

Background: Cleavage of the influenza HA precursor by host proteases is a critical step in the virus life cycle.
Results: The airway-secreted proteases kallikrein 5 and kallikrein 12 were found to cleave and activate influenza HA.
Conclusion: Each peptidase had distinct preferences for particular human-adapted influenza viruses.
Significance: Identification of these HA-cleaving peptidases aids in our understanding of host proteases involved in influenza infection.
Keywords: Influenza Virus, Kallikrein, Membrane Fusion, Protease, Serine Protease, Hemagglutinin
Abstract
A critical step in the influenza virus replication cycle is the cleavage activation of the HA precursor. Cleavage activation of influenza HA enables fusion with the host endosome, allowing for release of the viral genome into the host cell. To date, studies have determined that HA activation is driven by trypsin-like host cell proteases, as well as yet to be identified bacterial proteases. Although the number of host proteases that can activate HA is growing, there is still uncertainty regarding which secreted proteases are able to support multicycle replication of influenza. In this study, we have determined that the kallikrein-related peptidases 5 and 12 are secreted from the human respiratory tract and have the ability to cleave and activate HA from the H1, H2, and H3 subtypes. Each peptidase appears to have a preference for particular influenza subtypes, with kallikrein 5 cleaving the H1 and H3 subtypes most efficiently and kallikrein 12 cleaving the H1 and H2 subtypes most efficiently. Cleavage analysis using HA cleavage site peptide mimics revealed that the amino acids neighboring the arginine cleavage site affect cleavage efficiency. Additionally, the thrombolytic zymogens plasminogen, urokinase, and plasma kallikrein have all been shown to cleave and activate influenza but are found circulating mainly as inactive precursors. Kallikrein 5 and kallikrein 12 were examined for their ability to activate the thrombolytic zymogens, and both resulted in activation of each zymogen, with kallikrein 12 being a more potent activator. Activation of the thrombolytic zymogens may therefore allow for both direct and indirect activation of the HA of human-adapted influenza viruses by kallikrein 5 and kallikrein 12.
Introduction
Influenza remains a major cause of morbidity and mortality in the human population (1, 2). Influenza A is a member of the Orthomyxoviridae family and commonly circulates in waterfowl, which is thought to be the natural reservoir of the virus (3). In addition to avian species, influenza viruses of the H1, H2, and H3 subtypes can also circulate widely in mammalian species, such as humans, swine, and horses (4). Influenza virus contains eight segmented single-stranded RNA molecules that encode 11–12 proteins. Influenza is classified into subtypes by the distinct antigenicity of the HA and neuraminidase surface proteins. Seventeen HA and nine neuraminidase subtypes have been determined, where some of these subtypes have been transmitted to the human population either by direct transmission (H1N1) or by reassortment (H2N2 and H3N2) (5–7). As a result of transmission to the human population, a number of high morbidity and mortality pandemics have arisen over the past century. The 1918 Spanish influenza pandemic was the most devastating outbreak that resulted in upwards of 20 million deaths (8). In 2009, a new H1N1 influenza virus outbreak arose that has heightened concern for the potential of a high fatality pandemic.
Influenza virus infection is initiated by binding to host cell sialic acid receptors, with the human-adapted subtypes having a preference for α(2,6)-linked sialic acid and avian subtypes preferring α(2,3)-linked sialic acid, but with additional factors necessary for receptor binding (9–12). Once bound, the virus enters the cell by endocytosis, and the increasingly acidic environment of the endosome promotes conformational changes in the HA protein that expose the fusion peptide, allowing fusion between the viral and endosomal membranes (13, 14). The HA protein is synthesized as an inactive precursor and must be cleaved by host proteases to gain its fusogenic properties (15, 16). Specifically, HA cleavage occurs at the C-terminal side of a single arginine residue (Arg-343 in H1 numbering). However, the cleavage site-flanking region can also play a role, as demonstrated by a mutation at the P2 position of HA of the neurotropic mouse-adapted A/WSN/33 virus being a determinant for cleavability by plasmin (17).
Host proteases such as tryptase Clara and several members of the TMPRSS (transmembrane protease, serine) family have been shown to support cleavage activation of HA (18–23). Moreover, thrombolytic proteases such as plasmin, urokinase, and plasma kallikrein have also been shown to cleave and activate viral HA (24). There is, however, uncertainty regarding the secreted proteases normally circulating in the human respiratory tract that support multicycle replication of each of the human-adapted influenza subtypes. Gaining a full understanding of the host proteases involved in cleavage activation of HA would enable development of effective protease-based therapeutics.
