

Background: BLIP-II is a potent inhibitor of class A β-lactamases.
Results: BLIP-II residues contributing to binding are near the center of interface and influence off rates for inhibition.
Conclusion: BLIP-II uses common core residues to bind to several class A β-lactamases.
Significance: BLIP-II β-lactamase interaction domains could lead to β-lactamase inhibitors.
Keywords: Antibiotic Resistance, Drug Discovery, Enzyme Inhibitors, Enzyme Kinetics, Protein-Protein Interactions, Alanine Scanning Mutagenesis, Beta-Lactam Antibiotic, Beta-Lactamase, Binding Hot Spot, Molecular Recognition
Abstract
The interactions between β-lactamase inhibitory proteins (BLIPs) and β-lactamases have been used as model systems to understand the principles of affinity and specificity in protein-protein interactions. The most extensively studied tight binding inhibitor, BLIP, has been characterized with respect to amino acid determinants of affinity and specificity for binding β-lactamases. BLIP-II, however, shares no sequence or structural homology to BLIP and is a femtomolar to picomolar potency inhibitor, and the amino acid determinants of binding affinity and specificity are unknown. In this study, alanine scanning mutagenesis was used in combination with determinations of on and off rates for each mutant to define the contribution of residues on the BLIP-II binding surface to both affinity and specificity toward four β-lactamases of diverse sequence. The residues making the largest contribution to binding energy are heavily biased toward aromatic amino acids near the center of the binding surface. In addition, substitutions that reduce binding energy do so by increasing off rates without impacting on rates. Also, residues with large contributions to binding energy generally exhibit low temperature factors in the structures of complexes. Finally, with the exception of D206A, BLIP-II alanine substitutions exhibit a similar trend of effect for all β-lactamases, i.e., a substitution that reduces affinity for one β-lactamase usually reduces affinity for all β-lactamases tested.
Introduction
Cellular processes function through specific pathways that are mediated by protein-protein interactions. Therefore, understanding of the principles of binding specificity and affinity are essential to manipulate these interfaces to create diagnostics and disease treatments (1–3). Alanine scanning mutagenesis is a common approach to uncover the “hot spots” and specificity determinants of binding interfaces (4–6). Hot spots are interface residues that, when substituted with alanine, result in at least a 2.0 kcal/mol loss in binding energy as determined from the equilibrium dissociation constant (Kd) (4, 7, 8). In addition, contact residues have also been identified as specificity determinants if the substitution of the residue changes the binding affinity for one binding partner relative to another (8, 9). The relationship between specificity and affinity is an active area of research and requires model systems, such as the human growth hormone-receptor, barnase-barstar, and BLIP-β-lactamase, to provide insights (4, 5, 10, 11).
β-Lactamases are bacterial enzymes that provide resistance to β-lactam antibiotics, i.e., penicillins and cephalosporins, and constitute a major clinical threat (12). There are four classes of β-lactamases (classes A–D) with class A β-lactamases being the most prevalent among clinically relevant bacteria (12). The representative class A β-lactamases used in this study include the TEM-1, Bla1, KPC-2, and CTX-M-14 enzymes (13, 14). These enzymes share the same overall structure but have limited amino acid sequence identity (∼30–50%) and exhibit significantly different β-lactam substrate hydrolysis profiles (Fig. 1A).
FIGURE 1.

Alignment of the four class A β-lactamases used in this study and cartoon representation of BLIP-II-β-lactamase complexes. A, a sequence alignment of the loop-helix region (residues 99–114 based on the β-lactamase ambler numbering scheme) is shown (13). The sequence alignment was performed, and the identities were determined using ClustalW (34). The amino acid identity was determined when compared with TEM-1. However, all of the β-lactamases used in this study are not more than 52% identical to one another. B, the BLIP-II-TEM-1 (red; Protein Data Bank code 1JTD) and BLIP-II-Bla1 (cyan; Protein Data Bank code 3QHY) structures are shown with the catalytic serine depicted as a yellow sphere (27, 28). The loop-helix domain of β-lactamase is shown in blue.
β-Lactamase inhibitory proteins (BLIPs),2 including BLIP and BLIP-II, are inhibitors of class A β-lactamases and are produced from the soil bacterium Streptomyces (15–18). BLIP inhibits class A β-lactamases with a wide range of affinities (subnanomolar to micromolar) and has been the focus of many structural, thermodynamic, and kinetic investigations (8, 11, 19–23). However, the interactions between BLIP-II and class A enzymes are relatively poorly understood. BLIP-II exhibits no sequence or structural homology to BLIP but rather has a seven-bladed β-propeller fold, which is a common domain that typically functions as a scaffold or protein-protein interaction adaptor (Fig. 1B) (24–26). Despite the lack of sequence and structure homology, both BLIP-II and BLIP bind to a loop-helix region common to all class A β-lactamases (Fig. 1B) (20, 27). BLIP-II utilizes the numerous β-turns of the seven-bladed β-propeller structure to bind the loop-helix region and to sterically block the β-lactamase active site (27, 28).
BLIP-II is a more potent tightly binding inhibitor of β-lactamases than BLIP, with Ki values in the femtomolar to picomolar range despite utilizing a smaller interface. In addition, BLIP-II exhibits a much narrower range of binding affinity for β-lactamases (range of 2 orders of magnitude) when compared with BLIP (range of 6 orders of magnitude) (8, 27, 29). Pre-steady state kinetic experiments demonstrated that association rate constants for BLIP-II and BLIP binding to TEM-1 β-lactamase are similar to each other and to most protein-protein interactions, but it is the exceptionally slow dissociation rate constants of BLIP-II that make it one of the tightest protein-protein interactions known (27). This slow dissociation rate is not associated with significant changes to the structure of BLIP-II as shown by crystal structures of BLIP-II monomer and in complex with β-lactamase (27). However, the binding contributions of the individual BLIP-II contact residues to affinity and specificity of binding to class A β-lactamases remain largely unknown (8, 11, 22, 30, 33).
In this study, alanine scanning mutagenesis was used to examine the functional contribution of the individual BLIP-II residues at the binding interface with class A β-lactamases. Because of the tightly binding inhibition of the BLIP-II-β-lactamase interactions, kinetic analysis was used to measure the association and dissociation rate constants for wild-type and each alanine mutant to calculate the Kd. This approach allowed for a detailed examination of the contributions to binding kinetics of each residue to a collection of four diverse β-lactamases.
EXPERIMENTAL PROCEDURES
Construction of Alanine Mutants
The BLIP-II residues chosen for alanine substitutions were based on the previously performed cluster analysis of the BLIP-II-TEM-1 and BLIP-II-Bla1 interfaces (27). The BLIP-II interface residues were mutated to alanine by site-directed mutagenesis using the QuikChange method (Stratagene). The Pfu turbo polymerase (Stratagene) was used to replicate the pET-BLIP-II plasmid to introduce the desired point mutations. The DpnI restriction enzyme was then added to the solution to remove the parental strands. The product of the QuikChange reaction was introduced into XL-1 Blue Escherichia coli cells (Stratagene) by electroporation. DNA sequencing was used to confirm the presence of the designed mutations and to ensure that no extraneous mutations were present in the BLIP-II gene (Lonestar Labs).
