

Abstract
Recently, we reported the spectroscopic and kinetic characterizations of cytochrome P450 compound I in CYP119A1, effectively closing the catalytic cycle of cytochrome P450-mediated hydroxylations. In this minireview, we focus on the developments that made this breakthrough possible. We examine the importance of enzyme purification in the quest for reactive intermediates and report the preparation of compound I in a second P450 (P450ST). In an effort to bring clarity to the field, we also examine the validity of controversial reports claiming the production of P450 compound I through the use of peroxynitrite and laser flash photolysis.
Keywords: Cytochrome P450, Enzyme Catalysis, Enzyme Purification, Heme, Spectroscopy, P450 Compound I
Introduction
A significant amount of research in the field of bioinorganic chemistry is focused on discerning the intimate details of enzyme catalysis. Critical to these efforts is the preparation of reactive intermediates that form the stations of the catalytic cycle of an enzyme. The study of these transient species is driven by the hope that insights gleaned from their electronic, structural, and kinetic characterizations will guide the design of next-generation catalysts or point the way to inhibitors that could serve as drugs for a variety of maladies. Although a thorough characterization of all the intermediates in a catalytic cycle is required for a detailed dissection of the catalytic mechanism, there are certain species that have special significance and, as such, are considered high-value targets for characterization. These species are generally the highly reactive intermediates that are ultimately responsible for the most important, difficult, or chemically interesting transformation in the catalytic mechanism.
Recently, we reported the capture and characterization of one of the most highly sought intermediates in biological chemistry, P450 compound I (P450-I)4 (1). This iron(IV)-oxo (or ferryl) radical species (7 in Fig. 1) had long been thought to be the principal intermediate in cytochrome P450 catalysis, but due to its highly reactive nature, it had eluded definitive characterization for over 40 years. The existence of P450-I was postulated based on the observation of a “shunt pathway” (Fig. 1) allowing the oxidation of substrates through the use of oxygen donors such as hydrogen peroxide and meta-chloroperbenzoic acid. These oxidants were known to generate high-valent iron-oxo species in heme peroxidases (2, 3), suggesting that a similar intermediate might be involved in P450 catalysis. However, 4 decades worth of searching for the elusive P450-I had led to questions about not only its competence as a hydroxylating agent but also its role in P450 catalysis (4–6).
FIGURE 1.
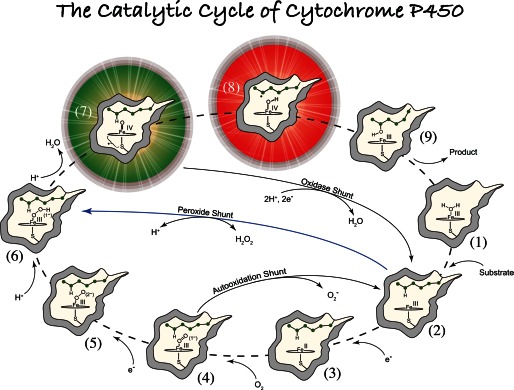
General paradigm for P450-catalyzed hydroxylations. The first step involves the binding of substrate to the resting low-spin ferric enzyme (1). This binding induces structural changes, which often, but not always, manifest themselves in the dissociation of the distally coordinated water and the conversion of the heme from low to high spin (2). These substrate-induced structural changes facilitate reduction of the ferric enzyme, allowing delivery of the first electron to generate the ferrous substrate-bound form of the enzyme (3). Dioxygen then binds to the ferrous heme, forming a species that is best described as a ferric superoxide complex (4). The subsequent reduction of this species forms a ferric peroxo species (5), which is protonated at the distal oxygen to generate a ferric hydroperoxo complex (6). The delivery of an additional proton to the distal oxygen cleaves the O–O bond, yielding compound I (7) and a water molecule. Compound I then abstracts hydrogen from substrate to yield compound II (8) and a substrate radical, which rapidly recombine to yield hydroxylated product and ferric enzyme (9). Hydroxylated product then dissociates, and water coordinates to the heme to regenerate the resting ferric enzyme (1).
Our investigations confirmed the existence and the reactive nature of the intermediate. P450-I is capable of hydroxylating unactivated C–H bonds with the remarkable rate constant of 1 × 107 m−1 s−1 (1). Kinetic isotope effects support a mechanism in which P450-I abstracts hydrogen from substrate, forming an iron(IV)-hydroxide complex that rapidly recombines with substrate to yield hydroxylated product (7–9 in Fig. 1) (1, 7, 8). An important and perhaps underappreciated consequence of this kinetic result is that it cements the role of P450 compound II (P450-II) (8 in Fig. 1) in P450 catalysis: the intermediate is directly involved in substrate oxidation (9, 10). In what follows, we highlight the developments that led to the capture and characterization of the elusive P450-I and review insights gained from our electronic and structural characterizations of compounds I and II.
This minireview also seeks to address recent (and controversial) reports of stable (i.e. relatively unreactive) preparations of P450-I (4, 6, 11–15). These reports are surprising, given the highly reactive and historically elusive nature of P450-I (1, 5). The authors of these studies have argued the need for a new/alternative oxidant in the catalytic cycle. These preparations of unreactive P450-I are reportedly obtained through an unusual process involving the use of peroxynitrite and laser flash photolysis (PN/LFP). We analyze the ability of the PN/LFP method to generate P450-I, hopefully bringing some clarity to the debate.
