/j.mehy.2004.02.051. [DOI] [PubMed] [Google Scholar]
- Chen AL, Chen TJ, Waite RL, Reinking J, Tung HL, Rhoades P, Downs BW, Braverman E, Braverman D, Kerner M, Blum SH, DiNubile N, Smith D, Oscar-Berman M, Prihoda TJ, Floyd JB, O’Brien D, Liu HH, Blum K. Hypothesizing that brain reward circuitry genes are genetic antecedents of pain sensitivity and critical diagnostic and pharmacogenomic treatment targets for chronic pain conditions. Med Hypotheses. 2009;72:14–22. doi: 10.1016/j.mehy.2008.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370:1671–1672. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- Ciraulo DA, Hitzemann RJ, Somoza E, Knapp CM, Rotrosen J, Sarid-Segal O, Ciraulo AM, Greenblatt DJ, Chiang CN. Pharmacokinetics and pharmacodynamics of multiple sublingual buprenorphine tablets in dose-escalation trials. J Clin Pharmacol. 2006;46:179–92. doi: 10.1177/0091270005284192. [DOI] [PubMed] [Google Scholar]
- Chiang CN, Hawks RL. Pharmacokinetics of the combination tablet of buprenorphine and naloxone. Drug Alcohol Depend. 2003;70(2 Suppl):S39–47. doi: 10.1016/s0376-8716(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Comings DE, Blum K. Reward deficiency syndrome: genetic aspects of behavioral disorders. Prog Brain Res. 2000;126:325–341. doi: 10.1016/S0079-6123(00)26022-6. [DOI] [PubMed] [Google Scholar]
- Comings DE, Muhleman D, Gysin R. Dopamine D2 receptor (DRD2) gene and susceptibility to posttraumatic stress disorder: a study and replication. Biol Psychiatry. 1996;40:368–372. doi: 10.1016/0006-3223(95)00519-6. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mallen P, Costa LG, Checkoway H. Genotype combinations for monoamine oxidase-B intron 13 polymorphism and dopamine D2 receptor TaqIB polymorphism are associated with ever-smoking status among men. Neurosci Lett. 2005;385:158–162. doi: 10.1016/j.neulet.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Cowan A. Buprenorphine: new pharmacological aspects. Int J Clin Pract. 2003;(Suppl133):3–8. [PubMed] [Google Scholar]
- Dackis C, O’Brien C. Neurobiology of addiction: treatment and public policy ramifications. Nat Neurosci. 2005;8:1431–1436. doi: 10.1038/nn1105-1431. [DOI] [PubMed] [Google Scholar]
- da Silva Lobo DS, Vallada HP, Knight J, Martins SS, Tavares H, Gentil V, Kennedy JL. Dopamine genes and pathological gambling in discordant sib-pairs. J Gambl Stud. 2007;23:421–33. doi: 10.1007/s10899-007-9060-x. [DOI] [PubMed] [Google Scholar]
- De Ridder D, Vanneste S, Kovacs S, Sunaert S, Dom G. Transient alcohol craving suppression by rTMS of dorsal anterior cingulate: an fMRI and LORETA EEG study. Neurosci Lett. 2011;496(1):5–10. doi: 10.1016/j.neulet.2011.03.074. [DOI] [PubMed] [Google Scholar]
- Dole VP, Nyswander ME. Rehabilitation of heroin addicts after blockade with methadone. N Y State J Med. 1966;66:2011–2017. [PubMed] [Google Scholar]
- Doehring A, Hentig N, Graff J, Salamat S, Schmidt M, Geisslinger G, Harder S, Lötsch J. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19:407–414. doi: 10.1097/FPC.0b013e328320a3fd. [DOI] [PubMed] [Google Scholar]
- Dow-Edwards DL, Milhorat TH, Freed LA, Gintzler AR. Regional brain glucose utilization during and following chronic naltrexone administration: preliminary observations in rat brain. Life Sci. 1989;44:571–577. doi: 10.1016/0024-3205(89)90619-x. [DOI] [PubMed] [Google Scholar]
