

Background: Little is known about the substrate specificity of the essential P450 CYP121 from Mycobacterium tuberculosis.
Results: CYP121 substrate analogues either display impaired binding to CYP121 or are poorly transformed by CYP121.
Conclusion: CYP121 is a highly specific P450.
Significance: This work contributes to the understanding of the function and the catalytic mechanism of CYP121 and provides novel data for the design of inhibitors.
Keywords: Crystallography, Cytochrome P450, Enzyme Structure, Nuclear Magnetic Resonance, Tuberculosis, Enzyme Specificity
Abstract
Cytochrome P450 CYP121 is essential for the viability of Mycobacterium tuberculosis. Studies in vitro show that it can use the cyclodipeptide cyclo(l-Tyr-l-Tyr) (cYY) as a substrate. We report an investigation of the substrate and reaction specificities of CYP121 involving analysis of the interaction between CYP121 and 14 cYY analogues with various modifications of the side chains or the diketopiperazine (DKP) ring. Spectral titration experiments show that CYP121 significantly bound only cyclodipeptides with a conserved DKP ring carrying two aryl side chains in l-configuration. CYP121 did not efficiently or selectively transform any of the cYY analogues tested, indicating a high specificity for cYY. The molecular determinants of this specificity were inferred from both crystal structures of CYP121-analog complexes solved at high resolution and solution NMR spectroscopy of the analogues. Bound cYY or its analogues all displayed a similar set of contacts with CYP121 residues Asn85, Phe168, and Trp182. The propensity of the cYY tyrosyl to point toward Arg386 was dependent on the presence of the DKP ring that limits the conformational freedom of the ligand. The correct positioning of the hydroxyl of this tyrosyl was essential for conversion of cYY. Thus, the specificity of CYP121 results from both a restricted binding specificity and a fine-tuned P450 substrate relationship. These results document the catalytic mechanism of CYP121 and improve our understanding of its function in vivo. This work contributes to progress toward the design of inhibitors of this essential protein of M. tuberculosis that could be used for antituberculosis therapy.
Introduction
Cytochrome P450 enzymes (P450s)5 constitute a superfamily of heme-containing proteins found in nearly all organisms from bacteria to humans (1). Most catalyze mixed function oxidations of a wide range of endogenous and xenobiotic substrates; a very large number of substrates and reactions have been described (2, 3). Nevertheless, it is widely accepted that there is a common catalytic cycle in which the substrate plays important roles. The substrate generally induces the low-to-high spin transition that facilitates the continuation of the cycle and may influence the nature of the reaction catalyzed (4). Some P450s have broad substrate and reaction specificity, whereas others are highly selective enzymes catalyzing regio- and stereospecific reactions. The characterization of substrate and reaction specificity of P450s would help to elucidate their biological functions and decipher the catalytic mechanism, with major implications for drug design and biocatalysis.
CYP121 from Mycobacterium tuberculosis is one of the most studied P450s from this human pathogen (5). The increasing number of deaths associated with M. tuberculosis over the last 25 years has highlighted the limitations of currently available anti-tuberculosis drugs. Novel antibiotics are required (6). Interest in CYP121 as a potential drug target followed the early discovery that CYP121 has strong affinity for several azole molecules that were also identified as effective antimycobacterial compounds (7–10). McLean et al. (11) showed that the cyp121 gene is essential for mycobacterial growth, reinforcing the idea that CYP121 could be a potential therapeutic target. The determination of the crystal structure of CYP121 in complex with fluconazole at 1.9 Å revealed a novel mode of azole binding to P450, with a nitrogen atom coordinating the heme iron through the sixth iron ligand. However, no molecules, other than azoles, that bind to CYP121 have been found, and initial attempts to identify ligands by testing compounds for their ability to induce heme iron spin transition were unsuccessful (9).
Recently, we showed that CYP121 catalyzes an unusual reaction by forming a C–C bond between the two tyrosyl side chains of the cyclodipeptide cyclo(l-Tyr-l-Tyr) (cYY; Fig. 1, 1) resulting in a novel chemical entity called mycocyclosin (Fig. 1, 2) (12, 13). This identification was based in part on the characterization of the activity of Rv2275, encoded by a gene associated in an operon-like structure with cyp121 (14, 15). Rv2275 uses charged tRNAs to synthesize cYY and several other cyclodipeptides. The identification of cYY was unexpected, because the x-ray crystal structure of CYP121 solved at atomic resolution revealed a large active site cavity that could accommodate bulky hydrophobic substrates (16). Accordingly, the x-ray structure of cYY-bound CYP121 solved at 1.4 Å resolution showed a single ligand molecule that only partially occupies the binding cavity (12). The protein conformation in this complex is very similar to that of ligand-free or fluconazole-bound CYP121. This suggested the absence of significant conformational change upon ligand binding, consistent with the previously described rigidity of the active site (12, 16). Our work also revealed unexpected structural features of substrate-bound P450 with the presence of a network of hydrogen bonds above the heme and between the substrate and residues of the active site and water molecules, including the sixth heme iron ligand. One hydroxyl of cYY, in particular, approaches the heme and participates in this network. According to these structural observations and the reaction catalyzed, we proposed a role for the two side chains of cYY in transformation. Nevertheless, the molecular determinants of the substrate involved in the catalytic cycle remain to be determined. The aim of this work was to identify the substrate determinants involved in the reaction and substrate specificity of CYP121. We used UV-visible spectrophotometry, x-ray crystallography, enzymatic assays, mass spectrometry, and solution NMR spectroscopy to characterize the binding to and transformation by CYP121 of a series of substrate analogues (Fig. 1).
FIGURE 1.

Chemical structures of compounds described in this study.
EXPERIMENTAL PROCEDURES
Chemicals
Chemicals were from Sigma-Aldrich unless otherwise stated. All chemicals were of the highest purity available.
Synthetic Chemistry Methods
Compounds 1, 4, 12, 13, and 14 were obtained from Bachem. Compounds 2, 3, 5, 6, and 7 were synthesized previously in our laboratory (12, 14). Compounds 8, 9, 10, 11, and 15 were chemically synthesized by adapting previously published procedures (17–21). The synthesis strategies are shown in Fig. 2.
FIGURE 2.

Synthetic schemes for 8, 9, and 10 (A), 11 (B), and 15 (C). A, cyclodipeptides were synthesized by liquid phase peptide synthesis by adapting previously described procedures (19). a, amino acids with side chains R1 and R2 and protecting groups P1 (Fmoc (fluorenylmethyloxycarbonyl) for synthesis of 8 or Boc (tert-butyloxycarbonyl) for synthesis of 9 and 10), P2 (acetonid for synthesis of 8 and tBu (tert-butyl) for synthesis of 9 and 10), and P3 (tBu) were incubated in N,N′-dicyclohexylcarbodiimide and triethylamine for 1 h at 4 °C and then overnight at room temperature, followed by overnight dessication; b (only for the synthesis of 8), dissolution in dimethylformamide/dichloromethane (1:1), 2 h at room temperature, evaporation to dryness; c, dissolution in HCO2H/TFA/H2O (9:0.5:0.5), 2 h at room temperature, evaporation to dryness; d, dissolution in butanol-1/toluene (9:1) or butanol-1 (synthesis of 9 and 10), reflux for 2 h, purification by RP-HPLC. B, the synthesis of 11 was adapted from previously published procedures (17). a, piperazine, triethylamine, and 1-(benzyloxy)-4-(chloromethyl)benzene (1:2.2:10) were stirred in tetrahydrofuran for 18 h at room temperature, evaporated to dryness, and subjected to chromatography on SiO2 in ethyl acetate/hexane (7:3, v/v); b, dissolution in methanol/ethanol (8:2, v/v), hydrogenation with 5 mg 10% palladium on carbon, drying under vacuum, purification by RP-HPLC. C, the synthesis of 15 was adapted from previous publications describing reductive amination (18, 20, 21) and liquid phase peptide synthesis (19). a, incubation in dimethylformamide/AcOH for 0.5 h at room temperature, cooled to 0 °C, NaBH3CN, 16 h at room temperature, evaporation under reduced pressure at temperature <40 °C; b, dissolution in methanol, 10% palladium on carbon overnight under 3.3 bars of H2, evaporation to dryness; c, dissolution in HCOOH, 2 h at room temperature with stirring, evaporation under reduced pressure (temperature <40 °C), addition of triethylamine and dry dimethylformamide and stirring for 16 h at room temperature, evaporation under reduced pressure, purification by RP-HPLC.