Closing the Cycle: The Quest for Compound I
The general paradigm for P450-catalyzed substrate hydroxylations is shown in Fig. 1 (16, 17). The first step involves the binding of substrate to the resting low-spin ferric enzyme (1). This binding induces structural changes, which often, but not always, (16), manifest themselves in the dissociation of the distally coordinated water and the conversion of the heme from low to high spin (2). These substrate-induced structural changes facilitate reduction of the ferric enzyme (18), allowing delivery of the first electron to generate the ferrous substrate-bound form of the enzyme (3). Dioxygen then binds to the ferrous heme, forming a species that is best described as a ferric superoxide complex (4). The subsequent reduction of this species forms a ferric peroxo species (5), which is protonated at the distal oxygen to generate a ferric hydroperoxo complex (6). The delivery of an additional proton to the distal oxygen cleaves the O–O bond, yielding compound I (7) and a water molecule. Compound I then abstracts hydrogen from substrate to yield compound II (8) and a substrate radical, which rapidly recombine to yield hydroxylated product and ferric enzyme (9). Hydroxylated product then dissociates, and water coordinates to the heme to regenerate the resting ferric enzyme (1).
P450-I has not been observed under turnover conditions, but it can be generated transiently via the peroxide shunt using oxidants such as meta-chloroperbenzoic acid (m-CPBA) (19–21). Traditionally, however, the yields in these experiments have been too low (<5%) to facilitate characterization by more advanced spectroscopic techniques. As a result, a number of ingenious experiments have been designed in attempts to circumvent the reactive nature of P450-I. These efforts have generally sought to make C–H bond activation (as opposed to compound I formation) rate-limiting. It is in this vein that researchers have employed cryogenic reduction techniques to deliver the reducing equivalent that triggers P450-I formation. In these experiments, the ferrous oxy form of the enzyme is reduced radiolytically, at cryogenic temperatures, by exposure to 60Co (22–25), 32P (26), or synchrotron radiation (27). Reduction of the oxy complex yields a ferric peroxo species, which can be annealed at higher temperatures to allow for proton delivery, cleavage of the O–O bond, and P450-I formation. This technique has been coupled with electron nuclear double resonance spectroscopy and x-ray crystallography in hopes of obtaining electronic and structural characterizations of the intermediate (22–27). These experiments provided clear evidence for cleavage of the dioxygen bond (i.e. the presence of hydroxylated product). However, P450-I did not accumulate to detectable amounts.
Investigators have also sought the use of flash-quench techniques, in which a laser pulse triggers the rapid reduction or oxidation of an active site of an enzyme. The idea with reductive flash-quench (as with cryogenic reduction) is to deliver the electron that triggers compound I formation. The source of electrons in these experiments is a photoactive redox agent that can be attached to the substrate via a hydrocarbon tether or covalently linked through modification of a non-native cysteine. Although electron injection by reductive flash-quench should be fast enough to make C–H bond activation rate-limiting, the successful generation of P450-I by this technique has yet to be reported. Instead, researchers have had limited success using the oxidative route. The rapid removal of one electron from the P450 active site effectively runs the catalytic cycle in reverse, generating compound II (an iron(IV)-hydroxide species) from ferric enzyme. As with reductive flash-quench, however, the technique has yet to yield P450-I (28, 29).
In efforts to prepare P450-I by slowing the decay of the intermediate, researchers have turned to the use of “slow” substrates. These substrates are compounds that have their targeted hydrogen atoms replaced by fluorines. In theory, this substitution should allow for preparation of the intermediate in high yield, as C–F bonds are not activated by P450-I. However, studies with these fluorinated compounds have found that P450-I either oxidizes alternative (non-fluorinated) positions on the substrate or decays through nonproductive uncoupling (30, 31).
Amazingly, despite these and other intense efforts (32), the capture and characterization of P450-I remained an unobtainable goal in biological chemistry. Indeed, a recent review on the enigmatic nature of P450-I noted that, despite 45 years of effort by the P450 community, the same questions remain: does P450-I exist, and how does it oxidize substrates? It was concluded that the quest for the elusive intermediate would require new and improved methods of preparation and detection combined with theoretical simulations (5).
Given this background, what is remarkable about the successful capture of P450-I is that the feat did not require any great advancement in technology. In the end, it did not require slow substrates, cryogenic reduction, or the use of flash-quench methods. Likewise, no improvements in rapid mixing or freezing techniques were necessary. The key to our success was simply enzyme purification (1). In what follows, observations that led to the eventual capture and characterization of P450-I will be discussed. In doing so, we hope to draw attention to what may be an underappreciated concern in the quest for reactive intermediates: the presence of endogenous substrates.