Protein Purification
The BLIP-II wild-type and alanine-substituted proteins were purified as previously described (27, 29). In short, the BLIP-II variants were purified using Talon metal affinity resin (Clontech) using a C-terminal His6 tag. The class A β-lactamases used in the study were purified as previously described (14, 27, 29, 35). The protein concentration of β-lactamases and BLIP-II were determined by a Bradford assay, the results of which were compared with a β-lactamase standard curve calibrated by quantitative amino acid analysis. In addition, the concentrations of BLIP-II variant proteins were also determined using 280-nm UV absorbance using an extinction coefficient of 66,920 m−1 cm−1 for the wild-type, N50A, D52A, T57A, F74A, L91A, N112A, D131A, D167A, S169A, D170A, D206A, F209A, I229A, F230A, E268A, R286A, and N304A variant proteins; an extinction coefficient of 61,420 m−1 cm−1 for the W53A, W152A, and W269A variant proteins; and an extinction coefficient of 65,430 m−1 cm−1 for the Y73A, Y113A, Y191A, Y208A, and Y248A variant proteins. Both methods (Bradford and UV absorbance) gave highly similar results (within 5%).
Enzymatic Determination of the Association Rate Constants
The association rate constants (kon) were determined using an enzymatic activity assay that has been described previously (27). All experiments were performed in 50 mm sodium phosphate, pH 7.0. The buffer was supplemented with 1 mg/ml BSA for all experiments except the KPC-2-BLIP-II association experiment, where it was added at a concentration of 0.03 mg/ml to reduce background. Aliquots (0.3 ml) were taken along the time course to measure the initial velocities of nitrocefin hydrolysis observed at optical density 482 nm. The initial velocities were used as readouts of free enzyme with the time 0 point being the maximum initial rate without BLIP-II addition. Nitrocefin was added at a concentration that yielded the maximum initial velocity of each enzyme. The nitrocefin concentrations in these experiments were 400, 200, 200, and 200 μm for TEM-1, Bla1, KPC-2, and CTX-M-14, respectively. The Km values of TEM-1, Bla1, KPC-2, and CTX-M-14 are 84, 19, 52, and 25 μm, respectively (14, 36–38). The β-lactamase concentrations were 0.5, 5, 1, and 1 nm for TEM-1, Bla1, KPC-2, and CTX-M-14, respectively. The BLIP-II concentration used was 3-fold higher than the β-lactamase concentration, allowing for the association rate constants to be determined by second order kinetics. We previously demonstrated that the association rate constants can be determined with different BLIP-II-β-lactamase ratios and different curve fittings to either pseudo-first versus second order kinetics with minimal differences (27). The difference between processing the data by pseudo-first or second order kinetics was small, but the errors for curve fitting were slightly better when the association rate constants were determined by second order kinetics. Therefore, the inhibition of β-lactamase activity over a time course was fitted to the following second order kinetic equation,
 |
where [E]t is the amount of free β-lactamase estimated by the enzymatic activity at time (t), [E]0 is the amount of free β-lactamase before the addition of BLIP-II, [B]0 is the initial BLIP-II concentration in the reaction, C is a fitting constant representing the background rate of nitrocefin hydrolysis, t is the time after mixing, and kon is the association rate constant of the interaction that is extrapolated from the data fitting. The background rate of nitrocefin hydrolysis was ∼10% of the total β-lactamase activity measured for each BLIP-II-β-lactamase pair.
For a few BLIP-II-β-lactamase variant combinations, complete inhibition was not observed because the BLIP-II substitution weakened the affinity to β-lactamase to the point that the equilibrium between the various components, BLIP-II-β-lactamase complex, free β-lactamase, and free BLIP-II, was reached. In these situations, the rate constants were determined by fitting the inhibition of β-lactamase activity over a time course to the following bimolecular binding differential equation accounting for a second order association and first order dissociation,
 |
where kon and koff are association and dissociation rate constants, [E]t, [B]t, and [E/B]t are the concentrations of free unbound β-lactamase protein, of free unbound BLIP-II protein, and of bound BLIP-II/β-lactamase complex at time (t), respectively. [E]t was experimentally determined as described above. [B]t and [E/B]t were then calculated based on known total concentrations of BLIP-II and β-lactamase proteins. The analytical solution (Equation 3) was derived using Maple 12 software (Maplesoft, Waterloo, Canada). Numerical fitting of the solution to the differential equation (Equation 3) yielded the kon and koff values.
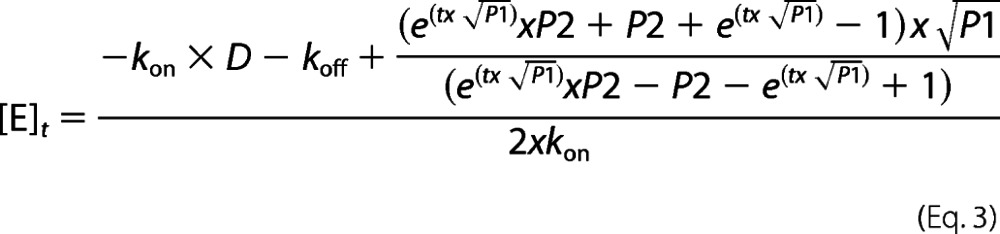 |
where D, P1, and P2 are intermediate variables defined as following:
 |
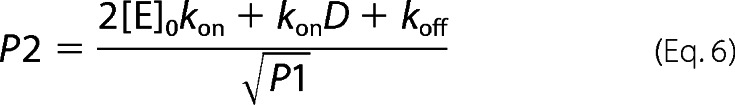 |
where [B]0 and [E]0 are total concentrations of BLIP-II and β-lactamase proteins in the experiments. kon, koff, and [E]0 are the association rate constant, dissociation rate constant, and initial concentration of active β-lactamase, respectively, which are determined by fitting Equation 3 to the data. The data processing was carried out using an Excel spreadsheet and the fitting of Equation 3 to the data utilizing the solver add-in of the Excel program. The experiments to determine the association rate constant were repeated in at least triplicate. The replicate experiments, however, did not utilize the exact same time points, and therefore all of the data sets were used to obtain the rate constant with an associated standard error, which was always less than 20%, through nonlinear regression.
