

Background: APOBEC3A is a potentially genotoxic DNA cytosine deaminase expressed in myeloid lineage cells.
Results: Exogenous APOBEC3A genotoxicity correlates with expression level and nuclear localization. Endogenous APOBEC3A is nontoxic and cytoplasmic, despite its capacity to be highly induced by interferon.
Conclusion: Cytoplasmic localization prevents endogenous APOBEC3A from accessing nuclear DNA.
Significance: Endogenous APOBEC3A is not genotoxic.
Keywords: DNA Damage, HIV, Innate Immunity, Interferon, Monocytes, APOBEC3A, DNA Cytosine Deamination, Cytoplasmic Localization, Gamma-H2AX, Genotoxicity
Abstract
APOBEC3A (A3A) is a myeloid lineage-specific DNA cytosine deaminase with a role in innate immunity to foreign DNA. Previous studies have shown that heterologously expressed A3A is genotoxic, suggesting that monocytes may have a mechanism to regulate this enzyme. Indeed, we observed no significant cytotoxicity when interferon was used to induce the expression of endogenous A3A in CD14+-enriched primary cells or the monocytic cell line THP-1. In contrast, doxycycline-induced A3A in HEK293 cells caused major cytotoxicity at protein levels lower than those observed when CD14+ cells were stimulated with interferon. Immunofluorescent microscopy of interferon-stimulated CD14+ and THP-1 cells revealed that endogenous A3A is cytoplasmic, in stark contrast to stably or transiently transfected A3A, which has a cell-wide localization. A3A constructs engineered to be cytoplasmic are also nontoxic in HEK293 cells. These data combine to suggest that monocytic cells use a cytoplasmic retention mechanism to control A3A and avert genotoxicity during innate immune responses.
Introduction
Myeloid lineage cells have critical roles in both innate and adaptive immune responses, including phagocytosis, antigen presentation, and cytokine production (reviewed in Ref. 1). Monocytes are produced by hematopoietic stem cells in the bone marrow and released into circulation. The monocytes migrate through the endothelium toward the tissues, where they differentiate into macrophages or dendritic cells. These professional phagocytic cells are stationed throughout the body to perform immune surveillance and thus come into contact with a variety of foreign molecules, including DNA.
APOBEC3A (A3A)5 is part of the APOBEC3 family of DNA cytosine deaminases, which in humans is composed of seven members: A, B, C, D, F, G, and H. A3A is the most active of these deaminases (2), but its expression is limited to myeloid lineage cells and therefore likely has a specialized function in these cells (3–6). A3A is strongly induced by type I interferon (10–1000-fold) (4, 6–8) and is also induced by other immunostimulatory molecules such as cytosolic DNA (6). This expression profile is consistent with A3A functioning in myeloid lineage cells as part of a foreign DNA clearance mechanism (6). By deaminating DNA cytosines to uracils, A3A can trigger further DNA damage and subsequent clearance. A3A has also been observed to deaminate 5-methyl cytosines in single-stranded DNA (2, 9, 10). The physiological relevance of this additional biochemical activity is not known, but it may benefit the cell by allowing A3A to recognize a broader range of foreign DNA substrates.
A3A may also have a role in restricting the replication of several transposons and viruses. For instance, A3A has been shown to limit the retrotransposition of L1 and Alu elements (3, 11–15). A3A has also been implicated in inhibiting the replication of certain DNA viruses, including parvovirus (16), hepatitis B virus (17), and papillomavirus (18), as well as the retroviruses human T-lymphotropic virus type-I (19) and HIV (4, 7, 20, 21).
Beyond these restriction activities, A3A has been suggested to have a role in cancer, due to its genotoxic capacity (22, 23). Overexpressed A3A has been shown to cause chromosomal DNA damage and mutation (22–26) as well as disruption of the cell cycle (22, 26). Expression of A3A in a cell threatens genomic integrity, suggesting that uncontrolled activation of this immune defense molecule would have undesirable consequences.
Alternatively, we hypothesize that myeloid lineage cells, which inducibly express A3A following interferon stimulation, have a mechanism to control A3A activity. We describe the induction of A3A expression with interferon in primary CD14+ monocytic cells and the monocytic cell line THP-1, with no evidence of decreased viability or DNA damage. Expression of A3A in HEK293 cells to lower levels than those observed in the CD14+ cells, however, caused marked genotoxicity. We show that endogenous A3A in CD14+ and THP-1 cells localized to the cytoplasm, in contrast to the cell-wide localization observed for exogenous A3A in a variety of heterologous cell types (3, 13, 14, 16, 21, 27–34). We further reveal that nuclear localization is necessary for A3A-mediated genotoxicity. We conclude that endogenous A3A is nongenotoxic, due to its retention in the cytoplasm. We suggest that endogenous A3A is unlikely to have a role in editing chromosomal DNA as its cytoplasmic localization would prohibit access to the nuclear DNA. This study supports a role for A3A in innate immunity by demonstrating that monocytic cells can safely express high levels of this potent DNA mutator.