- Elkader A, Sproule B. Buprenorphine: clinical pharmacokinetics in the treatment of opioid dependence. Clin Pharmacokinet. 2005;44:661–80. doi: 10.2165/00003088-200544070-00001. [DOI] [PubMed] [Google Scholar]
- Epstein LH, Temple JL, Neaderhiser BJ, Salis RJ, Erbe RW, Leddy JJ. Food reinforcement, the dopamine D2 receptor genotype, and energy intake in obese and nonobese humans. Behav Neurosci. 2007;121:877–889. doi: 10.1037/0735-7044.121.5.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Feigenbaum J, Yanai J. The role of dopaminergic mechanisms in mediating the central behavioral effects of morphine in rodents. Neuropsychobiology. 1984;11(2):98–105. doi: 10.1159/000118061. [DOI] [PubMed] [Google Scholar]
- Fang WB, Chang Y, McCance-Katz EF, Moody DE. Determination of naloxone and nornaloxone (noroxymorphone) by high-performance liquid chromatography-electrospray ionization- tandem mass spectrometry. J Anal Toxicol. 2009;33:409–17. doi: 10.1093/jat/33.8.409. [DOI] [PubMed] [Google Scholar]
- Garavan H, Pankiewicz J, Bloom A, Cho JK, Sperry L, Ross TJ, Salmeron BJ, Risinger R, Kelley D, Stein EA. Cue-induced cocaine craving: neuroanatomical specificity for drug users and drug stimuli. Am J Psychiatry. 2000;157:1789–1798. doi: 10.1176/appi.ajp.157.11.1789. [DOI] [PubMed] [Google Scholar]
- Gaulier JM, Marquet P, Lacassie E, Dupuy JL, Lachatre G. Fatal intoxication following self-administration of a massive dose of buprenorphine. J Forensic Sci. 2000;45:226–228. [PubMed] [Google Scholar]
- Gold MS. Opiate addiction and the locus coeruleus. The clinical utility of clonidine, naltrexone, methadone, and buprenorphine. Psychiatr Clin North Am. 1993;16:61–73. [PubMed] [Google Scholar]
- Gold MS, Dackis CA, Pottash AL, Sternbach HH, Annitto WJ, Martin D, Dackis MP. Naltrexone, opiate addiction, and endorphins. Med Res Rev. 1982;2:211–46. doi: 10.1002/med.2610020302. [DOI] [PubMed] [Google Scholar]
- Goldstein A. Heroin addiction: neurobiology, pharmacology, and policy. J Psychoactive Drugs. 1991;23:123–33. doi: 10.1080/02791072.1991.10472231. [DOI] [PubMed] [Google Scholar]
- Green AI, Zimmet SV, Strous RD, Schildkraut JJ. Clozapine for comorbid substance use disorder and schizophrenia: do patients with schizophrenia have a reward-deficiency syndrome that can be ameliorated by clozapine? Harv Rev Psychiatry. 1999;6:287–296. doi: 10.3109/10673229909017206. [DOI] [PubMed] [Google Scholar]
- Hans G. Buprenorphine--a review of its role in neuropathic pain. J Opioid Manag. 2007;3:195–206. doi: 10.5055/jom.2007.0005. [DOI] [PubMed] [Google Scholar]
- Harris DS, Jones RT, Welm S, Upton RA, Lin E, Mendelson J. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Depend. 2000;61:85–94. doi: 10.1016/s0376-8716(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Hervé S, Riachi G, Noblet C, Guillement N, Tanasescu S, Goria O, Thuillez C, Tranvouez JL, Ducrotte P, Lerebours E. Acute hepatitis due to buprenorphine administration. Eur J Gastroenterol Hepatol. 2004;16:1033–1037. doi: 10.1097/00042737-200410000-00013. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Hagan JJ. Novel pharmacotherapeutic approaches for the treatment of drug addiction and craving. Curr Opin Pharmacol. 2005;5:107–118. doi: 10.1016/j.coph.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Hume SP, Lingford-Hughes AR, Nataf V, Hirani E, Ahmad R, Davies AN, Nutt DJ. Low sensitivity of the positron emission tomography ligand [11C]diprenorphine to agonist opiates. J Pharmacol Exp Ther. 2007;322:661–7. doi: 10.1124/jpet.107.121749. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Pevnick JS, Griffith JD. Human pharmacology and abuse potential of the analgesic buprenorphine: a potential agent for treating narcotic addiction. Arch Gen Psychiatry. 1976;35:501–516. doi: 10.1001/archpsyc.1978.01770280111012. [DOI] [PubMed] [Google Scholar]