Amino acid derivatives were obtained from Bachem. They were used as received without further purification. All non-aqueous reactions were carried out under an atmosphere of argon in flame- or oven-dried glassware with magnetic stirring. Thin layer chromatography analyses were performed on Merck Silica Gel 60 F254 plates, and components were visualized by illumination with UV light or by staining with a potassium permanganate solution (1 g with 2 g of K2CO3 in 200 ml of water). Flash column chromatography was performed using Merck Geduran Si60 (40–63 μm). All compounds were purified by RP-HPLC (Discovery Bio Wide Pore C18 column, 250 × 10 mm, 5 μm). Purity was determined by analytical RP-HPLC (Atlantis dC18, 4.6 × 250 mm, 5 μm; >95%). High resolution mass spectra were recorded on a 4800 MALDI-TOF mass spectrometer as described previously, using 0.1–0.3 mm solutions of compound in 50% CH3CN (DMSO < 1‰) (14). Compounds were also characterized by NMR spectroscopy (see below).
High resolution mass spectra (m/z): 8, [MH]+ calculated for C18H19N2O5, 343.1294, found 343.1304. 9, [MH]+ calculated for C18H19N2O4, 327.1345, found 327.1364. 10, [MH]+ calculated for C18H19N2O4, 327.1345, found 327.1348. 11, [MH]+ calculated for C18H23N2O2, 299.1760, found 299.1757. 15, [MH]+ calculated for C18H19N2O4, 313.1552, found 313.1552.
Stock Solutions of Compounds
Compounds were dissolved in DMSO at the highest concentration that could be achieved. Concentrations were determined by UV-visible spectrophotometry after dilution of the stock solution in 100 mm potassium phosphate, pH 7.4, with the final DMSO concentration below 10%. For peptide compounds, the molar extinction coefficients of phenylalanine, tryptophan, and tyrosine were used. For 8, 11, and 15, molar extinction coefficients were found to be 3580 m−1 cm−1 at 279 nm, 1570 m−1 cm−1 at 271 nm, and 1795 m−1 cm−1 at 276 nm, respectively. Clear stock solutions of 3, 5, and 6 could only be obtained at concentrations up to 15–20, 40–50, and 60–70 mm, respectively. Other compounds were dissolved at concentrations of around 200 mm without difficulty. Stock solutions were conserved at −20 °C and were thawed at room temperature before use. RP-HPLC analysis of dilutions of these stock solutions revealed that purity was >99% (estimated from peak areas determined on chromatograms recorded at 220 nm). However, the solution of 12 (linear dipeptide YY) was found to change over time, with the appearance of a major contaminant; LC-MS/MS analysis indicated the presence of a high concentration of 1 (cyclic dipeptide cYY, between 5 and 10%). This was attributed to the formation of an intramolecular peptide bond, possibly favored by the presence of DMSO. To limit the formation of 1, stock solutions of 12 were prepared in DMSO extemporaneously and were used the same day. In these conditions, the amount of 1 in the samples was less than 1‰ as estimated by LC-MS/MS using a calibration curve with 1. In solutions of 13 and 14 (derivatives of linear dipeptide YY by acetylation of the primary amine and amidation of the carboxylic acid, respectively), 1 was detected only in trace quantities (<0.3‰).
CYP121 Expression and Purification
Wild type CYP121 was produced in Escherichia coli and purified as described previously (9, 12). Purified CYP121 had an Rz value (ratio of A416 nm to A280 nm) of >1.8. The purity was >95% as estimated by SDS-PAGE analysis. The concentration of CYP121 was determined by using the ϵ416 of 110 mm−1 cm−1 (11). Purified CYP121 (∼1 mm) was stored in 50 mm Tris-HCl, 1 mm EDTA (pH 7.2) at −80 °C.
Binding of Compounds to CYP121
Ligand binding to CYP121 was analyzed at 20 °C by spectral titration using a double-beam Uvikon 943 spectrophotometer and 1-cm path length quartz cells as described previously (12). The final DMSO concentration was kept under 1% (v/v). For 3 and 15, light scattering appeared during titration, resulting in large variations of Ks and ΔAbsmax values between the different assays. To limit the effects of light scattering on Ks determinations, ligand solution was added to the reference cuvette instead of DMSO. Plots appeared hyperbolic for all compounds showing binding to CYP121, and data were fitted to the equation, ΔAbs = (ΔAbsmax × [L])/(Ks + [L]), where ΔAbsmax is the overall maximal absorbance variation, and Ks is the apparent spectral dissociation constant.
For the determination of the Ks value of cYY for CYP121 in the presence of another ligand, the inhibitor ligand was added to the reference and sample cuvettes, the base line was recorded, CYP121 was added to the reference cuvette, and the titration with cYY was performed as above. The final DMSO concentration remained under 1.5% (v/v). Data were analyzed as above. All experiments were performed in triplicate at least, and results are expressed as means ± S.D.
Crystallization and Structure Determination of Ligand-bound CYP121
Ligand-bound CYP121 crystals were grown at 6 °C in a few days by the sitting drop method as described previously (purified CYP121 at 230 μm and ligand concentration around 1 mm in 100 mm NaMES at pH 5.0–5.5 and 1.6–2.2 M (NH4)2SO4). Crystals were cryoprotected in a solution containing 20% glycerol in the crystallization solution and cryocooled in liquid nitrogen. Diffraction data were collected at the European Synchrotron Radiation Facility or at SOLEIL (see Table 1 for details), processed with MOSFLM, and scaled with SCALA from the CCP4 package (22). The structures were refined with REFMAC (23) and BUSTER5 (24), using the coordinates of native CYP121 as the input entry (Protein Data Bank entry 1N40) (25), and manually corrected using COOT 0.6 (26). Ligands were built at the end of the refinement processes. The quality of the electron density was compatible with the molecule being completely built for complexes of CYP121 with 3, 8, and 15. In the case of CYP121 bound to 4 and CYP121 bound to 7, two alternate conformations were modeled on the basis of the electron density map. Their respective occupancies were adjusted to 0.25 (conformation 1) and 0.75 (conformation 2) for 4 and to 0.34 (conformation a) and 0.66 (conformation b) for 7, such that B factor values would be of the same order for each conformation. At the end of the refinement process, one additional run of BUSTER5 was performed after removal of the ligand molecules from the coordinate files, to calculate unbiased 2Fo − Fc electron density omit maps. Table 1 reports further information on x-ray analysis statistics.
TABLE 1.
Data collection and refinement statisticsa
| Ligand bound to CYP121 |
|||||
|---|---|---|---|---|---|
| 3 | 4 | 7 | 8 | 15 | |
| Protein Data Bank entry | 4ICT | 4IQ9 | 4IQ7 | 4IPW | 4IPS |
| Data collection | |||||
| Beam line | ID23 | ID23 | ID23 | ID23 | PX1 (Pilatus) |
| Wavelength (Å) | 0.933 | 0.933 | 0.933 | 0.933 | 0.8726 |
| Space group | P6522 | P6522 | P6522 | P6522 | P6522 |
| Resolution limits (Å) | 1.8 (1.9–1.8) | 1.4 (1.48–1.4) | 1.9 (2.0–1.9) | 1.4 (1.48–1.4) | 1.2 (1.27–1.2) |
| Rmerge | 0.152 (0.541) | 0.092 (0.346) | 0.146 (0.435) | 0.099 (0.285) | 0.042 (0.281) |
| Total no. of reflections | 296,243 (47,663) | 1,099,644 (100,350) | 206,187 (32,849) | 389,209 (33,474) | 2,918,439 (343,052) |
| No. of unique reflections | 44,879 (6445) | 93,373 (13,226) | 37,824 (5442) | 86,350 (10,768) | 274,037 (43,130) |
| Mean (I)/sd(I) | 9.6 (3.4) | 18.1 (4.4) | 10.4 (3.9) | 11.5 (2.8) | 32.65 (5.38) |
| Completeness (%) | 99.8 (100.0) | 99.8 (99.0) | 98.4 (99.6) | 92.5 (80.7) | 99.2 (96.4) |
| Multiplicity | 6.6 (7.4) | 11.8 (7.6) | 5.5 (6.0) | 4.5 (3.1) | 10.65 (7.95) |
| Refinement | |||||
| R factor | 0.1776 | 0.1582 | 0.1500 | 0.1566 | 0.1582 |
| Rfree | 0.2285 | 0.1794 | 0.1884 | 0.1824 | 0.1710 |
| Figure of merit | 0.8910 | 0.930 | 0.937 | 0.929 | 0.932 |
| Total no. of atoms | 3735 | 3989 | 3751 | 3868 | 3792 |
| No. of water molecules | 588 | 695 | 605 | 655 | 596 |
| Root mean square deviation | |||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.010 | 0.012 | 0.010 |
| Bond angles (degrees) | 1.196 | 1.00 | 0.99 | 1.06 | 1.01 |
| Average B factors (Å2): | |||||
| All atoms | 24.9 | 17.2 | 21.7 | 16.2 | 16.1 |
| Protein | 22.0 | 14.0 | 18.4 | 13.1 | 13.5 |
| Water | 39.3 | 31.6 | 37.0 | 30.6 | 29.5 |
| Heme | 18.3 | 9.6 | 12.8 | 8.9 | 10 |
| Ligand | 31.7 | 14.3/16.6 | 25.5 | 10.1 | 16.9 |
a Data in parentheses indicate the last resolution shell.