EXPERIMENTAL PROCEDURES
Constructs
pcDNA5TO-A3A-eGFP encodes A3A-GFP under the control of a CMV promoter that contains two tetracycline operator sites and has previously been reported (26). Standard PCR cloning and site-directed mutagenesis techniques were used to construct derivatives. pCI-neo-3×FLAG-TRB3 has been described (24).
Cells
T-REx 293 cells (Invitrogen), which stably express the tetracycline repressor, were transfected with the pcDNA5TO-A3A-eGFP construct and derivatives using TransIT-LT1 (Mirus). Stable clones were selected with hygromycin and blasticidin. Basal repression and doxycycline-induced expression of A3A were confirmed by flow cytometry for GFP fluorescence and by immunoblotting. T-REx 293 A3A and T-REx A3A-E72A stable clones were further engineered to stably express 3×FLAG-TRB3 or empty vector by transfection with pCI-neo-3×FLAG-TRB3 or pCI-neo-3×FLAG and selection with G418.
Primary human CD14+ monocytes were purified by negative selection with Rosette Sep human monocyte enrichment mixture (Stemcell Technologies) from fresh whole blood obtained the from Memorial Blood Center (St. Paul, MN). Purity (>90%) was confirmed by flow cytometry for CD14+ cells with CD14-FITC (Miltenyi Biotec).
THP-1 cells (35) were obtained from Dr. Andrea Cimarelli (Ecole Normale Supérieure de Lyon). A3A knockdown clones were obtained by transduction with pLKO-based lentiviral constructs (Open Biosystems) followed by puromycin resistance selection. Specific A3A knockdown was verified by immunoblotting and quantitative PCR.
Endogenous A3A was up-regulated by treating CD14+ or THP-1 cells with IFNα (300 units/ml Universal Type I IFNα; R&D Systems). RNA was isolated 6 h after induction; whole cell lysates for immunoblotting were harvested 24 h after induction.
Immunoblotting
γ-H2AX was detected with polyclonal rabbit anti-γ-H2AX (Bethyl Laboratories), H2AX was detected with polyclonal rabbit anti-H2AX (Bethyl Laboratories), tubulin was detected with monoclonal mouse anti-α-tubulin (Covance), HSP90 was detected with mouse anti-HSP90 (BD Biosciences), 3×FLAG-TRB3 was detected with monoclonal mouse anti-FLAG M2 (Sigma), and A3A was detected with rabbit polyclonal anti-A3A, as described previously (2).
mRNA Quantification
A3A mRNA was quantified by quantitative RT-PCR relative to the stable housekeeping transcript, TBP, using highly specific primers, as described previously (5).
MTS Viability Assay
Doxycycline was diluted in PBS in 96-well plates before adding to cells in appropriate growth medium. After 0, 24, 48, or 72 h, the medium was removed and replaced with 20 μl of MTS/phenazine methosulfate solution, prepared as described (Promega), and 100 μl of fresh growth medium. Absorbance was measured 2 h later at 490 nm in a Victor3 multilabel plate reader (PerkinElmer Life Sciences). The absorbance of the blank wells was subtracted from the experimental values, and the data were plotted relative to no doxycycline growth conditions.
Immunofluorescent Microscopy
Endogenous A3A was up-regulated by treating cells with interferon (IFN) as described above. THP-1 cells were additionally treated with phorbol 12-myristate 13-acetate (20 ng/ml; Sigma) to promote adherence to the microscope slide. Cells were fixed with 4% paraformaldehyde 24 h after induction. A3A was detected with rabbit anti-A3A (described above) and goat anti-rabbit FITC (Jackson ImmunoResearch Laboratories). hnRNP U was detected with monoclonal mouse anti-hnRNP U (Santa Cruz Biotechnology) and goat anti-mouse FITC. Hoechst dye was used to visualize nuclei. Cells were imaged with a DeltaVision deconvolution microscope (Applied Precision).
T-REx 293 A3A-GFP cells were induced with 100 pg/ml doxycycline for 24 h. Cells were fixed with 4% paraformaldehyde. 3×FLAG-TRB3 was detected with monoclonal mouse anti-FLAG M2 (Sigma) and donkey anti-mouse-TRITC.
Transfection Studies
T-REx 293 A3A and A3A-E72A cells were transfected with 3×FLAG-TRB3 or empty vector with TransIT-2020 (Mirus). After 24 h, the cells were counted, and (a) plated into doxycycline dilutions for the MTS viability assay, (b) plated into doxycycline or PBS for immunoblot analysis, or (c) plated into doxycycline or PBS for immunofluorescence microscopy.