Enzymatic Determination of the Dissociation Rate Constants
The dissociation rate constants were determined as previously described (27). The very slow dissociation rate constants (koff) were determined by measuring the recovery of the wild-type β-lactamase activity in the presence of an excess of an inactive TEM-1 variant (E166A) (27). This mutation prevents the deacylation step of the catalytic mechanism rendering the enzyme essentially inactive after one round of acylation. This allows for the mutant enzyme to remain in large excess and available to bind BLIP-II upon its dissociation from the wild-type β-lactamase being tested and also allows the amount of free, wild-type β-lactamase generated upon dissociation from BLIP-II to be measured by monitoring nitrocefin hydrolysis. We have shown previously that the TEM-1 Glu-166 residue is not at the binding interface with BLIP-II and that the E166A substitution does not affect binding affinity for BLIP-II as indicated by stopped flow tryptophan fluorescent spectrometric measurements (27). Therefore, the TEM-1 E166A enzyme binds tightly and sequesters BLIP-II to prevent the rebinding of BLIP-II to the wild-type β-lactamase. The experiment was initiated by incubating BLIP-II in 2-fold excess with the β-lactamase of interest for 1 h. The BLIP-II-β-lactamase complexes were then diluted into the ≥200 molar excess of TEM-1 E166A solution. The dissociation reaction was monitored by measuring initial velocities of nitrocefin hydrolysis at various time points to determine the amount of free wild-type enzyme. The concentrations of nitrocefin used were the same as those for the association rate experiments. The final enzyme concentrations were 50, 2.5, 50, and 10 nm for KPC-2, TEM-1, Bla1, and CTX-M-14, respectively. The buffer used in these experiments was 50 mm sodium phosphate, pH 7.0, supplemented with 1 mg/ml BSA.
The amount of active β-lactamase over the time course was fitted with first order kinetics to determine the kinetic parameters,
where [E]∞ is the amount of free β-lactamase when the dissociation had reached completion estimated by the enzyme activity when not inhibited by BLIP-II, [E]t is the amount of free β-lactamase estimated by enzyme activity at time (t), t is the time after mixing the BLIP-II-β-lactamase complex with the inactive TEM-1 E166A enzyme, C is the constant for curve fitting representing the background rate of nitrocefin hydrolysis (including the activity of the TEM-1 E166A enzyme), and koff is the dissociation rate constant obtained from fitting the data. Because of the long duration of the experiment, positive and negative controls of the β-lactamase being tested and the inactive TEM-1 E166A alone, respectively, were used to assess the stability of the β-lactamases during the assay. The background rate of nitrocefin hydrolysis, including the TEM-1 E166A background hydrolysis, was less than 10% of the total β-lactamase activity measured for each BLIP-II-β-lactamase pair. The experiments to determine the dissociation rate constant were repeated in at least triplicate. The replicate experiments, however, did not have the exact same time points, and therefore all of the data sets were combined into a single data set to obtain the rate constant with an associated standard error, which was always less than 20%, through nonlinear regression.
RESULTS
Association Rate Constants
To examine the contributions of BLIP-II interface residues for binding to β-lactamase, wild-type BLIP-II and 25 BLIP-II mutants were expressed and purified from E. coli. To date, crystal structures of BLIP-II bound to TEM-1 and to Bla1 β-lactamases have been determined (BLIP-II-TEM-1 and BLIP-II-Bla1) (Fig. 1) (27, 28). Previously, we used cluster analysis to computationally compare these interfaces (27, 40). The cluster analysis allowed for the identification of BLIP-II residues that formed H-bonds, electrostatic interactions, van der Waals contacts, and/or noncanonical interactions (such as cation-π) with β-lactamase. Altogether, 26 BLIP-II residues were identified to contact either TEM-1 and/or Bla1. However, the analysis of only 25 BLIP-II mutants is shown because the N51A BLIP-II mutant was unstable, and the binding data obtained was not reproducible.
Because of the tightly binding nature of BLIP-II and β-lactamase, traditional equilibrium-based determination of the dissociation constant (Kd) is not possible (27). Therefore, kinetic experiments were used to determine the association rate (kon) and dissociation rate (koff) constants to define the Kd of each BLIP-II variant and thereby assess the effect of each alanine substitution on binding toward each of the TEM-1, Bla1, KPC-2, and CTX-M-14 β-lactamases tested.
Association rate constants were determined for wild-type BLIP-II and 25 BLIP-II alanine mutants toward all four β-lactamases. This experiment was performed by following β-lactamase hydrolytic activity toward a chromogenic β-lactam substrate (nitrocefin) over a time course after the addition of 3-fold excess BLIP-II. These reactions were treated as irreversible under these conditions, and the data were fitted to the second order rate equation allowing for the determination of the association rate constant for wild-type BLIP-II and each of the BLIP-II variants toward these β-lactamases (Fig. 2). Overall, the BLIP-II alanine substitutions had a marginal effect on the association rate constants for binding to these β-lactamases. All of the alanine-substituted BLIP-II variants exhibited association rates that were within 10-fold of wild-type BLIP-II and each other for each β-lactamase. The association rates of the BLIP-II variants toward each β-lactamase ranged from 2.1 to 13.7 × 106 m−1 s−1 for TEM-1, from 0.8 to 2.4 × 106 m−1 s−1 for Bla1, from 2.5 to 21.2 × 106 m−1 s−1 for KPC-2, and from 1.7 to 6.7 × 106 m−1 s−1 for CTX-M-14 (Table 1).
FIGURE 2.

Representative time course of enzymatic activity measurements to determine the association rate constants. The plots of the Mobs (defined as [E]t/([E]t + [B]0 − [E]0)) of the experimentally measured instantaneous enzymatic activity [E]t versus time (t) after mixing the TEM-1 (0.5 nm, top left), Bla1 (5 nm, top right), KPC-2 (1 nm, bottom left), and CTXM-14 (1 nm, bottom right) with 3-fold higher concentrations of BLIP-II (wild type or the indicated mutant) as described under “Experimental Procedures.” The solid circles are the Mobs of the wild-type BLIP-II, and the solid triangles are the Mobs of the indicated mutant, whereas the solid curves are the fitting curves of the second order association kinetics according to Equation 1 (see “Experimental Procedures”) to determine the appropriate kon values (tabulated in Table 1). The experiments to determine the association rate constants were performed in at least triplicate, and all of the data sets were combined to obtain the rate constant with an associated standard error through nonlinear regression.
TABLE 1.