Turnover Experiments
CYP121 conversion of compounds was assessed in the presence of an electron transport chain constituted of spinach ferredoxin and spinach ferredoxin NADP+ reductase as described previously (12). The final DMSO concentration was kept at 1%, because preliminary experiments performed with final DMSO concentrations ranging from 1 to 5% indicated the absence of detectable effects on cYY transformation by CYP121 at concentrations below 5% (data not shown). Samples collected at various times were acidified and analyzed by LC-MS/MS as described previously (12). Control experiments in which CYP121 was omitted were conducted and analyzed as above. In these control experiments, the DMSO concentration was 1% (v/v).
Solution NMR Spectroscopy for Conformational Analysis
A Bruker Avance III spectrometer equipped with a TCI cryoprobe and operating at a 1H frequency of 500 MHz was used for NMR experiments. Spectra were recorded at 30 °C in DMSO-d6, in CD3OD, or in 90% H2O, 10% D2O (Eurisotop). 1H and 13C resonances were assigned through the analysis of one-dimensional 1H, one-dimensional 13C DEPTQ (distortionless enhancement by polarization transfer), two-dimensional 1H-1H COSY, two-dimensional 1H-1H ROESY (rotating frame NOE spectroscopy), two-dimensional 1H-13C HSQC (heteronuclear single quantum correlation), and two-dimensional 1H-13C HMBC (heteronuclear multiple-bond correlation). 1H and 13C chemical shifts were referenced to the DMSO solvent signal (δ 2.50 and 39.5 ppm, respectively). NMR data were processed and analyzed with Bruker TOPSPIN version 3.1 software.
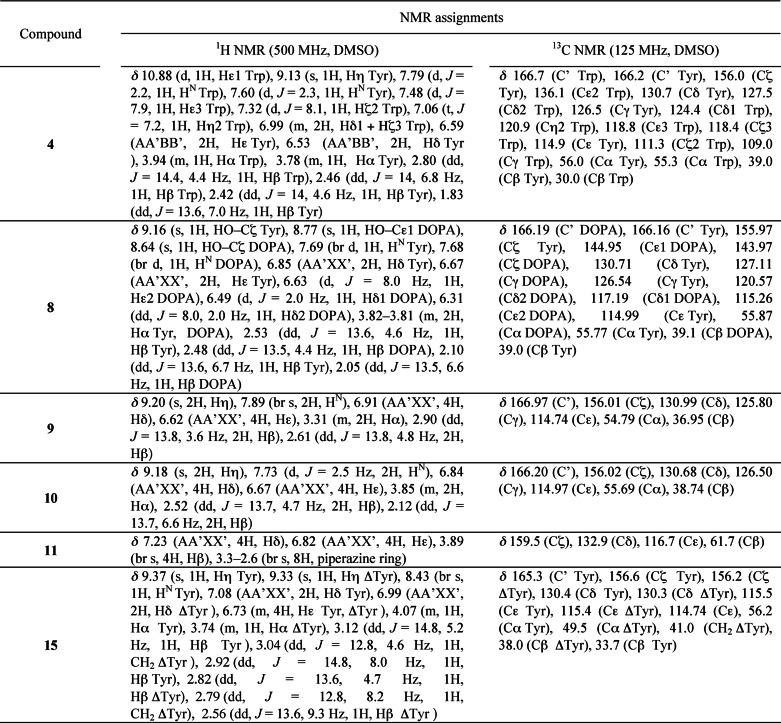
The conformational analysis was based on the measurement of homonuclear 3JHα-Hβ coupling constants on one-dimensional 1H spectra and heteronuclear 3JHα-Cγ and 3JHβ-CO coupling constants measured on phase-sensitive two-dimensional HMBC experiments. Using the Karplus curves obtained for these three vicinal coupling constants (27), the populations of χ1 rotamers and stereospecific assignment of Hβ methylenic protons could be obtained unambiguously. In particular, the 3JHα-Cγ coupling constant provided direct estimates of the gauche+ rotamer population, and 3JHβ-CO coupling constants allowed Hβ methylenic protons to be assigned stereospecifically. NMR assignments of the compounds are presented in Table 2.
TABLE 2.
NMR assignments for conformational analysis of the compounds in solution
RESULTS
Rationale for the Choice of cYY Analogues
The cYY analogues were chosen to be relevant to various issues: determining the specificity of CYP121, investigating its in vivo function, and providing information pertinent to drug design. Mycocyclosin 2, the major product of the reaction, was also included to study any possible end product inhibition of the reaction. CYP121 and the cyclodipeptide synthase Rv2275 are genetically linked, so we tested cyclodipeptides synthesized by Rv2275 because they may be natural substrates. They include cYF (3), cYW (4), cyclo(l-Tyr-l-Leu) (5), cyclo(l-Tyr-l-Met) (6), and cYA (7) (14, 28). We also included the cyclodipeptide cY-DOPA (8). This cyclodipeptide differs from cYY by only one additional hydroxyl positioned on one carbon atom involved in the C–C coupling catalyzed by CYP121. To evaluate the role of the chirality of tyrosine residues in cYY, we also investigated cyclo(l-Tyr-d-Tyr) and cyclo(d-Tyr-d-Tyr) (9 and 10, respectively). To study the relationship with CYP121 of compounds that carry two tyrosyl side chains anchored on a scaffold other than the DKP ring, we included a compound that carries the two tyrosyls on a piperazine scaffold (11), linear YY dipeptides with free or blocked N and C termini (12, 13, and 14), and a compound that differs from cYY by the reduction to CH2 of one of the keto functions of the DKP ring (15).
Cyclodipeptides with Two Aryl Side Chains in l-Configuration Bind Efficiently to CYP121
Binding of compounds to CYP121 was assessed in ligand titration experiments by UV-visible spectroscopy. Because the ligands studied are poorly soluble in water, we used stock solutions in DMSO. The addition of 1% DMSO to a CYP121 solution slightly diminished the intensity of the Soret band (<1%) without observable effect on λmax. Furthermore, binding constants of CYP121 for cYY were similar for final DMSO concentrations of 1 or 2% (19.4 ± 0.6 and 21.6 ± 1.2 μm, respectively). Therefore, the final DMSO concentration was kept under 1% in ligand titration experiments.
In addition, for ligands exhibiting no spectral variation upon addition to a solution of CYP121, their effects at a high concentration on the affinity of CYP121 for its substrate cYY were evaluated in inhibition experiments. In these assays, the DMSO concentration remained ≤1.5%. Data obtained in these two experiments are summarized in Table 3.
TABLE 3.
UV-visible spectroscopic characterization of ligand binding to CYP121
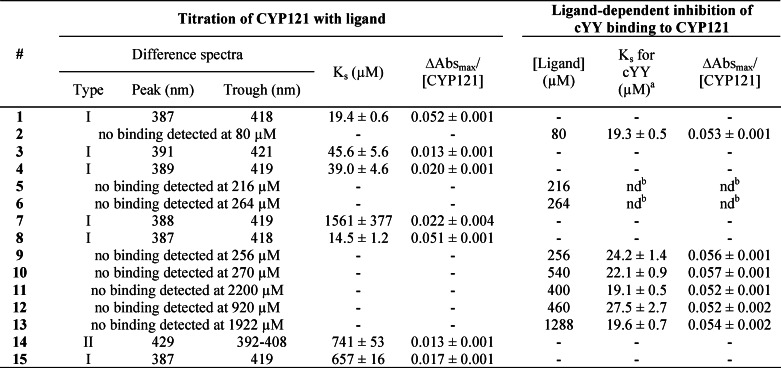
a The final DMSO concentration in the cuvette was between 1 and 1.5% (1.5% for 12).
b nd, not determined. Ks could not be determined because of light scattering after the addition of the compound to the cuvette even at lower concentrations of the compound or in the presence of 1% DMSO from the beginning of the titration.