RESULTS
Overexpressed Heterologous A3A Is Genotoxic, whereas Up-regulated Physiologic A3A Is Safely Expressed
A3A overexpression has been reported to be genotoxic (22, 23, 26). To characterize the extent of A3A toxicity, we employed a doxycycline-inducible system to regulate A3A expression levels. Stable clones were established by transfection of eGFP-tagged A3A (and catalytic mutant A3A-E72A) in pcDNA-5TO into the T-REx 293 cell line. Doxycycline allowed repression to be relieved in a dose-dependent manner, as seen by increasing mean GFP fluorescence by flow cytometry and by increasing band intensity by immunoblotting (Fig. 1A). We assessed the toxicity of A3A as compared with A3A-E72A in T-REx 293 cells by the MTS cellular viability assay, which measures the metabolism of a colorimetric substrate (Fig. 1B). Increasing expression of the catalytic mutant (A3A-E72A) did not result in any significant changes in viability, whereas viability strongly decreased with increasing wild-type A3A expression. A similar dose-dependent relationship was seen with untagged A3A (data not shown).
FIGURE 1.

Exogenously overexpressed A3A is toxic, whereas physiologically up-regulated A3A is not. A, mean GFP fluorescence with increasing doxycycline (Dox) concentrations for T-REx 293 doxycycline-inducible A3A-GFP and A3A-E72A-GFP cell lines. Inset: anti-A3A and anti-tubulin (TUB) immunoblots. B, MTS assay for cell viability following induction of A3A and A3A-E72A by increasing doxycycline concentrations. C, immunoblots for the indicated proteins in T-REx 293 A3A and A3A-E72A cell lines, incubated in the presence and absence of doxycycline induction. Ub, ubiquitin. D, A3A mRNA induction with type I interferon in primary CD14+ cells from three donors, THP-1 cell line, and two stable A3A shRNA knockdown THP-1 clones, relative to the housekeeping gene TBP. High base-line level of A3A expression in primary cells (especially the first donor) may be due to preexisting immune activation. E, MTS assay for the cell types used in panel D. Error bars in D and E indicate S.D.
To determine the cause of the observed toxicity, we examined the cells for γ-H2AX, an indicator of DNA damage (36). We observed induction of γ-H2AX, particularly the monoubiquitinated form, when A3A expression was induced in the T-REx 293 cells with doxycycline (Fig. 1C). In contrast, γ-H2AX was not induced with increased expression of the catalytic mutant.
To determine whether endogenous A3A is similarly cytotoxic, we utilized the monocytic cell line THP-1, in which A3A has been described to be strongly up-regulated upon type I IFN treatment (2). For comparison, we generated stable A3A shRNA knockdown clones, which expressed 20–40-fold lower A3A upon IFN treatment (Fig. 1D). Analysis of cell viability by the MTS assay revealed no significant changes between the parental line and the A3A knockdown clones, in either the presence or the absence of IFN (Fig. 1E).
To extend these endogenous A3A results to primary cells, we performed similar experiments with primary CD14+ cells purified from the whole blood of three donors. These cells have been shown to express the highest levels of A3A (3–6). IFN treatment caused a further induction of A3A expression, as seen by increased mRNA (Fig. 1D) and protein levels (Fig. 2A). Cellular viability, as measured by the MTS assay, only changed modestly (Fig. 1E).
FIGURE 2.

Heterologously induced A3A is toxic even at near physiologic levels. A, immunoblot of endogenous A3A after interferon induction of primary CD14+ cells and the THP-1 cell line, alongside the T-REx 293 A3A cell line treated with the indicated doxycycline (Dox) concentrations (pg/ml) for comparison. A low intensity LI-COR scan is shown to assess A3A levels in CD14+ cells, whereas a higher intensity scan is shown to visualize the level of A3A in the induced THP-1 cells. HSP90 is a loading control. B, MTS viability assay of T-REx 293 cells expressing A3A through a range that spans the maximal interferon-induced levels in primary CD14+ cells or the THP-1 line. Doxycycline concentration (pg/ml) is indicated. Error bars indicate S.D.
Near Physiologic Levels of Overexpressed A3A Are Still Toxic
To determine why endogenous A3A is not genotoxic, we performed a doxycycline titration on T-REx 293 cells inducibly expressing A3A and blotted for A3A with a recently developed rabbit anti-A3A polyclonal antibody, comparing doxycycline induced A3A with endogenously expressed A3A from THP-1 cells and primary monocytes (Fig. 2A). To our surprise, the induced level of A3A in THP-1 cells corresponded to a low level of doxycycline induction of A3A in the T-REx 293 cells (∼6.25–12.5 pg/ml). In contrast, the induced level of A3A in CD14+ cells was higher than the highest levels of A3A induction in the T-REx 293 cells. We repeated the MTS viability assay for our inducible cells and determined that at THP-1 physiologic levels of A3A, viability was constant, even after 72 h of induction (lower doxycycline concentrations in Fig. 2B). In contrast, at the highest levels of A3A induction in the T-REx 293 cells, which is still lower than what was observed in the CD14+ cells, dramatic cytotoxicity was observed (higher doxycycline concentrations in Fig. 2B).