Importance of Enzyme Purification in the Quest for Reactive Intermediates
In an effort to resolve a debate concerning the UV-visible spectrum of compound I in CYP119A1 (13, 20, 33), a P450 from the thermophilic organism Sulfolobus acidocaldarius (34–36), we noticed that the yield of P450-I in stopped-flow reactions was variable. Some enzyme preparations produced no compound I, whereas others would yield as much as 10%. Importantly, the protein used in these experiments was deemed to be of high purity based not only on its Reinheitszahl value (i.e. Rz or purity ratio of A416 nm/A280 nm) but also on an SDS-polyacrylamide gel. Purification was performed according to an established protocol, which involved heat denaturation and ammonium sulfate fractionation followed by size exclusion and ion exchange chromatographies (34).
Although the variable yields of P450-I in stopped-flow experiments were confusing, it seemed possible to justify them in terms of experimental variation/error (e.g. variable equivalents of protein and/or oxidant combined with variable levels of protein activity in different growth batches). A breakthrough came when a buffered solution of “purified” thermophilic enzyme turned cloudy after several days at ∼25 °C. Centrifugation of this suspension resulted in protein that could generate as much as 40% compound I. This observation suggested that there was a route to increased compound I yields. After careful analysis, it was determined that the enzyme used in our stopped-flow experiments had a mixture of contaminating small molecules bound to its active site. The presence of these endogenous species hindered compound I formation and expedited its decay.
GC/MS analysis of protein purified according to the established protocol (EP-CYP119A1) revealed that the enzyme contains a distribution of fatty acids (C12, C14, C16, and C18) bound at its active site. Although the binding of these endogenous substrates shows little, if any, spectroscopic signature, their presence can strongly (if not completely) suppress compound I formation (Fig. 2A). Over 80% of these contaminating fatty acids can be removed through additional ion exchange chromatography (1). The highly purified protein (HP-CYP119A1) obtained from this process shows an ∼10-fold increase/decrease in the rate constants for compound I formation/decay, allowing for the preparation of P450-I in high (∼75%) yields (Fig. 2B). Fig. 2C shows that there is very little difference between the UV-visible spectra of EP- and HP-CYP119A1. Removing the fatty acids from CYP119A1 red shifts the Soret maximum by ∼1 nm.
FIGURE 2.
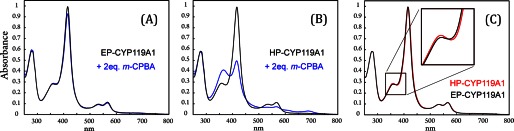
Effect of purification on compound I preparation. See text for a discussion of the EP (purified) and HP (highly purified) labels. A, spectrum of EP-CYP119A1 (black) and a spectrum taken (at maximum formation of P450-I) during the reaction of EP-CYP119A1 with 2 eq of m-CPBA at 4 °C (blue). Compound I is formed in very low yield. B, spectrum of HP-CYP119A1 (black) and a spectrum taken (at maximum formation of P450-I) during the reaction of HP-CYP119A1 with 2 eq of m-CPBA at 4 °C (blue). Compound I can be produced in up to ∼75% yield. C, overlay of HP-CYP119A1 (red) and EP-CYP119A1 (black).
Preparation of Compound I in a Second P450 (P450ST)
Given our success in preparing compound I in CYP119A1, we looked for another P450 system that might yield the elusive intermediate following extensive purification. We turned to the thermophilic enzyme P450ST (CYP119A2) from Sulfolobus tokodaii. This P450 has 64% sequence identity to CYP119A1 but was reported to have an unusual third Q-band in its visible absorption spectrum (37). We also observed an additional absorption in the Q-band region during the preliminary stages of protein purification (after heat denaturation and ammonium sulfate fractionation). However, this absorption was due to an unknown contaminating species, which was eliminated completely using tangential flow filtration.
Following tangential flow filtration, initial ion exchange chromatography results in P450ST that appears to be of high purity based on SDS-PAGE and its Rz of ∼1.65. However, P450ST at this stage of purification yields little, if any, compound I (≤10% depending on the growth batch). As with CYP119A1, GC/MS analysis revealed the presence of a distribution of fatty acids bound to the enzyme. The distribution in P450ST is slightly different, predominantly a mixture of C16 and C18. The majority of these endogenous substrates can be removed through an additional chromatographic step (1). This highly purified protein results in enzyme preparations that yield ∼65% P450ST-I. In Fig. 3, spectra obtained from EPR and Mössbauer measurements on this species are shown in comparison with those obtained for CYP119A1-I.
FIGURE 3.

Comparison of the EPR (left) and Mössbauer (right) spectra of CYP119A1-I and P450ST-I. Mössbauer spectra were recorded at 4.2 K with a 54-millitesla field oriented parallel to the γ-beam. Mössbauer spectra were obtained by subtracting contributions of ferric enzyme (30 and 35%, respectively) from the raw data. Fits of the Mössbauer data (shown in red) yield the following parameters: P450ST-I, ΔEQ = 0.85 mm/s and δ = 0.12 mm/s; and CYP119A1-I, ΔEQ = 0.90 mm/s and δ = 0.11 mm/s. The EPR spectra were obtained as reported previously (1). Fits of the P450ST-I EPR data indicate |J/D| = 1.3 and g = 1.95, 1.84, and 1.99, in good agreement with the values reported previously for CYP119A1-I (1).