Very little binding or no binding was observed for compounds comprising one aliphatic side chain, cyclo(l-Tyr-l-Leu) (5), cyclo(l-Tyr-l-Met) (6), and cYA (7). The binding constant of cYY for CYP121 in the presence of 5 and 6 could not be determined accurately because of light scattering (Table 3). However, spectral variations were observed upon the addition of a micromolar concentration of cYY to a CYP121 solution in the presence of 5 and 6 at 216 and 264 μm, respectively. Compounds cYF (3), cYW (4), and cY-DOPA (8), which have two aromatic side chains, exhibited significant binding to CYP121 with Ks values in the same range as those for cYY. These three compounds showed substrate-like binding with low to high spin transition as indicated by a peak around 387–390 nm and a trough around 417–420 nm on difference spectra. Binding of cY-DOPA to CYP121 was indistinguishable from that of cYY, except for a slightly lower Ks value. There were small differences in wavelength values between spectra for cYY binding and those for cYF and cYW binding. The intensity of the spin transition for these two compounds was significantly lower than that for cYY (75 and 61% lower, respectively). The Eadie-Hofstee representation of the data indicated the singularity of cYW binding to CYP121 (data not shown). Data for cYW binding appear to fit two straight lines of different slopes better than a single straight line, suggesting the possibility of two binding sites. There was similar, but weaker, evidence of two binding sites in the data for cYY and cYF but not for cY-DOPA. No evidence for binding of 9 or 10 to CYP121 was obtained, in either titration experiments or inhibition studies, indicating that changing the stereochemistry of one or two Cα atoms of cYY greatly impaired binding.
We then studied the role of the DKP ring with a range of molecules carrying the two tyrosyl side chains presented either on a linear peptide (12, 13, and 14) or on another cyclic scaffold (11 and 15). No binding was observed for 11 that possesses a non-peptide scaffold, in either titration experiments or inhibition studies. For the linear peptides, direct binding was detected only for 14 but with the Ks value being 38 times as high as that for cYY. Furthermore, type II binding was observed, indicating a heme coordination through a nitrogen atom. The possibility of 12 binding to CYP121 cannot be excluded, because a 40% increase of the Ks value of cYY for CYP121 was observed in inhibition studies. Nevertheless, a large amount of 12 was used (>20 fold the Ks value), suggesting that the binding, if there was any, was much weaker than that of cYY for CYP121. The reduction of one keto function of the DKP ring of cYY to CH2 greatly impaired binding to CYP121, with a 34-fold increase of the Ks value. Type I binding similar to that involving cYY was observed, but the calculated value for overall absorbance variation was significantly lower, suggesting a different binding type.
CYP121 Specifically Transforms cYY
Mycocyclosin 2 was identified as the major product of the activity of CYP121 on cYY 1 (12), but the formation of minor products was not addressed. To provide a more complete description, we reinvestigated the transformation of cYY by CYP121, focusing on minor products. We used tandem mass spectrometry coupled to reverse phase HPLC (LC-MS/MS) to analyze the reaction after a 1-h incubation (Table 4, substrate = 1). In addition to mycocyclosin 2, several other metabolites were found in small amounts (each <2%). Analysis of the MS2 spectra of the metabolites characterized by the [MH]+ ion at m/z 302.1 and 316.0 was mostly uninformative about the nature of these compounds. The metabolite characterized by a retention time of 30.8 min and a [MH]+ ion at m/z 325.0 displays an MS2 spectrum sharing similarities with that of mycocyclosin (data not shown). The MS2 spectrum of the metabolite with an [MH]+ ion at m/z 341.0 revealed the same neutral losses as those observed for mycocyclosin (data not shown), suggesting a possible hydroxylation of mycocyclosin during the reaction. Finally, a targeted search for metabolites at m/z 343.0 ± 0.5 (+16 mass increase relative to cYY) was unsuccessful, such that hydroxylation of cYY was not detected.
TABLE 4.
Metabolites detected in a CYP121 activity assay using compound 1, 3, 4, 7, 8, or 15 as substrate
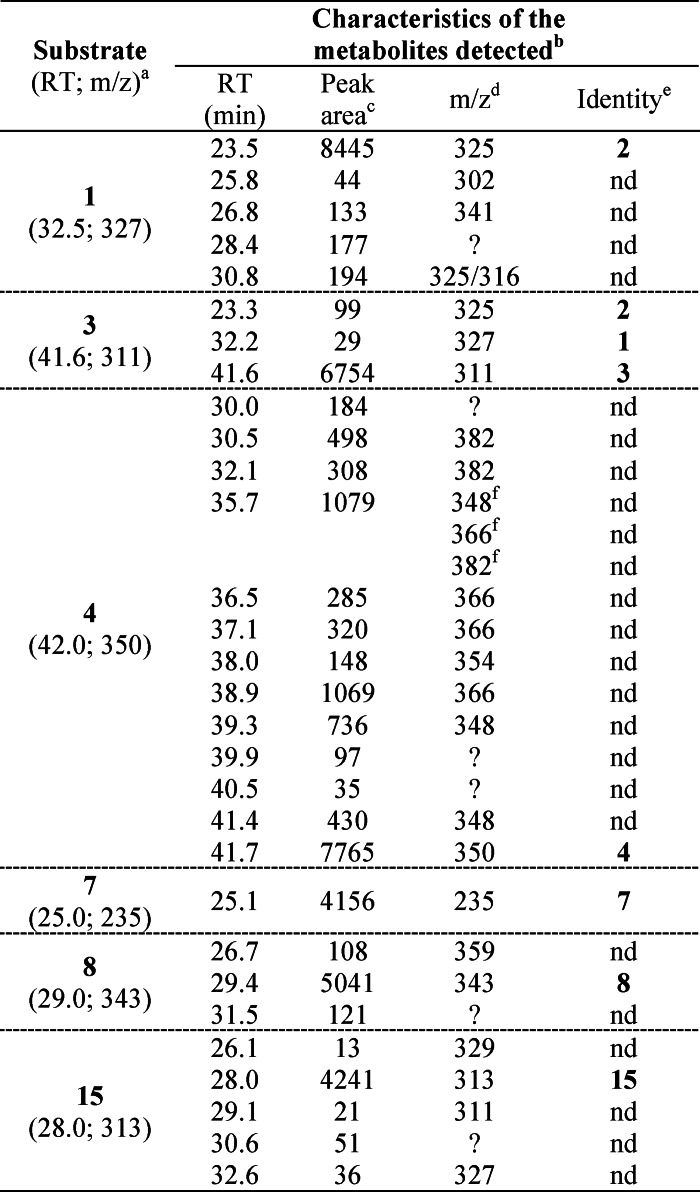
a The retention times in min (RT) and m/z values for the [MH]+ ion are indicated in parentheses.
b Reported metabolites were not detected in an assay in the absence of CYP121, apart from the remaining substrate and from the assay conducted with 4 (see “Results”).
c Peak areas were obtained from chromatograms recorded at 214 nm.
d m/z values correspond to the [MH]+ ion. Question marks indicate that no value could be obtained.
e Identity was attributed when retention time, m/z value, and MS2 spectra were identical to those of one of the compounds described in this study. nd, identity of the metabolite could not be determined.
f Metabolites with the [MH]+ ion at m/z 348, m/z 366, and m/z 382 were observed in the same peak characterized by a retention time of 35.7 min.
We next investigated the transformation by CYP121 of compounds inducing the low to high spin transition (cYF, cYW, cYA, cY-DOPA, and 15) (Fig. 3 and Table 4). Compound 4 (cYW) was the only substrate analog that was significantly consumed by CYP121 in activity assays; ∼50% of the initial compound remained after a 1-h incubation with CYP121, whereas 95% remained in the absence of CYP121. More than 10 different metabolites appearing during the reaction catalyzed by CYP121 are observed, but no major metabolite could be identified (Table 4). Analysis of ion current chromatograms revealed molecular weight differences of +16, +32, +4, and −2 relative to cYW. These results indicate the lack of specificity of CYP121 for metabolite formation from cYW. Some of these metabolites were also detected in an assay conducted in the absence of CYP121, but in much lower amounts, suggesting the propensity of cYW to be transformed.
FIGURE 3.

CYP121 activity on substrate analogues. The transformation by CYP121 of cYY (1) and of substrate analogues (3, 4, 7, 8, and 15) was assessed in coupled enzymatic reactions as described under “Experimental Procedures” and analyzed by LC-MS/MS. Chromatograms were recorded at 214 nm, and the percentage of remaining substrate analog in the reaction was estimated by measuring peak area. Results are presented for each compound according to the time of the reaction.