Endogenous A3A Is Cytoplasmic
To further explore the mechanism of protection from endogenous A3A, we investigated cellular localization using a rabbit anti-A3A polyclonal antibody (2). Surprisingly, in both CD14+ and THP-1 cells, A3A was observed to be cytoplasmic upon IFN induction (Fig. 3, A and B). A3A levels in the absence of induction were markedly lower with a faint cytoplasmic localization still detectable (not shown). This is in contrast to the observed cell-wide localization of overexpressed A3A in a variety of nonmyeloid lineage cell lines (3, 13, 14, 16, 21, 27–34) (see Fig. 4A below). As a control for nuclear permeabilization and staining, hnRNP U was seen to localize to the nucleus of the same cells (Fig. 3, C and D).
FIGURE 3.
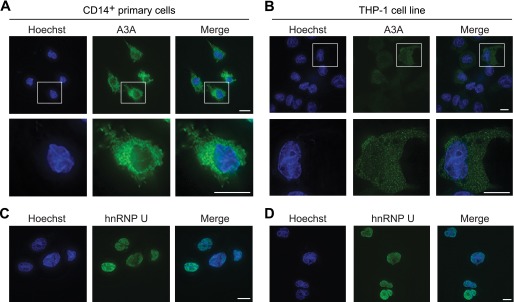
Endogenous A3A is predominantly cytoplasmic. A and B, representative immunofluorescent microscopy images of endogenous A3A in primary CD14+ cells and THP-1 cells, respectively, following treatment with IFN. Individual cells are shown enlarged below. Nuclear DNA is stained blue with Hoechst for compartment identification. Cells were imaged at a 600× magnification; the length of the white bar is 10 μm. C and D, representative immunofluorescent microscopy images of hnRNP U as a control for nuclear permeabilization and staining procedures. Nuclear DNA is stained blue with Hoechst for compartment identification. Cells were imaged at a 600× magnification; the length of the white bar is 10 μm.
FIGURE 4.

Heterologously induced cytoplasmic A3A is not toxic. A, representative live cell microscopy images of the indicated A3A-GFP constructs in HEK293 cells. Nuclear DNA is stained blue with Hoechst for compartment identification. Bright field view (red) is shown in the merge, with GFP and Hoechst. Cells were imaged at a 600× magnification; the length of the white bar is 10 μm. B, MTS assay for viability of T-REx 293 cell lines expressing the indicated A3A-eGFP constructs by induction with increasing concentrations of doxycycline (Dox). Error bars indicate S.D.
Cytoplasmic A3A Is Not Toxic
We created a series of doxycycline-inducible A3A-eGFP localization mutants and stably cloned them into the T-REx 293 cells to test the effect of A3A localization on cytotoxicity. GFP alone showed cell-wide localization, as did wild-type A3A, as observed previously (3, 13, 14, 16, 21, 27–34) (Fig. 4A). Fusion of the nuclear export sequence (NES) of the related protein AID (AID positions 186–198) onto the C terminus of A3A caused the localization to become primarily cytoplasmic, presumably by enabling active CRM1-dependent export of the protein from the nucleus, as seen with AID (37–39). A single point mutation in the NES (AID position L198S) abolishes NES activity and reverts the protein localization to cell-wide (38). The addition of the SV40 nuclear localization sequence (NLS) (×3) to the N terminus of the A3A-GFP fusion protein targeted the construct to the nucleus, causing the localization to become predominantly nuclear. In contrast, adding the large (116-kDa) β-galactosidase protein (encoded by lacZ) caused localization to become predominantly cytoplasmic, likely by making the construct too large to passively enter the nucleus. DiNoia and colleagues (40) used a similar strategy to block the active import of AID into the nuclear compartment.
We tested the effect of A3A localization on cell viability with the MTS assay. Constructs that could access the nucleus (either nuclear or cell-wide localization, i.e. A3A, A3A-NES-L198S and A3A-NLS) showed comparable, high levels of toxicity (Fig. 4B). In contrast, constructs that did not localize to the nucleus (A3A-NES and A3A-LacZ) showed little to no toxicity. As expected, the catalytic mutant, A3A-E72A, also showed no toxicity. These data demonstrate that nuclear localization is required for A3A to cause cytotoxicity and that this activity is completely dependent upon catalytic activity.