Dissociation rate constants (10−6 s−1)/association-rate constants (106m−1s−1) for BLIP-II alanine scanning mutants
| BLIP-II variantc |
koff/kon |
|||
|---|---|---|---|---|
| TEM-1 | Bla1 | KPC-2 | CTX-M-14 | |
| WT | 3.7/7.7 | 0.6/1.9 | 0.8/10.1b | 1.7/3.2 |
| N50A | 19/4.4 | 19.6/1.4 | 1.1/9.3 | 9.9/1.9 |
| D52A | 23.6/3.9 | 14.2/1.1 | 9.5/10.1 | 126.2/2.4 |
| W53A | 15.6/3.1 | 47.2/1.3 | 696.3/8.6 | 3961/4.1a |
| T57A | 6/7.7 | 0.7/2.2 | 1.2/15 | 5.3/4.2 |
| Y73A | 564.6/5.8 | 65.7/1.6 | 223.5/21.2 | 501.9/3.1 |
| F74A | 184.3/5.2 | 425.9/1.7 | 514.3/11.2 | 2649/3.3a |
| L91A | 8.4/5.7 | 1.9/1.6 | 6/11.1 | 62/3.4 |
| N112A | 20.1/5.4 | 5.6/1.3 | 5.8/12.3 | 31.5/2.2 |
| Y113A | 9.5/8.8 | 2.7/2.3 | 2.2/20.3 | 8.6/4 |
| D131A | 79.2/5.7 | 15.2/2.1 | 2.3/12.7 | 75.3/3.8 |
| W152A | 404.4/3.5 | 151/0.8 | 61.2/9.1 | 720.8/2.6a |
| D167A | 19.2/7.9 | 1.4/2.4 | 2/16.6 | 7.3/4.7 |
| S169A | 3.4/5.5 | 0.5/2 | 0.8/13.7 | 1.9/2.8 |
| D170A | 1.5/8 | 2.4/1.6 | 2.1/3.4 | 2.3/6.7 |
| Y191A | 533.8/4.3 | 162.2/1.7 | 48.2/11.3b | 160.3/1.7 |
| D206A | 1.1/13.7 | 215.3/1.9 | 0.5/5 | 1/5.3 |
| Y208A | 105.9/7.2 | 7.5/1.3 | 3.1/12 | 12.6/3 |
| F209A | 91/4.6 | 14/1.4 | 1.7/9.7 | 6.4/3.1 |
| I229A | 341.2/5.4 | 96.7/1.8 | 32.9/11.9 | 165.8/3.1 |
| F230A | 818/2.1 | 86.8/1.6 | 6/7.6 | 22.2/1.8 |
| Y248A | 17.9/9.2 | 1/2.3 | 9.9/14.8 | 7.8/4.7 |
| E268A | 103.9/9.5 | 105.3/2.2 | 51.3/11.2 | 108.9/5.2 |
| W269A | 38180/4.4a | 3168/1.1 | 3223/2.5a | 360.1/1.7a |
| R286A | 20/9.3 | 1.2/1.6 | 0.4/16.7 | 2.2/3.7 |
| N304A | 239.1/5.5 | 41.5/1.5 | 12.7/11 | 205/2.7 |
a Indicates that the association rate constant was determined using the bimolecular binding differential equation. The remaining constants were determined by the second order association rate equation.
b The data for BLIP-II wild-type and BLIP-II Y191A interactions with KPC-2 from Ref. 39.
c All of the determined association and dissociation rate constants have an associated standard error that is <20%.
Although the BLIP-II alanine substitutions do not greatly affect the association rate constants, these experiments did provide an early indication as to which variants greatly reduced the overall binding affinity. Some of the BLIP-II variants never achieved complete, 100% inhibition of the β-lactamase activity including W53A, F74A, W152A, and W269A toward CTX-M-14 and W269A toward TEM-1 and KPC-2. This indicates that the alanine substitution raised the overall Kd of the interaction near the protein concentrations at which the assay was performed. In these cases, as noted in Table 1, the association rate constant was determined using the bimolecular binding differential equation where the binding reaction is treated as reversible and reaches the steady state.
Impact of Alanine Substitutions on BLIP-II Dissociation Rate Constants and Kd
To examine the effects of the alanine substitutions on the dissociation rate constants and to determine the overall Kd of these BLIP-II variant interactions, an enzymatic recovery assay was used to measure BLIP-II-β-lactamase complex dissociation (27). The method involved first forming a BLIP-II-β-lactamase complex and then diluting the complex in a large excess of a deacylation deficient variant of TEM-1 (E166A) β-lactamase (27). This TEM variant exhibits very low levels of β-lactam hydrolysis but still binds to BLIP-II with similar affinity as wild-type TEM-1. These characteristics allow the dissociation reaction to be followed by determining the activity of the wild-type β-lactamase because as BLIP-II dissociates from the active β-lactamase, it binds to the large excess of inactive TEM β-lactamase and therefore does not rebind to the active enzyme. The data obtained were fit to the first order rate equation to determine the dissociation rate constant (Fig. 3).
FIGURE 3.

Enzymatic activity-based measurements of dissociation. Plots of the dissociation reactions between BLIP-II (wild type or alanine variants) and β-lactamase are shown. The fractional recovered enzymatic activities of TEM-1(2.5 nm complex, top left), Bla1 (50 nm complex, top right), KPC-2 (50 nm complex, bottom left), and CTX-M-14 (10 nm complex, bottom right) are plotted versus time, t, after mixing of the corresponding BLIP-II-β-lactamase complexes with ≥200 molar excess of inactive, deacylation deficient TEM-1 variant, E166A, competitor to absorb free BLIP-II. The solid circles are the normalized recovered activities from the corresponding wild-type complexes, and the solid triangles are from the labeled mutant complexes, whereas the solid curves are the fitting curves of the first order kinetics according Equation 7 (see “Experimental Procedures”) to determine the appropriate koff values (tabulated in Table 1). The experiments to determine the dissociation rate constants were performed in triplicate, and all of the data sets were combined and used to obtain the rate constant with an associated standard error through nonlinear regression.
Unlike the modest effects observed for the BLIP-II alanine substitutions on the association rate constants toward the four class A β-lactamases, the variants exhibited dramatically altered dissociation rate constants. Wild-type BLIP-II exhibited similar (within a 10-fold range) dissociation rate constants for the four β-lactamases tested with values ranging from 0.6 × 10−6 to 3.7 × 10−6 s−1. However, when the alanine substitutions were examined, the off rates changed by orders of magnitude depending on the residue position substituted (Table 1). Therefore, it is the dissociation rate constants that were more drastically affected by the alanine substitutions than the association rate constants. These dissociation rate constants (koff), together with the association rate constants (kon), were used to calculate the equilibrium dissociation constant (Kd) for each BLIP-II alanine mutant and β-lactamase combination. Taken together, these values allowed for the determination of the functional epitopes of BLIP-II binding to class A β-lactamases.