The rate of transformation of cYF (3) was very slow, with ∼98% of compound remaining after 1 h of incubation (100% in the control reaction without CYP121). Analysis of the ion current chromatograms obtained for the 60 min samples indicated the presence of two metabolites with retention time, m/z values, and MS2 spectra identical to those of cYY and mycocyclosin. This indicates that CYP121 may hydroxylate cYF, albeit at very low rate.
Transformation of cY-DOPA (8) by CYP121 was very slow; 91% of the compound remained after 1 h (100% in the control without CYP121). A first metabolite appeared during the reaction with a retention time ∼2 min lower and an m/z value for [MH]+ 16 units higher than the initial product, suggesting a possible hydroxylation. A second peak appeared at a retention time of 31.5 min for which no m/z value could be determined. No compound with a m/z value for the [MH+] ion at 341 (2-unit decrease) was observed, showing that the major reaction catalyzed by CYP121 on cYY did not occur with cY-DOPA.
For cYA (7), no new metabolite was detected upon incubation with CYP121, consistent with the absence of consumption observed (Fig. 3).
Conversion of 15 by CYP121 was very inefficient; 98% of the compound remained after 1 h (100% remained in the control without CYP121). Three additional compounds appeared specifically in the presence of CYP121, and each accounted for less than 1% of 15.
Crystal Structures of cYY Analog-bound CYP121 Reveal a Common Binding Mode
The crystal structures of CYP121 bound to cYF, cYW, cYA, cY-DOPA, and 15 were determined. Data collection and refinement statistics are summarized in Table 1. Independent of the nature of the molecule bound to CYP121, crystals were grown as previously described and were found to belong to the same crystal form as crystals of native CYP121 used for the atomic resolution of the CYP121 structure (16). The various structures were solved and refined at high resolution (1.2–1.9 Å resolution) such that the quality of the structures is compatible with fine structural analysis. The omit maps calculated from the data showed unambiguous density corresponding to the compound bound to CYP121 (Fig. 4). One single molecule was modeled in all cases except for cYW, for which two alternate conformations are observed. Each overall structure of ligand-bound CYP121 is highly similar to that of native CYP121 (Protein Data Bank entry 1N40) with root mean square deviations on equivalent Cα atoms of 0.177, 0.112, 0.169, 0.133, and 0.099 Å for complexes of CYP121 bound to cYF, cYW, cYA, cY-DOPA, and 15, respectively (determined for 381, 359, 374, 354, and 359 equivalent Cα atoms, respectively). Furthermore, for each structure solved, atoms of the side chains of the residues that constitute the active site superimpose well on the equivalent atoms in native CYP121 (Protein Data Bank entry 1N40).
FIGURE 4.

Bound ligands in CYP121 crystal structures. 2Fo − Fc omit maps at 1 σ level (blue mesh) of the ligand molecule found in 4ICT (A), 4IQ9 (B), 4IQ7 (C), 4IPW (D), and 4IPS (E) are shown. The ligands are superimposed as stick models with carbon atoms in yellow or orange (alternate conformations for B and C), oxygen atoms in red, and nitrogen atoms in blue.
In each structure, the ligand binds at the previously described cYY-binding site (Fig. 5). There was thus a common binding mode, similar to that identified for cYY. It involves van der Waals interactions of one aryl side chain with Phe168 and Trp182, and stacking of the DKP ring against the 310 helix and B′ helix with one of its carbonyls hydrogen-bonding the Nδ2 of Asn85, except for conformation 2 of cYW (Fig. 5). For each structure, the sixth ligand of the heme iron is not displaced and is positioned between 2.3 and 2.7 Å from the iron (Table 5). As in the cYY-CYP121 complex, the iron remains in the plane of the heme. Details of the interactions established by each compound are given in Fig. 6. The major specific features of each compound bound to CYP121 are as follows.
FIGURE 5.
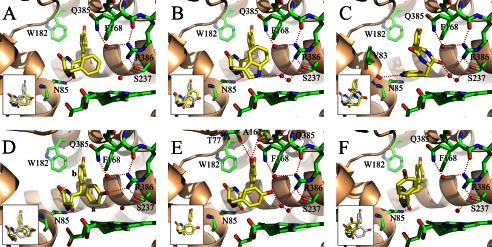
Crystal structures of CYP121 bound to 3 (A), 4 (B and C), 7 (D), 8 (E), and 15 (F). Detailed views of the active site are presented. The protein backbone is in a schematic representation colored in wheat. Residues of the active site and the heme are in a stick representation colored in green with oxygen, nitrogen, and iron atoms in red, blue, and orange, respectively. The ligand is in a stick representation with carbon, oxygen, and nitrogen atoms colored in yellow, red, and blue, respectively. Water molecules are shown as red spheres. Red dashed lines, possible hydrogen bonds. The two conformations observed for the tyrosyl side chain of 7 are noted as a and b in D. In each panel, the inset shows the superimposition of the CYP121-bound ligand (yellow carbons) over the CYP121-bound cYY (white carbons), obtained by superimposition of each overall structure presented herein with the crystal structure of cYY-bound CYP121 (Protein Data Bank entry 3G5H).
TABLE 5.
Distances between the heme iron and the fifth and sixth ligand in CYP121 crystal structures
| Bound ligand | Protein Data Bank entry | Resolution | Distances |
|
|---|---|---|---|---|
| S–Fe | Fe–6th ligand | |||
| Å | Å | |||
| None | 1N40 | 1.06 | 2.4 | 2.1 |
| None | 3G5F | 1.4 | 2.4 | 2.5 |
| 1 | 3G5H | 1.4 | 2.4 | 2.4 |
| 3 | 4ICT | 1.8 | 2.3 | 2.7 |
| 4 | 4IQ9 | 1.4 | 2.3 | 2.3 |
| 7 | 4IQ7 | 1.9 | 2.3 | 2.7 |
| 8 | 4IPW | 1.4 | 2.3 | 2.4 |
| 15 | 4IPS | 1.2 | 2.3 | 2.3 |
FIGURE 6.

Schematic representation of polar contacts (A) and non-polar contacts (B) made by ligands in the presence of CYP121. For compounds 4 and 7, the conformation is indicated in parentheses (1 and 2 for 4, a and b for 7). A, dashed lines indicate hydrogen bonds with interatomic distances given in Å. w, water molecule. L6, sixth ligand of the heme iron. B, dashed lines, non-covalent interactions (van der Waals, hydrophobic) between atoms closer than 5 Å. Lines starting from the center of an aromatic ring mean that each atom constituting the ring participates in non-covalent interactions. For clarity, the names of the atoms of CYP121 residues are omitted. The distances in Å between the heme iron (Fe) and the closest ligand atoms are indicated beside double arrows.
The position adopted by 3 (cYF) in the CYP121 binding site was with the tyrosyl residue pointing toward helices F and G and the phenylalanyl moiety pointing toward the heme and facing the DKP ring in positions highly similar to those observed for cYY (Fig. 5A). The ring approaching the heme, however, was closer to the heme iron; the distance between the Cϵ1 carbon atom and the iron is 5.3 Å (6.1 Å between the equivalent atoms in the CYP121-cYY complex). Furthermore, no hydrogen-bonded water molecule network connected the ligand and the protein at the oxygen binding site of the heme. This structure highlights the importance of the missing hydroxyl for establishment of the hydrogen-bond network above the heme and for the positioning of the substrate.
Two alternate conformations were observed at the same CYP121 binding site for 4 (cYW; Fig. 5, B and C). Conformation 1 (occupancy of 0.25) was very similar to that of cYY, the so-called canonical ligand conformation (Fig. 5B, inset). In this conformation, the tyrosyl side chain points toward helices F and G but does not occupy exactly the same position as those observed for cYY or cYF. The tryptophan moiety faces the DKP ring and is located above the heme. The Nϵ1 of tryptophanyl is the ligand atom closest to the heme iron, located at 5.6 Å. It interacts with the iron through the two water molecules WAT2 and WAT1 (distances between the heteroatoms of NH/WAT2/WAT1 and iron are 3.1, 2.7, and 2.3 Å, respectively). In this conformation, the tryptophanyl comes in close proximity to Phe168, with a distance of 3.2 Å between the CH2 carbon atom of cYW and the Cϵ2 carbon atom of Phe168. The χ1 angle of Phe168 was slightly different from that observed in native CYP121 (−69.8 and −75.1°, respectively), resulting in the side chain being positioned slightly away from the ligand binding cavity and closer to the Leu174 side chain. In the second conformation, the tyrosyl ring occupies roughly the same place, but the tryptophan and DKP rings interchange through a rotation of about 180° along the elongation axis of the compound (Fig. 5C). As a consequence, the DKP ring is located above the heme, with the oxygen atom of one carbonyl interacting with the iron through two tightly hydrogen-bonded water molecules, WAT2 and WAT1 (distances between the heteroatoms of C=O/WAT2/WAT1 and iron are 2.7, 2.6, and 2.3 Å, respectively). The tryptophan positions with the indole ring parallel to the heme, facing the DKP ring and packing against the 310 helix. Interestingly, the ionic interaction between one carbonyl of the DKP and Asn85-Nδ2 conserved in other structures was substituted by an ionic bond between the Nϵ1 of the tryptophan and the carbonyl of Val83.