The kallikrein-related peptidase family comprises 15 trypsin/chymotrypsin-like secreted serine proteases that are involved in a variety of functions that include vascular permeability and inflammation (25). A number of these peptidases are present in the respiratory tract at various concentrations, and interestingly, the cleavage specificity of some of the kallikrein-related peptidases is homologous to the cleavage site region of influenza virus HA (26). Specifically, the cleavage specificity of the kallikrein-related peptidases 5 (KLK5) and 12 (KLK12) closely resembles the cleavage site region of H1 subtype HA (IQSRX). High mRNA and protein levels of KLK5 have been observed in the skin, breast, ovary, and salivary gland (27, 28). KLK12 expression has been observed in a variety of tissues, including the stomach, brain, uterus, salivary gland, and tracheal epithelial cells (28, 29). Moreover, both peptidases are found at high concentrations in blood serum relative to the other members of the kallikrein family (30). Therefore, it is plausible that these peptidases have the ability to cleave and activate HA and be a part of the protease pool in the respiratory tract that facilitates influenza infection.
In this study, we investigated whether kallikrein 5 and kallikrein 12 have the ability to cleave and activate HA from the human-adapted influenza subtypes to gain a better understanding of the host proteases that can facilitate the multicycle replication of influenza virus in the human respiratory tract. Furthermore, we investigated whether kallikrein 5 and kallikrein 12 cleave and activate thrombolytic zymogens that have previously been shown to support multicycle replication of influenza virus.
EXPERIMENTAL PROCEDURES
Cells, Viruses, and Plasmids
293T, Vero, and Madin-Darby canine kidney cells (American Type Culture Collection) were maintained in DMEM (cellgro®) supplemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin (cellgro®), and 10 units/ml streptomycin (cellgro®). Plasmids encoding A/WSN/33 (H1N1), A/PR/8/34 (H1N1), and A/California/04/09 (H1N1) HA were generated following the methods described by Sun et al. (17). The gene encoding A/Japan/305/57 HA (H2N2) was synthesized by GeneArt and subcloned into pEF4 expression vector. The plasmids encoding A/Wyoming/3/03 HA (H3N2) and A/Wisconsin/67/05 HA (H3N2) were purchased from Sino Biological Inc. The plasmid encoding A/Aichi/2/68 HA (H3N2) was generously donated by David Steinhauer. The A/PR/8/34 HA (H1N1) and A/X-31 HA (H3N2) viruses were propagated in eggs and used to produce non-cleaved virus. Non-cleaved virus was generated by a single replication round in 293T cells, as they do not contain a functional protease capable of cleaving HA. The gene encoding A/WSN/33 HA was mutated by site-directed mutagenesis. The successful introduction of the R343V mutation and the absence of undesired mutations were assessed by sequencing of the entire gene. Peptides MCA-IPSIQSRGL-DNP (H1), MCA-IPSIQYRGL-DNP (H1), MCA-IPSIESRGL-DNP (H2), and MCA-VPEKQTRGL-DNP (H3) (where MCA is (7-methoxycoumarin-4-yl)acetyl, which acts as the donor, and DNP is N-2,4-dinitrophenyl, which acts as the acceptor) were purchased from RS Synthesis.
Proteases
Trypsin from bovine pancreas (tosylphenylalanyl chloromethyl ketone-treated) was obtained from Pierce. Human kallikrein 5 and kallikrein 12 were obtained from R&D Systems. Plasminogen and prekallikrein were obtained from EMD Biosciences. The mouse prourokinase gene (ATCC 63256) was obtained from American Type Culture Collection (31). The cloning, expression, and purification of prourokinase were followed as described by Hamilton et al. (23).
Analysis of HA Cleavage by KLK5 and KLK12
293T cells were grown in polylysine-coated 12-well plates and transfected with 0.8 μg of each HA-expressing plasmid using Lipofectamine 2000 (Invitrogen) for 12 h at 37 °C. The cells were then washed with PBS and incubated with 150 nm KLK5 and KLK12 for 1.5 h at 37 °C. Cells were also incubated in the absence of protease for 1.5 h and with 0.7 μm trypsin for 10 min at 37 °C. In preparation for Western blot analysis, cells were processed by cell surface biotinylation as described by Sun et al. (17). HA cleavage was analyzed by Western blotting using anti-A/PR/8/34 (H1), anti-A/Singapore (H2), and anti-A/Hong Kong/1/68 (H3) antibodies (NIAID Biodefense and Emerging Infections Research Resources Repository).