TRB3 Does Not Decrease A3A Cytotoxicity
TRB3 was recently implicated in negative regulation of A3A through direct binding (24). To determine whether TRB3 decreases A3A-mediated cellular toxicity in our experimental system, 3×FLAG-TRB3 was transfected into T-REx 293 A3A and A3A-E72A cells. Viability was assessed by the MTS assay over a range of doxycycline-induced A3A expression levels (Fig. 5A). As expected, TRB3 expression had no effect on the viability of cells expressing the catalytic mutant A3A-E72A. However, unexpectedly, TRB3 expression failed to attenuate the toxicity of wild-type A3A.
FIGURE 5.

Transient overexpression of TRB3 does not affect A3A toxicity or localization. A, MTS assay for viability of T-REx 293 A3A and A3A-E72A cell lines transiently transfected with 3×FLAG-TRB3 or empty vector. Error bars indicate S.D. B, immunoblots of A3A (anti-A3A) and TRB3 (anti-FLAG) from the cells in panel A treated with 0 and 100 pg/ml doxycycline (Dox). HSP90 is a loading control. C, representative immunofluorescent microscopy images of T-REx 293 cells induced with doxycycline for A3A and transfected with 3×FLAG-TRB3, alongside non-TRB3 transfected cells. Nuclear DNA is stained blue with Hoechst. Cells were imaged at a 600× magnification; the length of the white bar is 10 μm.
To determine whether TRB3 modulated the levels of A3A expression, we assayed protein levels by immunoblotting (Fig. 5B). Similar levels of A3A were expressed in the presence and absence of TRB3. The localization of A3A in the presence of TRB3 was assessed by immunofluorescent microscopy. As has been reported previously, TRB3 was observed in the nucleus of transfected cells (Fig. 5C) (24). A3A showed cell-wide localization in 293 cells, both in cells transfected with TRB3 and in adjacent nontransfected cells.
To confirm our findings, we also engineered T-REx 293 A3A and A3A-E72A cells to stably express 3×FLAG-TRB3. We again tested for modulation of A3A-mediated toxicity by TRB3 but observed no difference between TRB3-overexpressing clones and vector control-expressing clones (Fig. 6A), nor did TRB3 alter A3A protein levels (Fig. 6B). Thus, in our hands, TRB3 does not appear to be a negative regulator of A3A. We note that this negative result is consistent with the cytoplasmic localization of endogenous A3A described above.
FIGURE 6.

Stable expression of TRB3 does not affect A3A toxicity or localization. A, MTS assay for viability of three T-REx 293 A3A TRB3 stable clones as compared with vector stable clone and T-REx 293 A3A-E72A TRB3 and vector stable clones. Dox, doxycycline. Error bars indicate S.D. B, immunoblots of A3A (anti-A3A) and TRB3 (anti-FLAG) from the cells in panel D treated with 0 and 100 pg/ml doxycycline. HSP90 is a loading control.
DISCUSSION
A3A is a powerful DNA cytosine deaminase that has been identified as an interferon-inducible restrictor of foreign DNA, including plasmid DNA (6), methyl-C-containing sequences (2, 9, 10), retrotransposons (3, 11–15), and several viruses (4, 7, 16–21). However, A3A has also been shown to be genotoxic under conditions of overexpression (22, 23, 26) and to mutate genomic DNA (24, 25). CD14+ cells are known to express high levels of A3A upon type I IFN induction (4, 6, 7), suggesting that these cells must either have a mechanism for regulating A3A activity or be subject to its genotoxic effects.
Genotoxicity was determined to be proportional to A3A expression level by using the doxycycline-inducible T-REx 293 system to titrate A3A expression. As A3A levels increased, cell viability decreased. The catalytic mutant, however, did not decrease viability, demonstrating that DNA cytosine deamination activity is responsible for toxicity. A3A expression also caused increased levels of the DNA damage marker γ-H2AX. Interestingly, the increase was most obvious for the monoubiquitinated form of γ-H2AX, which is an important mediator of the DNA damage response (41–43). In contrast, endogenous A3A was not toxic, despite the high levels of A3A induction by type I IFN seen in primary CD14+ cells and the model monocyte cell line, THP-1.
The physiological level of induction seen in THP-1 cells and primary CD14+ cells was mimicked by inducing A3A with doxycycline in our T-REx 293 system to similar levels, as determined by immunoblot. A3A induction in the model monocytic cell line THP-1 corresponded to a low, sublethal level of A3A induction in our T-REx 293 cell line. In contrast, the level of A3A expression in primary CD14+ cells was higher than the highest level of induction in our T-REx 293 cells, corresponding to highly toxic heterologous A3A levels. However, no evidence of cytotoxicity was observed in primary cells.