We note that the observation of endogenous substrates in what might be considered “pure” protein is not unique to the systems reported here. The crystal structures of P450BSα and P450BSβ both contain fatty acids, even though no substrates were added to the purified enzyme (38, 39), and similar observations have been made for human microsomal CYP2CA (40). Furthermore, it seems unlikely that this phenomenon is unique to P450s. The rupture of the cell(s) of an organism during the course of enzyme purification releases myriad small molecules, many of which are poorly soluble. These species can find refuge in the hydrophobic cavities of enzymes, where they can interfere by blocking substrate access, altering proton transfer pathways, and modifying reduction potentials.
Compound II: An Underappreciated Partner in C–H Bond Activation
An important aspect of our work on P450-I is that it confirms that the rebound mechanism is operative in P450-mediated substrate hydroxylations, thereby cementing the role of compound II in C–H bond activation. This means that the fundamental physical parameters of compound II play a role in determining the cleavage of the C–H bond (9, 10). To understand this, it is helpful to consider the factors that govern C–H bond activation in this system.
Mayer has written extensively on hydrogen atom abstraction by metal-oxo complexes. Building on the work of Evans, Polanyi, and Bordwell, he has shown that ground state thermodynamics (the energy difference between the C–H bond broken and the O–H bond formed) is generally sufficient to understand reactivity in these systems (41, 42). The strength of the O–H bond in compound II is determined by the one-electron reduction potential of compound I and the pKa of compound II (Equation 1).
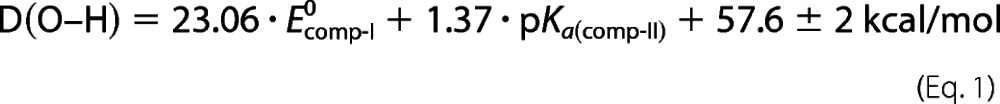 |
It has been shown that the P450 thiolate-ligated heme promotes basic iron(IV)-oxo or ferryl species. P450-II is best described as an iron(IV)-hydroxide complex)pKa > 8) (10, 43–47). Equation 1 and the elevated pKa observed in P450-II suggest a critical role for the unusual P450 thiolate ligand: to promote C–H bond activation at biologically viable reduction potentials (9, 10).
Peroxynitrite: A Panacea for Compound II Formation?
Interest in the preparation and characterization of P450-II leads one naturally to PN and the debate surrounding its use in P450s. In 2000, Ullrich and co-workers (48) reported the formation of what appeared to be a ferryl species during the PN-driven inactivation of P450BM3. The PN-generated species, which had a UV-visible spectrum similar to that of chloroperoxidase compound II (49), was relatively stable, decaying at an apparent rate of 0.08 s−1 (half-life of ∼9 s). These results suggested that PN could be used to prepare P450-II in high yield, and our group and others sought to use the technique for this purpose. Importantly, however, a thorough spectroscopic characterization of the PN-generated species revealed that it is not a ferryl intermediate (50).
The reaction of PN with P450s generates a ferric nitrosyl complex on the time scale of stopped-flow and freeze-quench reactions (50). Compound II does not accumulate to an appreciable degree. The Mössbauer, UV-visible, and resonance Raman spectra of the PN-generated intermediate match those of a ferric nitrosyl complex. Importantly, high-field Mössbauer measurements reveal that the PN-generated species is S = 0, as expected for an {FeNO}6 complex, but inconsistent with an authentic S = 1 iron(IV)-oxo (or hydroxide) species (50).
Unreactive Preparations of P450-I: Will the Real Compound I Please Stand up?
We now examine reports of PN/LFP being used to generate stable and unreactive preparations of P450-I (4, 6, 11–15). These reports have led to considerable confusion about the status of compound I and its role in P450 catalysis (51). The idea behind the PN/LFP method was that PN could be reacted with ferric enzyme to generate compound II, which could then be oxidized using LFP to yield compound I. The problem with this method is that there is no evidence that the reaction of PN with ferric P450 yields compound II on the time scale of the LFP experiments. As noted above, the reaction of PN with ferric P450 generates a ferric nitrosyl complex (50). It is not possible to generate compound I from a ferric nitrosyl complex using LFP.
The first report of the PN/LFP method being used to generate P450-I appeared in 2006, when the technique was purportedly used to prepare compound I in CYP119A1 (12). The PN/LFP-generated intermediate was reported to be relatively stable and unreactive toward lauric acid, even under saturating conditions. In a 2008 study, the rate constant for the reaction with lauric acid was revised to 0.8 s−1. It was in this study that proponents of the PN/LFP method first questioned the generally accepted UV-visible spectrum of P450-I (13). They did so because their PN/LFP-generated samples lacked a species possessing the well known spectrum of the intermediate.