In the structure of the CYP121-7 (cYA) complex, the DKP ring and the Cβ carbon atom of the tyrosyl side chain are well defined, but two alternative conformations of the tyrosyl side chain could be modeled (Fig. 5D). In one of the conformations, the tyrosyl points toward helices F and G similarly to the equivalent group in cYY. In the second conformation, the tyrosyl side chain points toward Phe280 facing the DKP ring. The hydroxyl is positioned just above the heme at 3.0 Å from one nitrogen of the guanidinium of Arg386, and it interacts with Gln385 through one water molecule. Its position is very close to that of the hydroxyl of cYY (the distance between the two hydroxyls is 0.8 Å) (Fig. 5D, inset). No electron density, however, was observed between cYA and the sixth ligand of the heme iron.
In the crystal structure of CYP121 in complex with 8 (cY-DOPA), the ligand positions at the same location as cYY (Fig. 5E). The only difference with cYY is associated with the additional hydroxyl group; it is involved in two direct hydrogen bonds with Thr77-Oγ1 (3.2 Å) and the oxygen atom of the carbonyl of Ala167 (3.1 Å). All of these observations are consistent with the spectral variations observed upon CYP121 titration with cY-DOPA.
Compound 15 positions in the CYP121 binding site with one tyrosyl pointing toward helices F and G and the ketopiperazine ring at a position similar to that observed for the DKP ring of other ligands (Fig. 5F). The major difference with other ligands involves the second tyrosyl that does not face the ketopiperazine ring; the χ1 angle associated with this tyrosine is 165.1°, whereas the angle in the case of cYY is 63.8°. As a consequence, this tyrosyl ring occupied a position that was not observed for any other compound studied. The weaker electron density and the higher B factor of the ring (19 Å2 instead of 14.7 Å2 for the tyrosyl ring pointing toward helices F and G) suggest that the occupancy of the ring may not be 100%. Similarly, the tyrosyl ring is located close to Met62, at 2.9–3.1 Å and 4.1 Å from the minor (30%) and major (70%) alternate conformations of Met62, respectively. This suggests that the major conformation for Met62 is the conformation with the tyrosyl ring of 15 occupying its visible position, whereas the minor conformation for Met62 is stable when this tyrosyl is elsewhere. Nevertheless, no clear alternate conformation of 15 was visible in the electron density. In particular, there was no electron density indicating that the tyrosyl ring might adopt the canonical position facing the piperazinone ring with the hydroxyl directed toward Arg386. At the oxygen-binding site, the network of hydrogen-bonded water molecules observed in the structure of cYY-bound CYP121 is largely disrupted. In particular, there was a large electron density bulb whose center was located 3.5 Å from the sixth ligand of the heme iron.
Solution NMR Spectroscopy of Ligands Reveals Rapid Equilibrium between Different Conformations
Changes in ligand conformation upon binding may make a large contribution to affinity (29). The compounds that we studied are particularly flexible, in part due to the rotation of the aryl side chains around the Cα–Cβ atom, and consequently, elucidation of the conformations these compounds adopt in solution may help to identify determinants of binding. Therefore, we used NMR spectroscopy to analyze the conformations in solution of 3, 4, 5, 6, 7, 8, 9, 10, and 15. In particular, the populations of the different χ1 rotamers of aromatic residues were determined from the vicinal homonuclear 3JHα-Hβ and heteronuclear 3JHα-Cγ and 3JHβ-CO coupling constants. The 3JHα-Hβ coupling constants measured in one-dimensional spectra for 1, 5, 7, and 15 differed only slightly between solutions in water and in DMSO, indicating that the solvent has little influence on the conformation of these compounds. The following conformational studies were therefore carried out mostly in DMSO because of the poor solubility of these compounds in water. Cyclic dipeptides 5, 6, and 7, each with an aliphatic residue, showed a major conformer for the tyrosyl side chain in solution (∼80% gauche+, ∼20% trans χ1 rotamers). This major gauche+ conformation corresponds to an orientation of the aromatic side chain above the DKP ring, indicating a favorable interaction between the aromatic and DKP rings, as described previously (30). The conformational preferences of the heterochiral compound 9 were similar, with the major conformer corresponding to the two aromatic rings pointing to the two faces of the DKP ring. In 1, 3, 4, 8, and 10, the two aromatic side chains have similar χ1 rotamer distributions and show a smaller gauche+ population (∼50% gauche+, ∼35% gauche−, ∼15% trans), indicating that the aromatic rings compete for orientation toward the DKP ring. There is no preference for tyrosyl, dihydroxyphenylalanyl, or tryptophanyl aromatic rings in the gauche+ conformer.
Conformational analysis of 15 indicated an equilibrium between two non-planar half-chair conformations of the piperazinone ring, as indicated by the vicinal coupling constants of the methylene group in the ring. The tyrosyl side chains do not have preferred conformations.
DISCUSSION
CYP121 is a P450 from M. tuberculosis that has drawn attention for several reasons. The organization of its active site and features of the reaction it catalyzes suggest novel mechanisms (12, 16), and it is also essential in vivo such that it is a potential target for the development of novel antimycobacterial agents (11, 28, 31). Here, we report an investigation of the substrate and reaction specificities of CYP121 involving synthesizing substrate analogues and studying their binding to and transformation by CYP121.
The major conclusion from these experiments is that CYP121 is a highly specific P450; it did not efficiently or selectively transform the analogues tested. We show that the molecular basis of this specificity is primarily the control of ligand binding. Indeed, only three of the 14 substrate analogues tested efficiently bound to CYP121 (3, 4, and 8). For these three compounds, the binding mode was essentially the same as that of cYY: the DKP ring facing the 310 helix with hydrogen bonding between one carbonyl and the side chain of Asn85 and one aryl side chain positioned for π-stacking interactions with Phe168 and Trp182 (see Fig. 3) (12). Solution NMR spectroscopic analysis of ligands indicated that the presence of only one aryl side chain anchored on the DKP ring (5, 6, and 7) favors a conformation detrimental for efficient binding with the aryl side chain facing the DKP ring. This preferential conformation was indeed observed for compound 7 bound to CYP121 (see Fig. 3D). One consequence of this preferential conformation is that π-stacking interactions with Phe168 and Trp182 are prevented. The binding of cyclodipeptides to CYP121 is also conditioned by the stereochemistry of the Cα atom; compounds 9 and 10, which are stereoisomers of the CYP121 substrate cYY, failed to bind. Solution NMR spectroscopy showed that the preferential conformation of 9 corresponds to the tyrosyl side chains facing the two sides of the DKP ring; this causes substantial steric hindrance to binding to CYP121. In the case of 10, for which both Cα atoms were in the d-configuration and there was no binding to CYP121, the tyrosyl side chains are predicted to position on the same side of the DKP ring as for cYY. One noticeable difference between 10 and cYY, however, is the positions of the C=O and NH functions of each amino acid in the DKP ring with respect to the corresponding Cα–Cβ bond (see Fig. 1). The crystal structure of cYY-bound CYP121 shows one hydrogen bond between one carbonyl of the DKP ring of cYY and the side chain of Asn85. According to these observations, it appears that 10 cannot bind to CYP121 in the same way as cYY with both the π-stacking interactions described above and hydrogen bonding between Asn85 and the DKP ring. Our results clearly indicate that the integrity of the DKP ring is a key feature of cyclodipeptide binding: its replacement (11), its opening (12, 13, and 14), or even the reduction of one of its keto functions (15) substantially reduced binding to CYP121. A comparison of the crystal structures of cYY-bound CYP121 and 15-bound CYP121 does not reveal obvious contacts between the ligand and the protein that could explain the 32-fold lower affinity of 15 (Table 3 and Fig. 6). The conformations of the ligands in solution may contribute to CYP121 binding. Indeed, solution NMR spectroscopy findings for the ligands lead us to suggest that the DKP ring in cYY restricts the conformational space of tyrosyl side chains, leading to conformations more favorable for CYP121 binding than those found in 15. This restricted chemical space for efficient binding to CYP121 is consistent with the previously observed rigidity of the CYP121 active site cavity (16).