- Judson BA, Goldstein A. Naltrexone treatment of heroin addiction: one-year follow-up. Drug Alcohol Depend. 1984;13:357–65. doi: 10.1016/0376-8716(84)90003-6. [DOI] [PubMed] [Google Scholar]
- Kosarac B, Fox AA, Collard CD. Effect of genetic factors on opioid action. Curr Opin Anaesthesiol. 2009;22:476–82. doi: 10.1097/ACO.0b013e32832e34c9. [DOI] [PubMed] [Google Scholar]
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int. 2001;121:65–69. doi: 10.1016/s0379-0738(01)00454-6. [DOI] [PubMed] [Google Scholar]
- Kirsch P, Reuter M, Mier D, Lonsdorf T, Stark R, Gallhofer B, Vaitl D, Hennig J. Imaging gene-substance interactions: the effect of the DRD2 TaqIA polymorphism and the dopamine agonist bromocriptine on the brain activation during the anticipation of reward. Neurosci Lett. 2006;405:196–201. doi: 10.1016/j.neulet.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Kreek MJ. Methadone-related opioid agonist pharmacotherapy for heroin addiction. History, recent molecular and neurochemical research and future in mainstream medicine. Ann N Y Acad Sci. 2000;909:186–216. doi: 10.1111/j.1749-6632.2000.tb06683.x. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, LaForge KS, Butelman E. Pharmacotherapy of addictions. Nat Rev Drug Discov. 2002;1:710–726. doi: 10.1038/nrd897. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the Brain Antireward System. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology 2010. 2010 Jan;35(1):217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesscher HM, Bailey A, Burbach JP, Van Ree JM, Kitchen I, Gerrits MA. Receptor-selective changes in mu-, delta- and kappa-opioid receptors after chronic naltrexone treatment in mice. Eur J Neurosci. 2003;17:1006–12. doi: 10.1046/j.1460-9568.2003.02502.x. [DOI] [PubMed] [Google Scholar]
- Lingford-Hughes A. Human brain imaging and substance abuse. Curr Opin Pharmacol. 2005;5:42–46. doi: 10.1016/j.coph.2004.10.002. [DOI] [PubMed] [Google Scholar]
- London ED, Cascella NG, Wong DF, Phillips RL, Dannals RF, Links JM, Herning R, Grayson R, Jaffe JH, Wagner HN., Jr Cocaine-induced reduction of glucose utilization in human brain. A study using positron emission tomography and [fluorine 18]-fluorodeoxyglucose. Arch Gen Psychiatry. 1990a;47:567–74. doi: 10.1001/archpsyc.1990.01810180067010. [DOI] [PubMed] [Google Scholar]
- London ED, Broussolle EP, Links JM, Wong DF, Cascella NG, Dannals RF, Sano M, Herning R, Snyder FR, Rippetoe LR, et al. Morphine-induced metabolic changes in human brain. Studies with positron emission tomography and [fluorine 18]fluorodeoxyglucose. Arch Gen Psychiatry. 1990b;47:73–81. doi: 10.1001/archpsyc.1990.01810130075010. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Lencz T, Correll CU, Kane JM. Genomics and the future of pharmacotherapy in psychiatry. Int Rev Psychiatry. 2007;19:523–530. doi: 10.1080/09540260701563460. [DOI] [PubMed] [Google Scholar]
- Mavridis M, Besson MJ. Dopamine-opiate interaction in the regulation of neostriatal and pallidal neuronal activity as assessed by opioid precursor peptides and glutamate decarboxylase messenger RNA expression. Neuroscience. 1999;92:945–66. doi: 10.1016/s0306-4522(99)00043-3. [DOI] [PubMed] [Google Scholar]