The likely reason for the lack of cytotoxicity became apparent when we determined the localization of endogenous A3A. Previous studies of A3A localization have shown that untagged protein in HeLa cells (27), GFP-tagged protein in HeLa (28–30) and HEK293T cells (29), or HA-tagged protein in HeLa (13, 14, 31, 32), HEK293 (3), HEK293T (21, 33, 34), and U20S cells (16) has a cell-wide localization. A3A was determined to enter the nucleus of HeLa cells by passive diffusion (29). Our group has recently developed antibodies that can detect endogenous A3A by immunofluorescence, enabling visualization of the endogenous protein. In contrast to the cell-wide localization of transiently or stably transfected A3A in nonphysiological cell types, endogenous A3A, both in THP-1 cells and in primary CD14+ cells, showed cytoplasmic localization. Exclusion from the nucleus would presumably prevent A3A from accessing the cellular DNA and thus protect the DNA from A3A-induced damage. We tested this hypothesis by creating A3A localization mutants and indeed determined that only the constructs that could localize to the nucleus (i.e. nuclear or cell-wide localization) showed similar cellular toxicity to wild-type A3A. In contrast, both A3A tagged with a nuclear export sequence and A3A tagged with a large protein to prevent entry into the nucleus showed cytoplasmic localization, similar to endogenous A3A, and both had negligible effects on cell viability. This was evident even at the highest levels of protein expression.
The other APOBEC3 proteins show a variety of steady-state localization patterns. A3B is observed to be nuclear in the majority of tested cell lines, including HeLa (14, 28, 29, 44, 45), U20S (29), TR146 (29), JSQ3 (29), MCF10A (29), HuH-7 (46), HepG2 (46), MDA-MB-453 (26), MDA-MB-468 (26), and HCC1569 (26), whereas it is cell-wide in 293T (29, 33) cells, which are believed to have an import defect (29). A3C is considered cell-wide in both HeLa (14, 28, 30) and 293T cells (31, 33, 34). A3D, A3F, and A3G are cytoplasmic in HeLa (14, 28, 30, 44, 45, 48–51) and 293T (3, 31, 33, 34, 48, 52) cells, with A3G additionally shown to be cytoplasmic in MDA-MB-453 (26), MDA-MB-468 (26), HCC1569 (26), HuH-7 (46), CEM-SS (51), H9 (48, 51), primary peripheral blood mononuclear cells (51), and primary T cells (48). The localization of A3H, interestingly, appears haplotype-dependent, with haplotype I being cell-wide in 293T (31) and HeLa (14, 30) cells and haplotype II being cytoplasmic in 293T and HeLa cells (28, 53). Indeed, Li and Emerman (53) suggest that the more active form of A3H, haplotype II, is actively retained in the cytoplasm.
A recent study reported that TRB3 is a negative regulator of A3A (24). However, in our inducible system, neither transient nor stable TRB3 overexpression had an effect on A3A-mediated cellular toxicity. Furthermore, TRB3 did not divert A3A localization from the nucleus, which, based on the evidence presented in this study, might have been a reasonable function for this putative negative regulator. Thus, because endogenous A3A is cytoplasmic (this study) and TRB3 is nuclear (Refs. 24, 54, and 55 and this study), we favor a model in which the inferred mechanism responsible for the cytoplasmic retention of A3A is TRB3-independent and remains to be discovered.
We conclude that cells that endogenously express A3A, i.e. CD14+ monocytic cells, are able to control its activity and protect their DNA from this powerful DNA cytosine deaminase by a cytoplasmic retention mechanism. This mechanism may be fundamentally distinct from that previously inferred for A3G as even heterologously expressed full-length A3G and the N-terminal half of A3G are cytoplasmic with the latter being of similar size to A3A (45, 47, 52). The mechanism may also be distinct from that of A3H haplotype II as A3A is cell-wide in HeLa and 293T cells, in contrast to A3H-hapII (53), suggesting that in these cells, the A3A cytoplasmic retention mechanism may be either lacking or defective. Cellular DNA is protected from A3A largely, and perhaps exclusively, based on the localization of A3A to the cytoplasm, as the cytoplasmic localization of exogenous A3A in the T-REx 293 system was sufficient to prevent cytotoxicity. Thus, CD14+ monocytic cells are able to safely express high levels of A3A and mediate innate immune functions such as foreign DNA clearance. Further work will identify the cellular machinery responsible for cytoplasmic localization and A3A regulation and determine how this regulation influences the innate immune activity of A3A. Together, the restricted expression profile of A3A in myeloid lineage cell types and the cytoplasmic localization of endogenous A3A in these cell types combine to strongly indicate that this protein is not normally genotoxic or a major factor in carcinogenesis.
Acknowledgments
We thank Andrea Cimarelli for THP-1, Simon Wain-Hobson for TRB3 in the pCI-neo-3×FLAG vector, and Javier Di Noia for pEGFP-N3-AID-LacZ (used to construct the A3A-LacZ construct).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI064046 and P01 GM091743 (to R. S. H.).