A residue is considered a part of the functional epitope of BLIP-II binding to β-lactamase when the alanine substitution results in a >100-fold decrease in binding affinity as defined by the calculated Kd values. By this definition, the functional epitope for BLIP-II binding to TEM-1 consists of seven residues: Tyr-73, Trp-152, Tyr-191, Ile-229, Phe-230, Trp-269, and Asn-304 (Fig. 4). Seven additional residues result in a 10–100-fold decrease in binding affinity upon mutation (Table 2). Eleven residues make up the functional epitope for BLIP-II binding to Bla1 by resulting in a >100-fold decrease in binding affinity upon mutation. These residues include Trp-53, Tyr-73, Phe-74, Trp-152, Tyr-191, Asp-206, Ile-229, Phe-230, Glu-268, Trp-269, and Asn-304 (Fig. 4). Six additional residues result in a 10–100-fold decrease in binding affinity for Bla1 upon mutation (Table 2). The functional epitope for BLIP-II binding to CTX-M-14 consists of nine residues: Asp-52, Trp-53, Tyr-73, Phe-74, Trp-152, Tyr-191, Ile-229, Trp-269, and Asn-304 (Fig. 4). Five additional residues result in a 10–100-fold decrease in binding affinity for CTX-M-14 upon substitution (Table 2). In contrast to the other β-lactamases, the functional epitope for BLIP-II binding to KPC-2 consists of only four residues: Trp-53, Tyr-73, Phe-74, and Trp-269 (Fig. 4). Six additional residues result in a 10–100-fold decrease in binding affinity for KPC-2 upon substitution (Table 2).
FIGURE 4.

Functional epitopes and specificity determinants of BLIP-II for binding class A β-lactamase. Representations of BLIP-II are shown (gray; Protein Data Bank code 3QI0). Residues that were mutated in this study are shown as spheres and are colored as a gradient: red (>1000- to 100-fold increase in Kd), orange (100- to 10-fold increase in Kd), yellow (10- to 5-fold increase in Kd), or green (<5-fold increase to a decrease in Kd).
TABLE 2.
Dissociation constants (pm) for BLIP-II alanine scanning mutants
| BLIP-II variant |
Kd |
|||
|---|---|---|---|---|
| TEM-1 | Bla1 | KPC-2 | CTX-M-14 | |
| WT | 0.48 | 0.30 | 0.08a | 0.54 |
| N50A | 4.32 | 13.53 | 0.12 | 5.20 |
| D52A | 6.12 | 13.52 | 0.95 | 53.70 |
| W53A | 5.04 | 37.73 | 80.59 | 972.92 |
| T57A | 0.79 | 0.33 | 0.08 | 1.26 |
| Y73A | 98.11 | 40.33 | 10.54 | 161.20 |
| F74A | 35.63 | 249.94 | 46.08 | 810.87 |
| L91A | 1.48 | 1.19 | 0.53 | 18.01 |
| N112A | 3.70 | 4.51 | 0.47 | 14.61 |
| Y113A | 1.08 | 1.16 | 0.11 | 2.15 |
| D131A | 13.79 | 7.28 | 0.18 | 19.57 |
| W152A | 116.84 | 179.98 | 6.75 | 278.19 |
| D167A | 2.43 | 0.57 | 0.12 | 1.57 |
| S169A | 0.62 | 0.26 | 0.06 | 0.68 |
| D170A | 0.19 | 1.52 | 0.62 | 0.35 |
| Y191A | 125.54 | 97.01 | 4.26a | 94.43 |
| D206A | 0.08 | 110.47 | 0.10 | 0.19 |
| Y208A | 14.71 | 5.69 | 0.26 | 4.22 |
| F209A | 19.71 | 9.75 | 0.17 | 2.05 |
| I229A | 63.08 | 53.18 | 2.76 | 53.15 |
| F230A | 384.94 | 53.85 | 0.79 | 12.61 |
| Y248A | 1.93 | 0.45 | 0.67 | 1.67 |
| E268A | 10.88 | 48.53 | 4.58 | 21.10 |
| W269A | 8738.51 | 2877.38 | 1296.95 | 218.25 |
| R286A | 2.15 | 0.74 | 0.03 | 0.61 |
| N304A | 43.54 | 28.37 | 1.15 | 76.49 |
a Data for BLIP-II interaction with KPC-2 from Ref. 39.
The results of the binding experiments identified several residues that decrease the BLIP-II-β-lactamase affinity by >100-fold depending on which β-lactamase was tested. The Y73A-, F74A-, and W269A-substituted inhibitors, however, exhibited a >100-fold decrease in affinity for all four β-lactamases tested, and the W53A inhibitor displayed >100-fold decreased affinity for three of the four β-lactamases tested. These BLIP-II residues are located near the center of the BLIP-II binding surface, suggesting that this region provides the largest contribution to binding energy (Fig. 4). This observation becomes more striking when the impact of the alanine substitutions on binding energy is mapped onto the BLIP-II surface (Fig. 4).
Amino Acid Residue Determinants of Binding Specificity
A specificity determinant is defined here as a residue that exhibits significantly different effects on binding affinity for different β-lactamases upon mutation to alanine. A striking feature of the comparison of binding affinities of BLIP-II alanine mutants is the relative lack of large effects on binding specificity. Most BLIP-II alanine substitutions exhibit a similar trend of effect for all β-lactamases, i.e., a substitution that reduces affinity for one β-lactamase usually reduces affinity for all β-lactamases tested (Fig. 5A). However, there are differences in the extent of reductions in affinity. For example, BLIP-II W53A exhibits a much larger reduction in binding affinity relative to wild type for binding to Bla1, KPC-2, and CTX-M-14 β-lactamases than it does for TEM-1 β-lactamase (Fig. 5A). Similarly, the BLIP-II N50A and D131A substitutions result in significant losses in binding affinity for the TEM-1, Bla1, and CTX-M-14 enzymes but have only a minimal effect on affinity for KPC-2 β-lactamase (Fig. 5A).
FIGURE 5.

Change in the energetic contributions of the BLIP-II interface residues upon alanine substitution. A, comparison of the ΔΔG values of the BLIP-II alanine variants for binding β-lactamases: TEM-1 (black), Bla1 (green), KPC-2 (red), and CTX-M-14 (purple). Relative binding free energy is defined as the difference in binding energy between wild-type BLIP-II and the alanine mutant (ΔΔG = −RT ln(Kd,wt/Kd,mut)). The upper significance control line is set at 2.7 kcal/mol, which indicates a 100-fold increase in Kd. The lower significance control line is set at 1.35 kcal/mol, which indicates a 10-fold decrease in Kd. B, plot of the temperature factor of BLIP-II interface residues versus the change in binding energy (ΔΔG (kcal/mol)) for the alanine substitution of the positions. The temperature factor for each residue was calculating by averaging the temperature factors for all of the atoms that make up the residue. The ΔΔG values are calculated from the Kd values in Table 2. B, the x axis indicates the temperature factor based on the BLIP-II-TEM-1 β-lactamase x-ray structure (Protein Data Bank 1JTD), and the y axis indicates the change in energy for binding for each mutant to TEM-1 β-lactamase. C, the x axis indicates the temperature factor based on the BLIP-II-Bla1 β-lactamase x-ray structure (Protein Data Bank 3QHY), and the y axis indicates the change in energy for binding for each mutant to Bla1 β-lactamase (27).