- McNicholas L. SAMHSA clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Treatment Improvement Protocol. 2004;40 DHHS Publication No. (SMA) 04–3939. [Google Scholar]
- Mei W, Zhang JX, Xiao Z. Acute effects of sublingual buprenorphine on brain responses to heroin-related cues in early-abstinent heroin addicts: an uncontrolled trial. Neuroscience. 2010;170(3):808–815. doi: 10.1016/j.neuroscience.2010.07.033. [DOI] [PubMed] [Google Scholar]
- Miller DK, Bowirrat A, Manka M, Miller M, Stokes S, Manka D, Allen C, Gant C, Downs BW, Smolen A, Stevens E, Yeldandi S, Blum K. Acute intravenous synaptamine complex variant KB220™ “normalizes” neurological dysregulation in patients during protracted abstinence from alcohol and opiates as observed using quantitative electroencephalographic and genetic analysis for reward polymorphisms: part 1, pilot study with 2 case reports. Postgrad Med. 2010;122:188–213. doi: 10.3810/pgm.2010.11.2236. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Reinforcement and addictive disorders. In: Nestler EJ, et al., editors. Molecular neuropharmacology: a foundation for clinical neuroscience. New York, NY: McGraw-Hill; 2009. [Google Scholar]
- O’Brien CP. Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry. 2005;162:1423–1431. doi: 10.1176/appi.ajp.162.8.1423. [DOI] [PubMed] [Google Scholar]
- Orman JS, Keating GM. Spotlight on buprenorphine/naloxone in the treatment of opioid dependence. CNS Drugs. 2009;23:899–902. doi: 10.2165/11203740-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Poisnel G, Dhilly M, Le Boisselier R, Barre L, Debruyne D. Comparison of five benzodiazepine-receptor agonists on buprenorphine-induced mu-opioid receptor regulation. J Pharmacol Sci. 2009;110:36–46. doi: 10.1254/jphs.08249fp. [DOI] [PubMed] [Google Scholar]
- Ponce G, Jimenez-Arriero MA, Rubio G, Hoenicka J, Ampuero I, Ramos JA, Palomo T. The A1 allele of the DRD2 gene (TaqI A polymorphisms) is associated with antisocial personality in a sample of alcohol-dependent patients. Eur Psychiatry. 2003;18:356–360. doi: 10.1016/j.eurpsy.2003.06.006. [DOI] [PubMed] [Google Scholar]
- Peng XQ, Li X, Li J, Ramachandran PV, Gagare PD, Pratihar D, Ashby CR, Jr, Gardner EL, Xi ZX. Effects of gabapentin on cocaine self-administration, cocaine-triggered relapse and cocaine-enhanced nucleus accumbens dopamine in rats. Drug Alcohol Depend. 2008;97:207–15. doi: 10.1016/j.drugalcdep.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrière H, Tatem L, Bories C, Pageaux GP, Blayac JP, Larrey D. Hepatitis after intravenous injection of sublingual buprenorphine in acute hepatitis C carriers: report of two cases of disappearance of viral replication after acute hepatitis. Ann Pharmacother. 2009;43:973–977. doi: 10.1345/aph.1L628. [DOI] [PubMed] [Google Scholar]
- Radwan GN, Setouhy ME, Mohamed MK, Hamid MA, Israel E, Azem SA, Kamel O, Loffredo CA. DRD2/ANKK1 TaqI polymorphism and smoking behavior of Egyptian male cigarette smokers. Nicotine Tob Res. 2007;9:1325–1329. doi: 10.1080/14622200701704889. [DOI] [PubMed] [Google Scholar]
- Ritchie T, Noble EP. Association of seven polymorphisms of the D2 dopamine receptor gene with brain receptor-binding characteristics. Neurochem Res 2003. 2003;28:73–82. doi: 10.1023/a:1021648128758. [DOI] [PubMed] [Google Scholar]