- A3A
- APOBEC3A
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- hnRNP U
- heterogeneous nuclear ribonucleoprotein U
- TRITC
- tetramethylrhodamine isothiocyanate
- NES
- nuclear export sequence
- AID
- activation-induced cytidine deaminase
- eGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Murray P. J., Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carpenter M. A., Li M., Rathore A., Lackey L., Law E. K., Land A. M., Leonard B., Shandilya S. M., Bohn M. F., Schiffer C. A., Brown W. L., Harris R. S. (2012) Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 287, 34801–34808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H., Lilley C. E., Yu Q., Lee D. V., Chou J., Narvaiza I., Landau N. R., Weitzman M. D. (2006) APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16, 480–485 [DOI] [PubMed] [Google Scholar]
- 4. Koning F. A., Goujon C., Bauby H., Malim M. H. (2011) Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J. Virol. 85, 13448–13452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Refsland E. W., Stenglein M. D., Shindo K., Albin J. S., Brown W. L., Harris R. S. (2010) Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stenglein M. D., Burns M. B., Li M., Lengyel J., Harris R. S. (2010) APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17, 222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berger G., Durand S., Fargier G., Nguyen X. N., Cordeil S., Bouaziz S., Muriaux D., Darlix J. L., Cimarelli A. (2011) APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 7, e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thielen B. K., McNevin J. P., McElrath M. J., Hunt B. V., Klein K. C., Lingappa J. R. (2010) Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J. Biol. Chem. 285, 27753–27766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wijesinghe P., Bhagwat A. S. (2012) Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 40, 9206–9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nabel C. S., Jia H., Ye Y., Shen L., Goldschmidt H. L., Stivers J. T., Zhang Y., Kohli R. M. (2012) AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 8, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bishop K. N., Holmes R. K., Sheehy A. M., Davidson N. O., Cho S. J., Malim M. H. (2004) Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14, 1392–1396 [DOI] [PubMed] [Google Scholar]
- 12. Bogerd H. P., Wiegand H. L., Doehle B. P., Lueders K. K., Cullen B. R. (2006) APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 34, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bogerd H. P., Wiegand H. L., Hulme A. E., Garcia-Perez J. L., O'Shea K. S., Moran J. V., Cullen B. R. (2006) Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muckenfuss H., Hamdorf M., Held U., Perkovic M., Löwer J., Cichutek K., Flory E., Schumann G. G., Münk C. (2006) APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 281, 22161–22172 [DOI] [PubMed] [Google Scholar]
- 15. Chiu Y. L., Witkowska H. E., Hall S. C., Santiago M., Soros V. B., Esnault C., Heidmann T., Greene W. C. (2006) High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 15588–15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narvaiza I., Linfesty D. C., Greener B. N., Hakata Y., Pintel D. J., Logue E., Landau N. R., Weitzman M. D. (2009) Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 5, e1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vartanian J. P., Guétard D., Henry M., Wain-Hobson S. (2008) Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320, 230–233 [DOI] [PubMed] [Google Scholar]
- 18. Suspène R., Guétard D., Henry M., Sommer P., Wain-Hobson S., Vartanian J. P. (2005) Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 8321–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ooms M., Krikoni A., Kress A. K., Simon V., Münk C. (2012) APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 86, 6097–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng G., Greenwell-Wild T., Nares S., Jin W., Lei K. J., Rangel Z. G., Munson P. J., Wahl S. M. (2007) Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110, 393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berger A., Münk C., Schweizer M., Cichutek K., Schüle S., Flory E. (2010) Interaction of Vpx and apolipoprotein B mRNA-editing catalytic polypeptide 3 family member A (APOBEC3A) correlates with efficient lentivirus infection of monocytes. J. Biol. Chem. 285, 12248–12254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landry S., Narvaiza I., Linfesty D. C., Weitzman M. D. (2011) APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 12, 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suspène R., Aynaud M. M., Guétard D., Henry M., Eckhoff G., Marchio A., Pineau P., Dejean A., Vartanian J. P., Wain-Hobson S. (2011) Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 4858–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aynaud M. M., Suspène R., Vidalain P. O., Mussil B., Guétard D., Tangy F., Wain-Hobson S., Vartanian J. P. (2012) Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J. Biol. Chem. 287, 39182–39192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shinohara M., Io K., Shindo K., Matsui M., Sakamoto T., Tada K., Kobayashi M., Kadowaki N., Takaori-Kondo A. (2012) APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burns M. B., Lackey L., Carpenter M. A., Rathore A., Land A. M., Leonard B., Refsland E. W., Kotandeniya D., Tretyakova N., Nikas J. B., Yee D., Temiz N. A., Donohue D. E., McDougle R. M., Brown W. L., Law E. K., Harris R. S. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goila-Gaur R., Khan M. A., Miyagi E., Kao S., Strebel K. (2007) Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hultquist J. F., Lengyel J. A., Refsland E. W., LaRue R. S., Lackey L., Brown W. L., Harris R. S. (2011) Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lackey L., Demorest Z. L., Land A. M., Hultquist J. F., Brown W. L., Harris R. S. (2012) APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 419, 301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LaRue R. S., Jónsson S. R., Silverstein K. A., Lajoie M., Bertrand D., El-Mabrouk N., Hötzel I., Andrésdóttir V., Smith T. P., Harris R. S. (2008) The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 9, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinomoto M., Kanno T., Shimura M., Ishizaka Y., Kojima A., Kurata T., Sata T., Tokunaga K. (2007) All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 35, 2955–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lovsin N., Peterlin B. M. (2009) APOBEC3 proteins inhibit LINE-1 retrotransposition in the absence of ORF1p binding. Ann. N.Y. Acad. Sci. 1178, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marin M., Golem S., Rose K. M., Kozak S. L., Kabat D. (2008) Human immunodeficiency virus type 1 Vif functionally interacts with diverse APOBEC3 cytidine deaminases and moves with them between cytoplasmic sites of mRNA metabolism. J. Virol. 82, 987–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niewiadomska A. M., Tian C., Tan L., Wang T., Sarkis P. T., Yu X. F. (2007) Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 81, 9577–9583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsuchiya S., Yamabe M., Yamaguchi Y., Kobayashi Y., Konno T., Tada K. (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26, 171–176 [DOI] [PubMed] [Google Scholar]
- 36. Rogakou E. P., Boon C., Redon C., Bonner W. M. (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ito S., Nagaoka H., Shinkura R., Begum N., Muramatsu M., Nakata M., Honjo T. (2004) Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. U.S.A. 101, 1975–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McBride K. M., Barreto V., Ramiro A. R., Stavropoulos P., Nussenzweig M. C. (2004) Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J. Exp. Med. 199, 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brar S. S., Watson M., Diaz M. (2004) Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J. Biol. Chem. 279, 26395–26401 [DOI] [PubMed] [Google Scholar]
- 40. Patenaude A. M., Orthwein A., Hu Y., Campo V. A., Kavli B., Buschiazzo A., Di Noia J. M. (2009) Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat. Struct. Mol. Biol. 16, 517–527 [DOI] [PubMed] [Google Scholar]
- 41. Pan M. R., Peng G., Hung W. C., Lin S. Y. (2011) Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 286, 28599–28607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu C. Y., Kang H. Y., Yang W. L., Wu J., Jeong Y. S., Wang J., Chan C. H., Lee S. W., Zhang X., Lamothe B., Campos A. D., Darnay B. G., Lin H. K. (2011) Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J. Biol. Chem. 286, 30806–30815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen W. T., Alpert A., Leiter C., Gong F., Jackson S. P., Miller K. M. (2013) Systematic identification of functional residues in mammalian histone H2AX. Mol. Cell. Biol. 33, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stenglein M. D., Harris R. S. (2006) APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 281, 16837–16841 [DOI] [PubMed] [Google Scholar]
- 45. Stenglein M. D., Matsuo H., Harris R. S. (2008) Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J. Virol. 82, 9591–9599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bonvin M., Achermann F., Greeve I., Stroka D., Keogh A., Inderbitzin D., Candinas D., Sommer P., Wain-Hobson S., Vartanian J. P., Greeve J. (2006) Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43, 1364–1374 [DOI] [PubMed] [Google Scholar]
- 47. Bennett R. P., Diner E., Sowden M. P., Lees J. A., Wedekind J. E., Smith H. C. (2006) APOBEC-1 and AID are nucleo-cytoplasmic trafficking proteins but APOBEC3G cannot traffic. Biochem. Biophys. Res. Commun. 350, 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wichroski M. J., Robb G. B., Rana T. M. (2006) Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D. (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103 [DOI] [PubMed] [Google Scholar]
- 50. Gallois-Montbrun S., Holmes R. K., Swanson C. M., Fernández-Ocaña M., Byers H. L., Ward M. A., Malim M. H. (2008) Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J. Virol. 82, 5636–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gallois-Montbrun S., Kramer B., Swanson C. M., Byers H., Lynham S., Ward M., Malim M. H. (2007) Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 81, 2165–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bennett R. P., Presnyak V., Wedekind J. E., Smith H. C. (2008) Nuclear Exclusion of the HIV-1 host defense factor APOBEC3G requires a novel cytoplasmic retention signal and is not dependent on RNA binding. J. Biol. Chem. 283, 7320–7327 [DOI] [PubMed] [Google Scholar]
- 53. Li M. M., Emerman M. (2011) Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J. Virol. 85, 8197–8207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu M., Xu L. G., Zhai Z., Shu H. B. (2003) SINK is a p65-interacting negative regulator of NF-κB-dependent transcription. J. Biol. Chem. 278, 27072–27079 [DOI] [PubMed] [Google Scholar]
- 55. Ord D., Ord T. (2003) Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp. Cell Res. 286, 308–320 [DOI] [PubMed] [Google Scholar]