Two BLIP-II residue positions where alanine substitutions did result in a switch in direction of affinity (i.e., the substitution increased affinity for one enzyme while decreasing affinity for another) are Asp-170 and Asp-206. The BLIP-II D206A substitution had the most dramatic effect because it increased binding affinity for TEM-1 by ∼6-fold, had no significant effect on binding affinity toward KPC-2 or CTX-M-14, but resulted in a ∼370-fold decrease in binding affinity for Bla1 (Table 2 and Fig. 5A). The BLIP-II D170A substitution exhibited more modest effects on specificity than Asp-206 with essentially unchanged affinity for TEM-1 and CTX-M-14 but reduced affinity for the Bla1 and KPC-2 β-lactamases.
DISCUSSION
Although protein-protein interactions govern the majority of the processes of the cell, the ability to manipulate these large surfaces for disease treatment remains elusive (1, 41, 42). However, the concept that hot spots provide the majority of the driving force for an interaction presents a more tractable problem (4, 7, 43). The alanine scanning approach taken here identified the hot spot residues for the interaction of BLIP-II with four diverse class A β-lactamases where the association rate constants (kon) were only modestly affected in comparison with the dissociation rate constants (koff).
The protein-protein association rate consists of a diffusion step for the partners to form a transient complex followed by rearrangement of the complex to the final state (44, 45). Protein association reactions that occur extremely rapidly (108–109 m−1 s−1), such as barnase-barstar, utilize long range complementary electrostatic interactions to orient the protein surfaces relative to one another to speed up formation of the transient complex (46). Most protein interactions have been estimated from theoretical studies to have a “basal” or diffusion-controlled association rate of 105–106 m−1 s−1 (47, 48). The association rate constants for the BLIP-II-β-lactamase interactions described in Table 1 are at the upper end of this range with values ranging from 1.9 to 10.1 × 106 m−1 s−1. The observation that alanine substitutions of BLIP-II residues do not greatly alter the association rate is consistent with the idea that the BLIP-II-β-lactamase interactions reflect basal or diffusion limit range of association and do not make extensive use of long range electrostatic steering interactions to orient the encounter complex. This suggests that residue substitutions utilizing long range electrostatic attraction could further improve the BLIP-II affinity for β-lactamases and overcome the diffusion-limited process.
Hot spot residues are commonly defined by a >2.0 kcal/mol loss in binding energy caused by the alanine substitution (7, 49). The composition of hot spots is biased toward aromatic residues and, in particular, tryptophan and tyrosine residues (7). In addition, hot spot residues are most often located at the center of the protein-protein interaction interface, and the residues surrounding them have been proposed to form an “O-ring” (4, 7). The O-ring hypothesis is based on observations from alanine mutagenesis that hot spots are often surrounded by hydrophilic residues that do not significantly impact binding when mutated and are suggested to enhance the strength of interactions by excluding bulk water from the interface (4, 7).
The structure of the seven-bladed β-propeller fold of BLIP-II consists of concentric rings of amino acid residues consistent with the O-ring model of a binding surface (Fig. 4). In addition, the BLIP-II residues that constitute the hot spots share several features with previously characterized hot spots. First, the BLIP-II residues that comprise the hot spots are biased toward aromatic residues, in particular, tryptophan and tyrosine, with Trp-53, Tyr-73, Phe-74, Trp-152, Tyr-191, and Tyr-191 making large contributions to binding affinity for all of the β-lactamases tested (Table 2). Upon examination of the BLIP-II-β-lactamase structures, it is apparent that BLIP-II hot spot residues Tyr-73 and Phe-74 could contribute significantly to the stability of the complex because they bind with numerous residues on the β-lactamase (Fig. 6A). The BLIP-II W269A substitution resulted in dramatic reductions in binding affinity for all of the β-lactamases tested. This interaction is at the center of the interface, and the alanine substitution would result in an unfavorable cavity in the interface (Fig. 6B). Trp-53 is located on a BLIP-II loop that sterically blocks the β-lactamase active site with the help of BLIP-II residues Asn-50 and Asp-52. Trp-53, however, makes only limited side chain contacts with one or two residues of β-lactamase, and yet the BLIP-II D53A substitution exhibits greatly decreased binding affinity for β-lactamases (Fig. 6C). Second, as seen in Fig. 4, the BLIP-II residues making the largest contributions to binding energy for all four β-lactamases are largely clustered near the center of the binding interface with less important residues found in the outer ring of the BLIP-II binding surface, which is consistent with the O-ring hypothesis for organization of the interface (7). In fact, the seven-bladed β-propeller fold, which consists of an inner ring and an outer ring, provides an ideal structure for the O-ring model in that the outer rim is indeed an O-shaped ring of largely hydrophilic residues. Thus, the structure of BLIP-II and related proteins seems an ideal scaffold for mediating protein-protein interactions. This organization of the BLIP-II scaffold may explain why BLIP-II binds class A β-lactamases 100–1000-fold tighter than BLIP, despite the fact that BLIP-II has a smaller interface size (2187 Å2 for TEM-1) than BLIP (2636 Å2 for TEM-1).
FIGURE 6.

Structural representations of BLIP-II-TEM-1 (red; Protein Data Bank 1JTD) and BLIP-II-Bla1 (cyan; Protein Data Bank 3QHY) interactions (27, 28). The selected interactions of the BLIP-II hot spots and specificity determinants with TEM-1 and Bla1 β-lactamase are shown including Phe-73 and Tyr-74 (A), Trp-269 (B), Trp-53 (C), and Asp-206 (D).
The results of this study also indicated that residues near the center of the interface that contribute strongly to binding generally possess lower temperature factors in the BLIP-II-TEM-1 and BLIP-II-Bla1 β-lactamase x-ray structures than residues at the periphery of the binding interface (Fig. 5, B and C). The relative lack of motion of residues at the center of interface may be indicative of a favorable entropic contribution of these residues to binding, which could also partially explain why these positions form the hot spot. A plot of the temperature factors for the alanine-substituted residues versus the change in binding energy for the mutants reveals a correlation (Fig. 5, B and C). The correlation is weakened, however, by a few outlier points. For example, the W269A substitution is well off of the linear regression fit line for both BLIP-TEM-1 and BLIP-II-Bla1 because of the very large drop in binding energy for the W269A mutant. In addition, the D206A substitution falls well below the regression line for BLIP-II-TEM-1 but well above the line for BLIP-II Bla1 (Fig. 5, B and C). This is interesting in that the D206A substitution exhibits the largest change in β-lactamase binding specificity among all of the mutants studied. It is possible that differences in binding specificity for BLIP-II D206A could be due to differences in entropic contributions to binding TEM-1 versus Bla1 β-lactamases.