- Ritchie T, Noble EP. [3H]naloxone binding in the human brain: alcoholism and the TaqI A D2 dopamine receptor polymorphism. Brain Res. 1996;718:193–197. doi: 10.1016/0006-8993(96)00068-6. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Blough BE, Baumann MH. Dual dopamine/serotonin releasers as potential medications for stimulant and alcohol addictions. AAPS J. 2007;9:E1–10. doi: 10.1208/aapsj0901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradgoli Najafabadi M, Ohadi M, Joghataie MT, Valaie F, Riazalhosseini Y, Mostafavi H, Mohammadbeigi F, Najmabadi H. Association between the DRD2 A1 allele and opium addiction in the Iranian population. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:39–41. doi: 10.1002/ajmg.b.30117. [DOI] [PubMed] [Google Scholar]
- Schottenfeld RS, Chawarski MC, Pakes JR, Pantalon MV, Carroll KM, Kosten TR. Methadone versus buprenorphine with contingency management or performance feedback for cocaine and opioid dependence. Am J Psychiatry. 2005;162:340–349. doi: 10.1176/appi.ajp.162.2.340. [DOI] [PubMed] [Google Scholar]
- Spangler R, Wittkowski KM, Goddard NL, Avena NM, Hoebel BG, Leibowitz SF. Opiate-like effects of sugar on gene expression in reward areas of the rat brain. Brain Res Mol Brain Res. 2004;124:134–142. doi: 10.1016/j.molbrainres.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Seeman P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin Schizophr Relat Psychoses. 2010;4:56–73. [PubMed] [Google Scholar]
- Spitz MR, Shi H, Yang F, Hudmon KS, Jiang H, Chamberlain RM, Amos CI, Wan Y, Cinciripini P, Hong WK, Wu X. Case-control study of the D2 dopamine receptor gene and smoking status in lung cancer patients. J Natl Cancer Inst. 1998;90:358–363. doi: 10.1093/jnci/90.5.358. [DOI] [PubMed] [Google Scholar]
- Stock C, Shum JH. Bupreorphine:a new pharmacotherapy for opioid addictions treatment. J Pain Palliat Care Pharmacother. 2004;18:35–54. [PubMed] [Google Scholar]
- Swan GE, Jack LM, Valdes AM, Ring HZ, Ton CC, Curry SJ, McAfee T. Joint effect of dopaminergic genes on likelihood of smoking following treatment with bupropion SR. Health Psychol. 2007;26:361–368. doi: 10.1037/0278-6133.26.3.361. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Han SD, Lucas LR. Chronic passive exposure to aggression decreases D2 and 5-HT 1B receptor densities. Physiol Behav. 2010;99(5):562–570. doi: 10.1016/j.physbeh.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA. GABAergic transmission modulates ethanol excitation of ventral tegmental area dopamine neurons. Neuroscience. 2011;13;172:94–172103. doi: 10.1016/j.neuroscience.2010.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracqui A, Tournoud C, Flesch F, Kopferschmitt J, Kintz P, Deveaux M, Ghysel MH, Marquet P, Pépin G, Petit G, Jaeger A, Ludes B. Acute poisoning during substitution therapy based on high-dosage buprenorphine. 29 clinical cases--20 fatal cases. Presse Med. 1998;27:557–61. [PubMed] [Google Scholar]
- Umbricht A, Montoya ID, Hoover DR, Demuth KL, Chiang CT, Preston KL. Naltrexone shortened opioid detoxification with buprenorphine. Drug Alcohol Depend. 1999;56:181–190. doi: 10.1016/s0376-8716(99)00033-2. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services. Substance Abuse and Mental Health Services Administration (SAMHSA) [Accessed February 26, 2003];Opioid agonist therapy (OAT) with methadone or LAAM. Available at: http://dpt.samhsa.gov/001025o.ptx.htm.