We show that, in addition to this specificity of binding, CYP121 exhibits further stringent requirements for catalysis; four of the five compounds (3, 7, 8, and 14) that are able to bind to CYP121 are transformed only poorly or not at all. For example, the relationships of cYF (3) and cYA (7) with CYP121 illustrate the requirement for not only the presence (cYF) but also the correct positioning (cYA) of the hydroxyl above the heme for efficient transformation. Its absence or incorrect positioning impairs the low to high spin transition concomitant with the modification of the hydrogen bond network involving the sixth ligand of the heme iron, showing the importance of this network to the heme iron spin state. This is consistent with previous observations suggesting a possible change in spin state for CYP101A through modification of the pKa value of the sixth ligand, influencing its OH−/H2O equilibrium (32–34). In the case of CYP121, the loss of the hydrogen bond network may have a similar effect, with the associated reduction of spin transition. The importance of spin transition in the P450 catalytic cycle is associated with an increase of the heme iron oxidation/reduction potential that may be necessary for rapid reduction of P450s and the continuation of the catalytic cycle (4, 35). In the case of CYP121, the lower spin transition observed when 3 or 7 is bound cannot alone account for the very low transformation rate, because the spin transition remains relatively high (25 and 42% of that observed with cYY, respectively). More likely, the hydroxyl group of cYY specifically favors the reaction and the nature of the transformation. Indeed, no intramolecular C–C bond could be detected for 3 transformation, and the only product identified was hydroxylated 3. Arene hydroxylation by P450 reactive species compound I implies that the π-system is initially attacked by the activated oxygen of compound I (36, 37). In the case of 3, the iron is too far from the site of hydroxylation for such a reaction, and this may explain the low efficiency. It was suggested recently that cis-trans isomerization of the Val78–Pro79 peptide bond in CYP121 could trigger repositioning of the substrate, bringing it close to the heme iron (38). If so, it is clear that this mechanism is not sufficient to allow hydroxylation of 3. Finally, the poor transformation of 3 is consistent with a CYP121 mechanism initiating with either hydrogen abstraction or electron transfer; the observed distances between the ligand and the iron are compatible, and density functional theoretical calculations predict that such mechanisms would have high energy intermediates, making them unlikely in the case of a benzene moiety (36). The phenol moiety of cYY is much more favorable for such hydrogen abstraction or electron transfer than the benzene moiety of 3 (39).
One exception to the poor transformation of the compounds tested was 4 (cYW), which was significantly metabolized by CYP121. More than 10 different products were detected, so the reaction was not specific. This absence of specificity is probably due to the presence of a tryptophan residue, because such residues are particularly sensitive to oxidation under various conditions (40). The reactive species OH• and H2O2 react with tryptophan residues to form various oxidation products (41–44). These reactive species can be formed during the P450 catalytic cycle when O2 reduction is not coupled to substrate oxidation (45). Furthermore, indoyl radicals, which may be formed during the reaction with CYP121 by hydrogen abstraction, can react to form various oxidation products (46). Nevertheless, CYP121 and 4 are clearly not tailored to react together efficiently. Thus, CYP121 is a selective P450, specifically catalyzing the oxidation of cYY.
This demonstration of the selectivity of CYP121 has implications for its function in vivo. Indeed, the genetic association between the CYP121 gene and rv2275, which encodes a cyclodipeptide synthase producing cYY and also various amounts of 3, 4, 5, 6, and 7 (10.8, 0.8, 0.7, 1.0, and 10.9%, respectively, of the total cyclodipeptides synthesized) raises questions about the possible transformation of these cyclodipeptides by CYP121 and the function of CYP121 in vivo (12–14). The selectivity of the reaction catalyzed by CYP121 suggests that the Rv2275/CYP121 tandem mostly synthesizes and transforms cYY in vivo. This transformation occurs almost exclusively through C–C coupling with very few minor products. In particular, we were unable to detect any hydroxylation of cYY. However, other P450s can hydroxylate phenol to catechol (47–50), and the positioning of cYY in the CYP121 active site is consistent with such a reaction (12). Interestingly, compound 8, a hydroxylated cYY, binds more efficiently than cYY to CYP121 but is poorly transformed. Consequently, if CYP121 were able to synthesize 8 by hydroxylation of cYY, it appears that the product of the reaction would inhibit the enzymatic activity. Mycocyclosin (2), the natural product of CYP121 activity on cYY, shows no such end product inhibition. These results are consistent with CYP121 being essential for viability in vivo (11).
The cyp121 gene is essential for mycobacterial growth, so inhibiting CYP121 is a promising approach to treating tuberculosis (11, 31). Our findings contribute to the development of inhibitors with potential clinical applications. We demonstrate the stringent requirement for the DKP ring for cyclodipeptide binding to CYP121. Fortuitously, the DKP ring is an attractive scaffold for medicinal chemistry and is commonly found in compound libraries (51). Our results suggest that a DKP ring bearing two aryl side chains, as in 8, may serve for anchoring various other structures to the CYP121 binding cavity. This strategy could be used to identify novel chemical functions for binding to CYP121, circumventing the high specificity of binding of CYP121.
This work was supported by the Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA), CNRS, and grants from the Région Ile-de-France. The Service d'Ingénierie Moléculaire des Protéines is a member of the Laboratory of Excellence LERMIT.
- P450
- cytochrome P450 enzyme
- cYY
- cyclo(l-Tyr-l-Tyr)
- DKP
- diketopiperazine
- RP-HPLC
- reverse phase HPLC
- Ks
- apparent spectral dissociation constant
- cYF
- cyclo(l-Tyr-l-Phe)
- cYW
- cyclo(l-Tyr-l-Trp)
- cYA
- cyclo(l-Tyr-l-Ala)
- cY-DOPA
- c(l-Tyr-l-3,4-dihydroxyphenylalanine)
- rms
- root mean squared
- RT
- retention time
- amu
- atomic mass unit.