As described above, the BLIP-II hot spot residues for binding β-lactamases are dominated by aromatic amino acids. In contrast, the residues that alter binding specificity, Asp-170 and Asp-206, are charged. This finding is similar to observations made with other protein-protein interactions (23, 31, 50, 51). In the BLIP-β-lactamase system, the BLIP Glu-73–Lys-74 motif has been shown to be a specificity determinant for interactions with several different β-lactamases (8, 32, 35). Another example is the cross-reactivity between bovine and human growth hormone and the human growth hormone receptor that occurs when the human growth hormone Arg-43 and receptor Asp-171 salt bridge is eliminated (50). These examples support the notion of aromatic and charged residues in providing overall binding energy and specificity to protein-protein interactions, respectively.
Because alanine scanning mutagenesis has now been performed on both tightly binding inhibitors, BLIP and BLIP-II, it is of interest to compare their interactions with β-lactamase. BLIP-II is a femtomolar to picomolar affinity binder to all class A β-lactamases tested. The broad specificity of BLIP-II is also reflected in the finding that alanine substitutions, with the exception of BLIP-II D206A, do not greatly alter binding specificity. In contrast, BLIP contains three specificity-determining regions (8). The fact that BLIP displays a wide range of binding affinities for class A β-lactamases and that its specificity can be altered readily suggests the BLIP scaffold is highly malleable for altering binding specificity for class A β-lactamases, whereas the BLIP-II scaffold is well suited for binding the entire class A β-lactamase family.
Detection and inhibition of β-lactamases are of great clinical interest. It seems unlikely BLIP-II could be used as a therapeutic agent because it would likely elicit an immune response. A BLIP-II-based peptide is possible, but it would be significantly less potent than wild-type BLIP-II. Nevertheless, such a peptide could serve as a starting point for further development. BLIP and BLIP-II, however, could be used as diagnostic reagents to detect β-lactamases in bacterial strains. For example, the tightly binding characteristics of BLIP-II could be used to determine whether a class A β-lactamase is present, and engineered versions of BLIP with narrow binding specificity could be used to distinguish among class A enzymes. BLIP and BLIP-II are potent binders of β-lactamases and could detect the low concentrations of these enzymes in clinical samples.
This work was supported, in whole or in part, by National Institutes of Health Grant AI32956 (to T. P.) and a training fellowship from the Biomedical Discovery Training Program of the Gulf Coast Consortia supported by National Institutes of Health Grant 1 T90 DA022885-04.
- BLIP
- β-lactamase inhibitory protein.
REFERENCES
- 1. Jubb H., Higueruelo A. P., Winter A., Blundell T. L. (2012) Structural biology and drug discovery for protein-protein interactions. Trends Pharm. Sci. 33, 241–248 [DOI] [PubMed] [Google Scholar]
- 2. Kuzu G., Keskin O., Gursoy A., Nussinov R. (2012) Constructing structural networks of signaling pathways on the proteome scale. Curr. Opin. Struct. Biol. 22, 367–377 [DOI] [PubMed] [Google Scholar]
- 3. Schreiber G., Keating A. E. (2011) Protein binding specificity versus promiscuity. Curr. Opin. Struct. Biol. 21, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clackson T., Wells J. A. (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267, 383–386 [DOI] [PubMed] [Google Scholar]
- 5. Cunningham B. C., Wells J. A. (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244, 1081–1085 [DOI] [PubMed] [Google Scholar]
- 6. DeLano W. L. (2002) Unraveling hot spots in binding interfaces. Progress and challenges. Curr. Opin. Struct. Biol. 12, 14–20 [DOI] [PubMed] [Google Scholar]
- 7. Bogan A. A., Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z., Palzkill T. (2004) Dissecting the protein-protein interface between β-lactamase inhibitory protein and class A β-lactamases. J. Biol. Chem. 279, 42860–42866 [DOI] [PubMed] [Google Scholar]
- 9. Dutta S., Gullá S., Chen T. S., Fire E., Grant R. A., Keating A. E. (2010) Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 398, 747–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petrosino J., Rudgers G., Gilbert H., Palzkill T. (1999) Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J. Biol. Chem. 274, 2394–2400 [DOI] [PubMed] [Google Scholar]
- 11. Reichmann D., Cohen M., Abramovich R., Dym O., Lim D., Strynadka N. C., Schreiber G. (2007) Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J. Mol. Biol. 365, 663–679 [DOI] [PubMed] [Google Scholar]
- 12. Fisher J. F., Meroueh S. O., Mobashery S. (2005) Bacterial resistance to β-lactam antibiotics. Compelling opportunism, compelling opportunity. Chem. Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 13. Ambler R. P., Coulson A. F., Frère J.-M., Ghuysen J.-M., Joris B., Forsman M., Levesque R. C., Tiraby G., Waley S. G. (1991) A standard numbering scheme for the class A β-lactamases. Biochem. J. 276, 269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishii Y., Galleni M., Ma L., Frère J.-M., Yamaguchi K. (2007) Biochemical characterisation of the CTX-M-14 β-lactamase. Int. J. Antimicrob. Agents 29, 159–164 [DOI] [PubMed] [Google Scholar]
- 15. Doran J. L., Leskiw B. K., Aippersbach S., Jensen S. E. (1990) Isolation and characterization of a β-lactamase-inhibitory protein from Streptomyces clavuligerus and cloning and analysis of the corresponding gene. J. Bacteriol. 172, 4909–4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gretes M., Lim D. C., de Castro L., Jensen S. E., Kang S. G., Lee K. J., Strynadka N. C. (2009) Insights into positive and negative requirements for protein-protein interactions by crystallographic analysis of the β-lactamase inhibitory proteins BLIP, BLIP-I, and BLP. J. Mol. Biol. 389, 289–305 [DOI] [PubMed] [Google Scholar]
- 17. Kang S. G., Park H. U., Lee H. S., Kim H. T., Lee K. J. (2000) New β-lactamase inhibitory protein (BLIP-I) from Streptomyces exfoliatus SMF19 and its roles on the morphological differentiation. J. Biol. Chem. 275, 16851–16856 [DOI] [PubMed] [Google Scholar]
- 18. Park H. U., Lee K. J. (1998) Cloning and heterologous expression of the gene for BLIP-II, a β-lactamase-inhibitory protein from Streptomyces exfoliatus SMF19. Microbiology 144, 2161–2167 [DOI] [PubMed] [Google Scholar]
- 19. Reichmann D., Rahat O., Albeck S., Meged R., Dym O., Schreiber G. (2005) The modular architecture of protein-protein binding interfaces. Proc. Natl. Acad. Sci. U.S.A. 102, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strynadka N. C., Jensen S. E., Alzari P. M., James M. N. (1996) A potent new mode of β-lactamase inhibition revealed by the 1.7Å X-ray crystallographic structure of the TEM-1-BLIP complex. Nat. Struct. Biol. 3, 290–297 [DOI] [PubMed] [Google Scholar]