The spectrum of P450-I had been previously reported by Ishimura et al. (P450cam) (19) and Sligar et al. (CYP119A1) (20). In both cases, the intermediate was generated in low yield by reacting ferric enzyme with m-CPBA, and the spectrum of P450-I was obtained using global analysis techniques. Both P450-I spectra show great similarity to the well known spectrum of chloroperoxidase compound I (52), a thiolate-ligated heme protein that has often served as a model system for P450. Importantly, nothing resembling these spectra was produced from the PN/LFP experiments. As a result, proponents of the PN/LFP technique argued that the previously reported P450-I spectra were flawed, suggesting that they were obtained through the improper use of global analysis fitting routines. They argued that the results obtained from global analysis techniques are highly dependent upon the initial guess for rate constants and that previous investigators used P450-I decay rates that were too high because they were biased by knowledge of the chloroperoxidase compound I spectrum (13). Proponents of the PN/LFP technique claimed that the real spectrum of P450-I was almost identical to that of ferric enzyme. Indeed, in a 2009 article (4), they wrote, “The UV spectra, and especially the Soret absorbances, of the P450 compounds I are similar to the UV-visible spectra of resting enzymes. This similarity is undoubtedly one of the major difficulties in attempting to detect Compound I under turnover conditions or in reactions of the enzymes with chemical oxidants.”
Over the course of 2009, several articles on the ferric-looking P450-I intermediate were published, some even reporting its quantitative production in P450BM3 and CYP119A1 (4, 6, 14, 15). These species were reported to be reactive but not overly so: oxidizing benzyl alcohol at a rate of 4 s−1 at 22 °C. The sluggish reactivity of the PN/LFP-generated species led to the call for an alternative oxidant in the catalytic cycle, one that was more oxidizing than the sluggish PN/LFP-generated P450-I.
In an effort to provide clarity to the debate, we sought to obtain the UV-visible spectrum of P450-I from stopped-flow data via model-independent methods (33). Singular value decomposition and target testing provided a parameter-free P450-I UV-visible spectrum that was in excellent agreement with those reported previously by Ishimura et al. (19) and Sligar et al. (20). Importantly, no assumptions about reaction kinetics were required to obtain the spectrum. The implications are clear. The PN/LFP-generated species is not P450-I. This result was confirmed during our most recent investigation of P450-I, in which we were able to prepare the intermediate in ∼75% yield, obtaining its definitive characterization via Mössbauer, EPR, and UV-visible spectroscopies (1).
In 2010, a forum in Inorganic Chemistry (33) noted, “What is required is a thorough spectroscopic characterization of the LFP-generated species. Given the reported quantitative yields and exceedingly long lifetimes, the LFP-generated species would seem ripe for spectroscopic characterization. Mössbauer, electron paramagnetic resonance, and resonance Raman spectroscopies, in particular, would provide significant insights into the nature of the LFP-generated species. We await these spectroscopic reports.”
To date, no convincing spectroscopic evidence has been presented for the existence of a PN/LFP-generated P450-I species. Still, reports of the method's use continue to appear in the literature, with the most recent report appearing during the writing of this minireview (53). We briefly examine a troubling (yet insightful) aspect of this recent work. In it, Newcomb and co-workers (53) presented the UV-visible spectrum of the species generated by the PN/LFP method. They assigned this spectrum to P450-I. To support this claim, they overlaid a P450-I reference spectrum, obtained from the reaction of m-CPBA with ferric enzyme. Fig. 4A shows the data and reference spectrum extracted from Fig. 1C of Ref. 53. At first glance, it appears that there is good agreement between the P450-I reference spectrum and the spectrum of the PN/LFP-generated species, but, upon closer inspection, it is clear that the PN/LFP-generated species lacks Q-band absorbance features that are the hallmarks of P450-I.
FIGURE 4.

A, data (black) and shifted reference spectrum (gray) from Fig. 1C of Ref. 53. Spectra were extracted using an analog-to-digital program. B, comparison of the spectrum of the PN/LFP-generated species in CYP119A1 (black) (data from Fig. 1C of Ref. 53) with an authentic CYP119A1 P450-I reference spectrum (red) (1).
The Q-bands are the absorption bands that can be seen in the reference spectrum between ∼500 and 700 nm, but they are clearly absent in the raw data (Fig. 4A). The most prominent of the P450-I Q-bands is known to appear at ∼690 nm and is thought to be responsible for the green color of P450-I (1, 19, 20). It is also noteworthy that the 690 nm band cannot be seen in the spectrum shown in Fig. 1B of Ref. 53, which the authors reported could be decomposed into 50% P450-I, 15% P450-II, and 35% ferric enzyme. The lack of a 690 nm band in the raw data of Ref. 53 is troubling.
Additionally, the P450-I reference spectrum used in Ref. 53 appears to be blue-shifted by ∼12 nm. To examine the matter further, an analog-to-digital program (GraphClick v3.0.2, Arizona Software) was used to extract the spectra from Fig. 1C of Ref. 53. Upon plotting the extracted raw data, one observes that its maximum absorbance is centered at ∼355 nm. This is a clear indication that the spectral assignment in Ref. 53 is in error. P450-I has a maximum absorbance at ∼367 nm (1, 19, 20, 33, 52). This can be seen in Fig. 4B, in which the data from Fig. 1C of Ref. 53 are plotted with an actual (unshifted) P450-I spectrum (shown in red) (1). It is clear that the spectrum produced by the PN/LFP method does not belong to P450-I.
In closing the subject, it is interesting to note the use of a shifted P450-I spectrum by Newcomb and co-workers (53) as their reference and not the ferric-like spectrum that they have argued for previously. One wonders about the implications for their previous work (4, 6, 11–15). Furthermore, given that the PN/LFP technique does not yield P450-I, it would appear to be difficult to follow P450-I reaction kinetics using this method (4, 6, 11–15).