REFERENCES
- 1. Nelson D. R. (2009) The cytochrome p450 homepage. Hum. Genomics 4, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650 [DOI] [PubMed] [Google Scholar]
- 3. Ortiz de Montellano P. R., De Voss J. J. (2005). in Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd Ed (Ortiz de Montellano P. R., ed) pp. 183–245, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 4. Denisov I. G., Makris T. M., Sligar S. G., Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 [DOI] [PubMed] [Google Scholar]
- 5. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 6. van den Boogaard J., Kibiki G. S., Kisanga E. R., Boeree M. J., Aarnoutse R. E. (2009) New drugs against tuberculosis. Problems, progress, and evaluation of agents in clinical development. Antimicrob. Agents Chemother. 53, 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmad Z., Sharma S., Khuller G. K. (2005) In vitro and ex vivo antimycobacterial potential of azole drugs against Mycobacterium tuberculosis H37Rv. FEMS Microbiol. Lett. 251, 19–22 [DOI] [PubMed] [Google Scholar]
- 8. Ahmad Z., Sharma S., Khuller G. K., Singh P., Faujdar J., Katoch V. M. (2006) Antimycobacterial activity of econazole against multidrug-resistant strains of Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 28, 543–544 [DOI] [PubMed] [Google Scholar]
- 9. McLean K. J., Cheesman M. R., Rivers S. L., Richmond A., Leys D., Chapman S. K., Reid G. A., Price N. C., Kelly S. M., Clarkson J., Smith W. E., Munro A. W. (2002) Expression, purification and spectroscopic characterization of the cytochrome P450 CYP121 from Mycobacterium tuberculosis. J. Inorg. Biochem. 91, 527–541 [DOI] [PubMed] [Google Scholar]
- 10. McLean K. J., Marshall K. R., Richmond A., Hunter I. S., Fowler K., Kieser T., Gurcha S. S., Besra G. S., Munro A. W. (2002) Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology 148, 2937–2949 [DOI] [PubMed] [Google Scholar]
- 11. McLean K. J., Carroll P., Lewis D. G., Dunford A. J., Seward H. E., Neeli R., Cheesman M. R., Marsollier L., Douglas P., Smith W. E., Rosenkrands I., Cole S. T., Leys D., Parish T., Munro A. W. (2008) Characterization of active site structure in CYP121. A cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 283, 33406–33416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belin P., Le Du M. H., Fielding A., Lequin O., Jacquet M., Charbonnier J. B., Lecoq A., Thai R., Courçon M., Masson C., Dugave C., Genet R., Pernodet J. L., Gondry M. (2009) Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 106, 7426–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vetting M. W., Hegde S. S., Blanchard J. S. (2010) The structure and mechanism of the Mycobacterium tuberculosis cyclodityrosine synthetase. Nat. Chem. Biol. 6, 797–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gondry M., Sauguet L., Belin P., Thai R., Amouroux R., Tellier C., Tuphile K., Jacquet M., Braud S., Courçon M., Masson C., Dubois S., Lautru S., Lecoq A., Hashimoto S., Genet R., Pernodet J. L. (2009) Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat. Chem. Biol. 5, 414–420 [DOI] [PubMed] [Google Scholar]
- 15. Roback P., Beard J., Baumann D., Gille C., Henry K., Krohn S., Wiste H., Voskuil M. I., Rainville C., Rutherford R. (2007) A predicted operon map for Mycobacterium tuberculosis. Nucleic Acids Res. 35, 5085–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leys D., Mowat C. G., McLean K. J., Richmond A., Chapman S. K., Walkinshaw M. D., Munro A. W. (2003) Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 Å reveals novel features of cytochrome P450. J. Biol. Chem. 278, 5141–5147 [DOI] [PubMed] [Google Scholar]
- 17. Bogatcheva E., Hanrahan C., Nikonenko B., Samala R., Chen P., Gearhart J., Barbosa F., Einck L., Nacy C. A., Protopopova M. (2006) Identification of new diamine scaffolds with activity against Mycobacterium tuberculosis. J. Med. Chem. 49, 3045–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Borch R. F., Bernstein M. D., Dupont Durst H. (1971) Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 93, 2897–2904 [Google Scholar]
- 19. Jeedigunta S., Krenisky J. M., Kerr R. G. (2000) Diketopiperazines as advanced intermediates in the biosynthesis of ecteinascidins. Tetrahedron 56, 3303–3307 [Google Scholar]
- 20. Martinez J., Bali J. P., Rodriguez M., Castro B., Magous R., Laur J., Lignon M. F. (1985) Synthesis and biological activities of some pseudo-peptide analogues of tetragastrin. The importance of the peptide backbone. J. Med. Chem. 28, 1874–1879 [DOI] [PubMed] [Google Scholar]
- 21. Mizutani H., Takayama J., Honda T. (2005) Enantiospecific total synthesis of TAN1251C and TAN1251D. Synlett 2, 0328–0330 [Google Scholar]
- 22. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 24. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Smart O. S., Vonrhein C., Womack T. O. (2009) BUSTER, version 2.8.0. Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 25. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidt J. M. (2007) Asymmetric Karplus curves for the protein side-chain 3J couplings. J. Biomol. NMR 37, 287–301 [DOI] [PubMed] [Google Scholar]
- 28. Belin P., Moutiez M., Lautru S., Seguin J., Pernodet J. L., Gondry M. (2012) The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways. Nat. Prod. Rep. 29, 961–979 [DOI] [PubMed] [Google Scholar]
- 29. Bissantz C., Kuhn B., Stahl M. (2010) A medicinal chemist's guide to molecular interactions. J. Med. Chem. 53, 5061–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kopple K. D., Marr D. H. (1967) Conformations of cyclic peptides. The folding of cyclic dipeptides containing an aromatic side chain. J. Am. Chem. Soc. 89, 6193–6200 [DOI] [PubMed] [Google Scholar]
- 31. Hudson S. A., McLean K. J., Surade S., Yang Y. Q., Leys D., Ciulli A., Munro A. W., Abell C. (2012) Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem. Int. Ed. Engl. 51, 9311–9316 [DOI] [PubMed] [Google Scholar]
- 32. Jung C. (2007) Leakage in cytochrome P450 reactions in relation to protein structural properties. in The ubiquitous roles of cytochrome P450 proteins, Metal ions in life sciences, Vol. 3 (Sigel A., Sigel H., Sigel R. K. O., eds) pp. 187–234, John Wiley & Sons, Ltd., Chichester, UK [Google Scholar]
- 33. Raag R., Poulos T. L. (1989) The structural basis for substrate-induced changes in redox potential and spin equilibrium in cytochrome P-450CAM. Biochemistry 28, 917–922 [DOI] [PubMed] [Google Scholar]
- 34. Raag R., Poulos T. L. (1991) Crystal structures of cytochrome P-450CAM complexed with camphane, thiocamphor, and adamantane. Factors controlling P-450 substrate hydroxylation. Biochemistry 30, 2674–2684 [DOI] [PubMed] [Google Scholar]
- 35. Guengerich F. P., Johnson W. W. (1997) Kinetics of ferric cytochrome P450 reduction by NADPH-cytochrome P450 reductase. Rapid reduction in the absence of substrate and variations among cytochrome P450 systems. Biochemistry 36, 14741–14750 [DOI] [PubMed] [Google Scholar]
- 36. de Visser S. P., Shaik S. (2003) A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 125, 7413–7424 [DOI] [PubMed] [Google Scholar]
- 37. Korzekwa K. R., Swinney D. C., Trager W. F. (1989) Isotopically labeled chlorobenzenes as probes for the mechanism of cytochrome P-450 catalyzed aromatic hydroxylation. Biochemistry 28, 9019–9027 [DOI] [PubMed] [Google Scholar]
- 38. Pochapsky T. C., Kazanis S., Dang M. (2010) Conformational plasticity and structure/function relationships in cytochromes P450. Antioxid. Redox Signal. 13, 1273–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huynh M. H., Meyer T. J. (2007) Proton-coupled electron transfer. Chem. Rev. 107, 5004–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Simat T. J., Steinhart H. (1998) Oxidation of free tryptophan and tryptophan residues in peptides and proteins. J. Agric. Food Chem. 46, 490–498 [DOI] [PubMed] [Google Scholar]
- 41. Jayson G. G., Scholes G., Weiss J. (1954) Formation of formylkynurenine by the action of x-rays on tryptophan in aqueous solution. Biochem. J. 57, 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kell G., Steinhart H. (1990) Oxidation of tryptophan by H2O2 in model systems. J. Food. Sci. 55, 1120–1123 [Google Scholar]
- 43. Maskos Z., Rush J. D., Koppenol W. H. (1992) The hydroxylation of tryptophan. Arch. Biochem. Biophys. 296, 514–520 [DOI] [PubMed] [Google Scholar]
- 44. Uchida K., Enomoto N., Itakura K., Kawakishi S. (1990) Formation of diastereoisomeric 3a-hydroxypyrroloindoles from a tryptophan residue analog mediated by iron (II)-EDTA and l-ascorbate. Arch. Biochem. Biophys. 279, 14–20 [DOI] [PubMed] [Google Scholar]
- 45. Coon M. J. (2005) Cytochrome P450. Nature's most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol. 45, 1–25 [DOI] [PubMed] [Google Scholar]
- 46. Grossweiner L. I. (1984) Photochemistry of proteins. A review. Curr. Eye Res. 3, 137–144 [DOI] [PubMed] [Google Scholar]
- 47. Koop D. R., Laethem C. L., Schnier G. G. (1989) Identification of ethanol-inducible P450 isozyme 3a (P450IIE1) as a benzene and phenol hydroxylase. Toxicol. Appl. Pharmacol. 98, 278–288 [DOI] [PubMed] [Google Scholar]
- 48. Powley M. W., Carlson G. P. (2001) Cytochrome P450 isozymes involved in the metabolism of phenol, a benzene metabolite. Toxicol. Lett. 125, 117–123 [DOI] [PubMed] [Google Scholar]
- 49. Schlosser P. M., Bond J. A., Medinsky M. A. (1993) Benzene and phenol metabolism by mouse and rat liver microsomes. Carcinogenesis 14, 2477–2486 [DOI] [PubMed] [Google Scholar]
- 50. Stiborová M., Suchá V., Miksanová M., Páca J., Jr., Páca J. (2003) Hydroxylation of phenol to catechol by Candida tropicalis. Involvement of cytochrome P450. Gen. Physiol. Biophys. 22, 167–179 [PubMed] [Google Scholar]
- 51. Horton D. A., Bourne G. T., Smythe M. L. (2002) Exploring privileged structures. The combinatorial synthesis of cyclic peptides. Mol. Divers. 5, 289–304 [DOI] [PubMed] [Google Scholar]