- 21. Strynadka N. C., Jensen S. E., Johns K., Blanchard H., Page M., Matagne A., Frère J.-M., James M. N. (1994) Structural and kinetic characterization of a β-lactamase-inhibitor protein. Nature 368, 657–660 [DOI] [PubMed] [Google Scholar]
- 22. Wang J., Zhang Z., Palzkill T., Chow D.-C. (2007) Thermodynamic investigation of the role of contact residues of β-lactamase-inhibitory protein for binding to TEM-1 β-lactamase. J. Biol. Chem. 282, 17676–17684 [DOI] [PubMed] [Google Scholar]
- 23. Yuan J., Huang W., Chow D. C., Palzkill T. (2009) Fine mapping of the sequence requirements for binding of β-lactamase inhibitory protein (BLIP) to TEM-1 β-lactamase using a genetic screen for BLIP function. J. Mol. Biol. 389, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fülöp V., Jones D. T. (1999) β propellers. Structural rigidity and functional diversity. Curr. Opin. Struct. Biol. 9, 715–721 [DOI] [PubMed] [Google Scholar]
- 25. Guruprasad K., Dhamayanthi P. (2004) Structural plasticity associated with the β-propeller architecture. Int. J. Biol. Macromol. 34, 55–61 [DOI] [PubMed] [Google Scholar]
- 26. Hadjebi O., Casas-Terradellas E., Garcia-Gonzalo F. R., Rosa J. L. (2008) The RCC1 superfamily. From genes, to function, to disease. Biochim. Biophys. Acta 1783, 1467–1479 [DOI] [PubMed] [Google Scholar]
- 27. Brown N. G., Chow D.-C., Sankaran B., Zwart P., Prasad B. V., Palzkill T. (2011) Analysis of the binding forces driving the tight binding between β-lactamase inhibitory protein II (BLIP-II) and class A β-lactamases. J. Biol. Chem. 286, 32723–32735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lim D., Park H. U., De Castro L., Kang S. G., Lee H. S., Jensen S., Lee K. J., Strynadka N. C. (2001) Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat. Struct. Biol. 8, 848–852 [DOI] [PubMed] [Google Scholar]
- 29. Brown N. G., Palzkill T. (2010) Identification and characterization of β-lactamase inhibitor protein-II (BLIP-II) interactions with β-lactamases using phage display. Protein Eng. Des. Sel. 23, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanes M. S., Reynolds K. A., McNamara C., Ghosh P., Bonomo R. A., Kirsch J. F., Handel T. M. (2011) Specificity and cooperativity at β-lactamase position 104 in TEM-1/BLIP and SHV-1/BLIP interactions. Proteins 79, 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kühlmann U. C., Pommer A. J., Moore G. R., James R., Kleanthous C. (2000) Specificity in protein-protein interactions. The structural basis for dual recognition in endonuclease colicin-immunity protein complexes. J. Mol. Biol. 301, 1163–1178 [DOI] [PubMed] [Google Scholar]
- 32. Yuan J., Chow D. C., Huang W., Palzkill T. (2011) Identification of a β-lactamase inhibitory protein variant that is a potent inhibitor of staphylococcus PC1 β-lactamase. J. Mol. Biol. 406, 730–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Palzkill T. (2003) Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J. Biol. Chem. 278, 45706–45712 [DOI] [PubMed] [Google Scholar]
- 34. Thompson J. D., Gibson T. J., Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX, in Current Protocols in Bioinformatics [DOI] [PubMed] [Google Scholar]
- 35. Hanes M. S., Jude K. M., Berger J. M., Bonomo R. A., Handel T. M. (2009) Structural and biochemical characterization of the interaction between KPC-2 β-lactamase and β-lactamase inhibitor protein. Biochemistry 48, 9185–9193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown N. G., Pennington J. M., Huang W., Ayvaz T., Palzkill T. (2010) Multiple global suppressors of protein stability defects facilitate the evolution of extended-spectrum TEM β-lactamases. J. Mol. Biol. 404, 832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Materon I. C., Queenan A. M., Koehler T. M., Bush K., Palzkill T. (2003) Biochemical characterization of β-lactamases Bla1 and Bla2 from Bacillus anthracis. Antimicrob. Agents Chemother. 47, 2040–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stachyra T., Levasseur P., Péchereau M. C., Girard A. M., Claudon M., Miossec C., Black M. T. (2009) In vitro activity of the β-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J. Antimicrob. Chemother. 64, 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown N. G., Chow D.-C., Palzkill T. (2013) BLIP-II is a highly potent inhibitor of the Klebsiella pneumoniae carbapenemase (KPC-2). Antimicrob. Agents Chemother., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Potapov V., Reichmann D., Abramovich R., Filchtinski D., Zohar N., Ben Halevy D., Edelman M., Sobolev V., Schreiber G. (2008) Computational redesign of a protein-protein interface for high affinity and binding sepcificity using modular architecture and naturally occurring template fragments. J. Mol. Biol. 384, 109–119 [DOI] [PubMed] [Google Scholar]
- 41. Thiel P., Kaiser M., Ottmann C. (2012) Small-molecule stabilization of protein-protein interactions. An underestimated concept in drug discovery? Angew. Chem. Int. Ed. Engl. 51, 2012–2018 [DOI] [PubMed] [Google Scholar]
- 42. Surade S., Blundell T. L. (2012) Structural biology and drug discovery of difficult targets. The limits of ligandability. Chem. Biol. 19, 42–50 [DOI] [PubMed] [Google Scholar]
- 43. Scott D. E., Ehebauer M. T., Pukala T., Marsh M., Blundell T. L., Venkitaraman A. R., Abell C., Hyvönen M. (2013) Using a fragment-based approach to target protein-protein interactions. Chembiochem. 14, 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schreiber G., Haran G., Zhou H. X. (2009) Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alsallaq R., Zhou H.-X. (2008) Electrostatic rate enhancement and transient complex of protein-protein association. Proteins 71, 320–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Janin J. (1997) The kinetics of protein-protein recognition. Proteins 28, 153–161 [DOI] [PubMed] [Google Scholar]
- 47. Schlosshauer M., Baker D. (2004) Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci. 13, 1660–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou H.-X. (1997) Enhancement of protein-protein association rate by interaction potential. Accuracy of prediction based on local Boltzmann factor. Biophys. J. 73, 2441–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moreira I. S., Fernandes P. A., Ramos M. J. (2007) Hot spots-A review of the protein-protein interface determinant amino acid residues. Proteins 68, 803–812 [DOI] [PubMed] [Google Scholar]
- 50. Clackson T., Ultsch M. H., Wells J. A., de Vos A. M. (1998) Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol. 277, 1111–1128 [DOI] [PubMed] [Google Scholar]
- 51. Curtis M. D., James R. (1991) Investigation of the specificity of the interaction between colicin E9 and its immunity protein by site-directed mutagenesis. Mol. Microbiol. 5, 2727–2733 [DOI] [PubMed] [Google Scholar]