Conclusion and Outlook
Enzyme purification is key to the preparation of reactive intermediates. Fatty acids have been found to occupy the active sites of a number of purified P450s. In CYP119, the presence of these small molecules hinders the formation and expedites the decay of compound I. Removal of these endogenous substrates through extensive enzyme purification allows for the preparation of P450-I in high yield. Importantly, P450-I prepared in this manner can be reacted directly with substrate, allowing researchers to avoid the limitations of steady-state kinetics. The use of transient techniques has the potential to provide unprecedented insight into a number of P450-mediated oxidations. However, the subset of P450s that is amenable to this treatment is unknown. Significant effort has been devoted only toward the thermophilic enzymes discussed here.
Finally, consider that this is not the first time that endogenous reductants have masked or interfered with the formation of high-valent intermediates in heme systems. Indeed, the first intermediate identified in the reaction of horseradish peroxidase with hydrogen peroxide was compound II (54). It was not until 4 years later, in 1941, that the observation of compound I was reported (54). In 1976, Dunford and Stillman (54) wrote, “Work on all peroxidases has been hampered by the spontaneous decay of both compound I and compound II, particularly the former, a problem which can be circumvented today only by careful work on pure enzyme samples.” Given this history and the observation of bound substrates in the crystal structures of purified P450s, it is interesting to think that the stage was set for the isolation of P450-I at least a decade ago. Hindsight is always 20/20.
This is the second article in the Thematic Minireview Series on Cytochrome P450.
- P450-I
- P450 compound I
- P450-II
- P450 compound II
- PN/LFP
- peroxynitrite/laser flash photolysis
- m-CPBA
- meta-chloroperbenzoic acid
- EP-CYP119A1
- established protocol-purified CYP119A1
- HP-CYP119A1
- highly purified CYP119A1.
REFERENCES
- 1. Rittle J., Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science 330, 933–937 [DOI] [PubMed] [Google Scholar]
- 2. Moss T. H., Ehrenberg A., Bearden A. J. (1969) Mössbauer spectroscopic evidence for electronic configuration of iron in horseradish peroxidase and its peroxide derivatives. Biochemistry 8, 4159–4162 [DOI] [PubMed] [Google Scholar]
- 3. Schonbaum G. R., Lo S. (1972) Interaction of peroxidases with aromatic peracids and alkyl peroxides. Product analysis. J. Biol. Chem. 247, 3353–3360 [PubMed] [Google Scholar]
- 4. Sheng X., Zhang H., Im S. C., Horner J. H., Waskell L., Hollenberg P. F., Newcomb M. (2009) Kinetics of oxidation of benzphetamine by compounds I of cytochrome P450 2B4 and its mutants. J. Am. Chem. Soc. 131, 2971–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jung C. (2011) The mystery of cytochrome P450 compound I: a mini-review dedicated to Klaus Ruckpaul. Biochim. Biophys. Acta 1814, 46–57 [DOI] [PubMed] [Google Scholar]
- 6. Wang Q., Sheng X., Horner J. H., Newcomb M. (2009) Quantitative production of compound I from a cytochrome P450 enzyme at low temperatures. Kinetics, activation parameters, and kinetic isotope effects for oxidation of benzyl alcohol. J. Am. Chem. Soc. 131, 10629–10636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groves J. T., McClusky G. A. (1976) Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 98, 859–861 [Google Scholar]
- 8. Groves J. T., McClusky G. A., White R. E., Coon M. J. (1978) Aliphatic hydroxylation by highly purified liver microsomal cytochrome-P-450–evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 81, 154–160 [DOI] [PubMed] [Google Scholar]
- 9. Green M. T. (2009) C–H bond activation in heme proteins: the role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol. 13, 84–88 [DOI] [PubMed] [Google Scholar]
- 10. Green M. T., Dawson J. H., Gray H. B. (2004) Oxoiron(IV) in chloroperoxidase compound II is basic: implications for P450 chemistry. Science 304, 1653–1656 [DOI] [PubMed] [Google Scholar]
- 11. Chen X., Su Z., Horner J. H., Newcomb M. (2011) Oxidation of 10-undecenoic acid by cytochrome P450BM-3 and its compound I transient. Org. Biomol. Chem. 9, 7427–7433 [DOI] [PubMed] [Google Scholar]