- Van den Oever MC, Lubbers BR, Goriounova NA, Li KW, Van der Schors RC, Loos M, et al. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology. 2010;35:2120–33. doi: 10.1038/npp.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol Learn Mem. 2002;78:610–624. doi: 10.1006/nlme.2002.4099. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. The addicted human brain: insights from imaging studies. J Clin Invest. 2003a;111:1444–1451. doi: 10.1172/JCI18533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56(Suppl 1):3–8. doi: 10.1016/j.neuropharm.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Ma Y, Fowler JS, Zhu W, Maynard L, Telang F, Vaska P, Ding YS, Wong C, Swanson JM. Expectation enhances the regional brain metabolic and the reinforcing effects of stimulants in cocaine abusers. J Neurosci. 2003b;23:11461–11468. doi: 10.1523/JNEUROSCI.23-36-11461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Jayne M, Telang F, Swanson JM. Effects of expectation on the brain metabolic responses to methylphenidate and to its placebo in non-drugabusing subjects. Neuroimage. 2006a;32:1782–1792. doi: 10.1016/j.neuroimage.2006.04.192. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M, Ma Y, Pradhan K, Wong C. Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement. J Neurosci. 2007;27:12700–12706. doi: 10.1523/JNEUROSCI.3371-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley AY, Alperen JK, Cheng DM, Botticelli M, Castro-Donlan C, Samet JH, Alford DP. Office-based management of opioid dependence with buprenorphine: clinical practices and barriers. J Gen Intern Med. 2008;23:1393–1398. doi: 10.1007/s11606-008-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SL, Gilson SF, Jasinski DR, Stapleton JM, Phillips RL, Dannals RF, Schmidt J, Preston KL, Grayson R, Bigelow GE, et al. Buprenorphine reduces cerebral glucose metabolism in polydrug abusers. Neuropharmacology. 1994;10:157–170. doi: 10.1038/npp.1994.18. [DOI] [PubMed] [Google Scholar]
- Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol. 2005;5:9–19. doi: 10.1016/j.coph.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Wilson SJ, Sayette MA, Fiez JA. Prefrontal responses to drug uses: a neurocognitive analysis. Nat Neurosci. 2004;7:211–214. doi: 10.1038/nn1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E, Li K, Palepu A, Marsh DC, Schechter MT, Hogg RS, Montaner JS, Kerr T. Sociodemographic disparities in access to addiction treatment among a cohort of Vancouver injection drug users. Subst Use Misuse. 2005;40:1153–67. doi: 10.1081/JA-200042287. [DOI] [PubMed] [Google Scholar]
- Xiao C, Zhang J, Krnjeviæ K, Ye JH. Effects of ethanol on midbrain neurons: role of opioid receptors. Alcohol Clin Exp Res. 2007;31:1106–1113. doi: 10.1111/j.1530-0277.2007.00405.x. [DOI] [PubMed] [Google Scholar]
- Xu K, Lichtermann D, Lipsky RH, Franke P, Liu X, Hu Y, Cao L, Schwab SG, Wildenauer DB, Bau CH, Ferro E, Astor W, Finch T, Terry J, Taubman J, Maier W, Goldman D. Association of specific haplotypes of D2 dopamine receptor gene with vulnerability to heroin dependence in 2 distinct populations. Arch Gen Psychiatry. 2004;61:597–606. doi: 10.1001/archpsyc.61.6.597. [DOI] [PubMed] [Google Scholar]
- Yassen A, Olofsen E, Romberg R, Sarton E, Danhof M, Dahan A. Mechanism-based pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of buprenorphine in healthy volunteers. Anesthesiology. 2006;104:1232–42. doi: 10.1097/00000542-200606000-00019. [DOI] [PubMed] [Google Scholar]
- Yoburn BC, Shah S, Chan K, Duttaroy A, Davis T. Supersensitivity to opioid analgesics following chronic opioid antagonist treatment: relationship to receptor selectivity. Pharmacol Biochem Behav. 1995;51:535–539. doi: 10.1016/0091-3057(94)00375-s. [DOI] [PubMed] [Google Scholar]
- Yücel M, Lubman DI, Harrison BJ, Fornito A, Allen NB, Wellard RM, Roffel K, Clarke K, Wood SJ, Forman SD, Pantelis C. A combined spectroscopic and functional MRI investigation of the dorsal anterior cingulate region in opiate addiction. Mol Psychiatry. 2007;12:611, 691–702. doi: 10.1038/sj.mp.4001955. [DOI] [PubMed] [Google Scholar]
- Zhou SF, Liu JP, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev. 2009;41:89–295. doi: 10.1080/03602530902843483. [DOI] [PubMed] [Google Scholar]