- 12. Newcomb M., Zhang R., Chandrasena R. E. P., Halgrimson J. A., Horner J. H., Makris T. M., Sligar S. G. (2006) Cytochrome P450 compound I. J. Am. Chem. Soc. 128, 4580–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheng X., Horner J. H., Newcomb M. (2008) Spectra and kinetic studies of the compound I derivative of cytochrome P450 119. J. Am. Chem. Soc. 130, 13310–13320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sheng X., Zhang H., Hollenberg P. F., Newcomb M. (2009) Kinetic isotope effects in hydroxylation reactions effected by cytochrome P450 compounds I implicate multiple electrophilic oxidants for P450-catalyzed oxidations. Biochemistry 48, 1620–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan X., Wang Q., Horner J. H., Sheng X., Newcomb M. (2009) Kinetics and activation parameters for oxidations of styrene by compounds I from the cytochrome P450BM-3 (CYP102A1) heme domain and from CYP119. Biochemistry 48, 9140–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denisov I. G., Makris T. M., Sligar S. G., Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 [DOI] [PubMed] [Google Scholar]
- 17. Ortiz de Montellano P. R. (ed) (2005) Cytochrome P450 Structure, Mechanism, and Biochemistry, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 18. Whitehouse C. J. C., Yang W., Yorke J. A., Tufton H. G., Ogilvie L. C. I., Bell S. G., Zhou W., Bartlam M., Rao Z., Wong L. L. (2011) Structure, electronic properties and catalytic behaviour of an activity-enhancing CYP102A1 (P450BM3) variant. Dalton Trans. 40, 10383–10396 [DOI] [PubMed] [Google Scholar]
- 19. Egawa T., Shimada H., Ishimura Y. (1994) Evidence for compound I formation in the reaction of cytochrome-P450cam with m-chloroperbenzoic acid. Biochem. Biophys. Res. Commun. 201, 1464–1469 [DOI] [PubMed] [Google Scholar]
- 20. Kellner D. G., Hung S. C., Weiss K. E., Sligar S. G. (2002) Kinetic characterization of compound I formation in the thermostable cytochrome P450 CYP119. J. Biol. Chem. 277, 9641–9644 [DOI] [PubMed] [Google Scholar]
- 21. Spolitak T., Dawson J. H., Ballou D. P. (2005) Reaction of ferric cytochrome P450cam with peracids. Kinetic characterization of intermediates on the reaction pathway. J. Biol. Chem. 280, 20300–20309 [DOI] [PubMed] [Google Scholar]
- 22. Davydov R., Hoffman B. M., Valentine A. M., Lippard S. J., Sligar S. G., Ikeda-Saito M. (1999) EPR and ENDOR studies on cryoreduced metalloproteins. J. Inorg. Biochem. 74, 110 [Google Scholar]
- 23. Davydov R., Macdonald I. D. G., Makris T. M., Sligar S. G., Hoffman B. M. (1999) EPR and ENDOR of catalytic intermediates in cryoreduced native and mutant oxy-cytochromes P450cam: mutation-induced changes in the proton delivery system. J. Am. Chem. Soc. 121, 10654–10655 [Google Scholar]
- 24. Davydov R., Perera R., Jin S., Yang T. C., Bryson T. A., Sono M., Dawson J. H., Hoffman B. M. (2005) Substrate modulation of the properties and reactivity of the oxy-ferrous and hydroperoxo-ferric intermediates of cytochrome P450cam as shown by cryoreduction-EPR/ENDOR spectroscopy. J. Am. Chem. Soc. 127, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 25. Davydov R., Razeghifard R., Im S. C., Waskell L., Hoffman B. M. (2008) Characterization of the microsomal cytochrome P4502B4 O2 activation intermediates by cryoreduction and electron paramagnetic resonance. Biochemistry 47, 9661–9666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Denisov I. G., Makris T. M., Sligar S. G. (2001) Cryotrapped reaction intermediates of cytochrome P450 studied by radiolytic reduction with phosphorus-32. J. Biol. Chem. 276, 11648–11652 [DOI] [PubMed] [Google Scholar]
- 27. Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 28. Ener M. E., Lee Y. T., Winkler J. R., Gray H. B., Cheruzel L. (2010) Photooxidation of cytochrome P450-BM3. Proc. Natl. Acad. Sci. U.S.A. 107, 18783–18786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilker J. J., Dmochowski I. J., Dawson J. H., Winkler J. R., Gray H. B. (1999) Substrates for rapid delivery of electrons and holes to buried active sites in proteins. Angew Chem. Int. Ed. 38, 89–92 [Google Scholar]
- 30. Eble K. S., Dawson J. H. (1984) Novel reactivity of cytochrome P-450-CAM. Methyl hydroxylation of 5,5-difluorocamphor. J. Biol. Chem. 259, 14389–14393 [PubMed] [Google Scholar]
- 31. Kadkhodayan S., Coulter E. D., Maryniak D. M., Bryson T. A., Dawson J. H. (1995) Uncoupling oxygen transfer and electron transfer in the oxygenation of camphor analogs by cytochrome P450-CAM. Direct observation of an intermolecular isotope effect for substrate C-H activation. J. Biol. Chem. 270, 28042–28048 [DOI] [PubMed] [Google Scholar]
- 32. Cherepanov A. V., de Vries S. (2004) Microsecond freeze-hyperquenching: development of a new ultrafast micro-mixing and sampling technology and application to enzyme catalysis. Biochim. Biophys. Acta 1656, 1–31 [DOI] [PubMed] [Google Scholar]
- 33. Rittle J., Younker J. M., Green M. T. (2010) Cytochrome P450: the active oxidant and its spectrum. Inorg. Chem. 48, 3610–3617 [DOI] [PubMed] [Google Scholar]
- 34. McLean M. A., Maves S. A., Weiss K. E., Krepich S., Sligar S. G. (1998) Characterization of a cytochrome P450 from the acidothermophilic archaea Sulfolobus solfataricus. Biochem. Biophys. Res. Commun. 252, 166–172 [DOI] [PubMed] [Google Scholar]
- 35. Rabe K. S., Kiko K., Niemeyer C. M. (2008) Characterization of the peroxidase activity of CYP119, a thermostable P450 from Sulfolobus acidocaldarius. ChemBioChem. 9, 420–425 [DOI] [PubMed] [Google Scholar]
- 36. Wright R. L., Harris K., Solow B., White R. H., Kennelly P. J. (1996) Cloning of a potential cytochrome P450 from the archaeon Sulfolobus solfataricus. FEBS Lett. 384, 235–239 [DOI] [PubMed] [Google Scholar]
- 37. Oku Y., Ohtaki A., Kamitori S., Nakamura N., Yohda M., Ohno H., Kawarabayasi Y. (2004) Structure and direct electrochemistry of cytochrome P450 from the thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. J. Inorg. Biochem. 98, 1194–1199 [DOI] [PubMed] [Google Scholar]
- 38. Lee D. S., Yamada A., Sugimoto H., Matsunaga I., Ogura H., Ichihara K., Adachi S., Park S. Y., Shiro Y. (2003) Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J. Biol. Chem. 278, 9761–9767 [DOI] [PubMed] [Google Scholar]
- 39. Fujishiro T., Shoji O., Nagano S., Sugimoto H., Shiro Y., Watanabe Y. (2011) Crystal structure of H2O2-dependent cytochrome P450SPα with its bound fatty acid substrate insight into the regioselective hydroxylation of fatty acids at the alpha position. J. Biol. Chem. 286, 29941–29950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schoch G. A., Yano J. K., Wester M. R., Griffin K. J., Stout C. D., Johnson E. F. (2004) Structure of human microsomal cytochrome P4502C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 279, 9497–9503 [DOI] [PubMed] [Google Scholar]
- 41. Mayer J. M. (1998) Hydrogen atom abstraction by metal-oxo complexes: understanding the analogy with organic radical reactions. Acc. Chem. Res. 31, 441–450 [Google Scholar]
- 42. Mayer J. M. (2004) Proton-coupled electron transfer: a reaction chemist's view. Annu. Rev. Phys. Chem. 55, 363–390 [DOI] [PubMed] [Google Scholar]
- 43. Stone K. L., Behan R. K., Green M. T. (2006) Resonance Raman spectroscopy of chloroperoxidase compound II provides direct evidence for the existence of an iron(IV)-hydroxide. Proc. Natl. Acad. Sci. U.S.A. 103, 12307–12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stone K. L., Hoffart L. M., Behan R. K., Krebs C., Green M. T. (2006) Evidence for two ferryl species in chloroperoxidase compound II. J. Am. Chem. Soc. 128, 6147–6153 [DOI] [PubMed] [Google Scholar]
- 45. Behan R. K., Hoffart L. M., Stone K. L., Krebs C., Green M. T. (2006) Evidence for basic ferryls in cytochromes P450. J. Am. Chem. Soc. 128, 11471–11474 [DOI] [PubMed] [Google Scholar]
- 46. Green M. T. (2006) Application of Badger's rule to heme and non-heme iron-oxygen bonds: an examination of ferryl protonation states. J. Am. Chem. Soc. 128, 1902–1906 [DOI] [PubMed] [Google Scholar]
- 47. Behan R. K., Green M. T. (2006) On the status of ferryl protonation. J. Inorg. Biochem. 100, 448–459 [DOI] [PubMed] [Google Scholar]
- 48. Daiber A., Herold S., Schöneich C., Namgaladze D., Peterson J. A., Ullrich V. (2000) Nitration and inactivation of cytochrome P450BM-3 by peroxynitrite–stopped-flow measurements prove ferryl intermediates. Eur. J. Biochem. 267, 6729–6739 [DOI] [PubMed] [Google Scholar]
- 49. Lambeir A. M., Dunford H. B., Pickard M. A. (1987) Kinetics of the oxidation of ascorbic-acid, ferrocyanide and para-phenolsulfonic acid by chloroperoxidase compound-I and compound-II. Eur. J. Biochem. 163, 123–127 [DOI] [PubMed] [Google Scholar]
- 50. Behan R. K., Hoffart L. M., Stone K. L., Krebs C., Green M. T. (2007) Reaction of cytochrome P450BM3 and peroxynitrite yields nitrosyl complex. J. Am. Chem. Soc. 129, 5855–5859 [DOI] [PubMed] [Google Scholar]
- 51. Ortiz de Montellano P. R. (2010) Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110, 932–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Palcic M. M., Rutter R., Araiso T., Hager L. P., Dunford H. B. (1980) Spectrum of chloroperoxidase compound-I. Biochem. Biophys. Res. Commun. 94, 1123–1127 [DOI] [PubMed] [Google Scholar]
- 53. Su Z., Horner J. H., Newcomb M. (2012) Cytochrome P450 119 compounds I formed by chemical oxidation and photooxidation are the same species. Chemistry 10.1002/chem.201202254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dunford H. B., Stillman J. S. (1976) On the function and mechanism of action of peroxidases. Coord. Chem. Rev. 19, 187–251 [Google Scholar]