Determination of the HA Residue of Cleavage by Kallikreins
293T cells were grown in polylysine-coated 12-well plates and transfected with 0.8 μg of a plasmid encoding native A/WSN/33 (WT) HA and a plasmid encoding the R343V mutant of A/WSN/33 HA using the method described above. The cells were then washed with PBS and incubated with 150 nm KLK5 and 750 nm KLK12 for 1.5 h at 37 °C. The cells were also incubated in the absence of protease for 1.5 h and with 0.7 μm trypsin for 10 min at 37 °C as controls. Cleavage was assessed by Western blot analysis using the method described above.
Cell-Cell Fusion Assay
Vero cells were grown in 24-well plates containing a glass coverslip and transfected with 0.5 μg of A/PR/8/34 HA-, A/Japan/305/57 HA-, and A/Aichi/2/68 HA-expressing plasmids using Lipofectamine 2000 for 12 h at 37 °C. The cells were then washed with PBS and incubated with 200 nm KLK5 and KLK12 for 3 h at 37 °C. Cells were also incubated in the absence of protease for 3 h and with 1 μm trypsin for 15 min at 37 °C. The cells were then washed with PBS; treated with 5 mm HEPES, 5 mm MES, 5 mm succinate, and 150 mm NaCl (pH 5.0) for 2 min; and subsequently incubated for 1 h in DMEM at 37 °C. Cell-cell fusion was analyzed by immunofluorescence staining using each anti-HA antibody described above coupled with Alexa Fluor 488 (green)-coupled anti-goat antibody, and the nuclei were stained with Hoechst 33258 (blue; Invitrogen).
Virus Infection Assay
A/PR/8/34 HA and A/X-31 HA viruses were used as prototype H1 and H3 subtype strains, respectively. The reassortant A/X-31 has both HA and neuraminidase genes derived from the A/Hong Kong/1/68 (Aichi strain) virus, and the remaining genes were derived from the A/PR/8/34 H1 virus (32). To produce non-cleaved virus, 293T cells were grown in a 6-cm dish, washed with PBS, and incubated with egg-derived A/PR/8/34 HA (H1N1) and A/X-31 HA (H3N2) (1 pfu/cell) in RPMI 1640 medium for 1 h at 37 °C. The virus solution was then removed, and the cells were washed with PBS and incubated in 3 ml of minimum essential media for 3 days at 37 °C. The resulting virus solution was harvested and treated with 150 nm KLK5 and KLK12 for 1 h at 37 °C. Each virus was also incubated in the absence of protease and with 0.5 μm trypsin for 1 h at 37 °C. Madin-Darby canine kidney cells were grown in a 24-well plate containing a glass coverslip, washed with PBS, and incubated with the virus solution from each condition for 4 h at 37 °C. Virus infection was determined by immunofluorescence staining using rabbit anti-influenza nucleoprotein primary antibody and Alexa Fluor 488-coupled anti-rabbit antibody, and the nuclei were stained with Hoechst 33258 (blue).
Cleavage of Synthetic Peptides by Kallikreins
In a 50-μl total reaction volume, 20 nm each kallikrein and 100 μm each synthetic peptide shown above were incubated for 1 h at 37 °C. The change in fluorescence at 390 nm was observed over the course of the reaction, and a Vmax was obtained by determining the slope of the linear portion of the curve (SPECTRAmax Gemini-XS, Molecular Devices). The Vmax from each reaction was plotted using KaleidaGraph (Synergy Software).
Activation of Thrombolytic Zymogens by Kallikreins
Kallikrein 5 and kallikrein 12 (0.1 ng/μl each) were incubated with 0.2 μg/μl plasminogen, prekallikrein (plasma), and prourokinase in 50 mm Tris and 10 mm CaCl2 (pH 7.4) for 3 h at 37 °C. Zymogen without kallikrein treatment and each kallikrein in the absence of zymogen were incubated at 37 °C for 3 h as controls. Each sample was then incubated with the fluorogenic substrate GGGR-7-amino-4-methylcoumarin (EMD Chemicals) at 150 μm and monitored for the increase in fluorescence at 440 nm over the course of 1 h at 37 °C (SPECTRAmax Gemini-XS). The change in fluorescence over the course of each reaction was plotted using KaleidaGraph, and the Vmax was determined following the method described above.
KLK5 and KLK12 Concentration in Nasal Wash Samples
Nasal wash samples from healthy adults were obtained from subjects participating in a study of live attenuated influenza vaccine prior to vaccination. For sample collection, 5 ml of saline was flushed though each nasal cavity, and the sample was collected by gravity. Four individual nasal samples for KLK5 and eight individual nasal samples for KLK12 were incubated overnight on a Immulon 4HBX plate (Thermo Scientific). Serial dilutions of KLK5 and KLK12 were also incubated on the plate to produce a standard concentration curve. The concentration of each kallikrein was measured by standard ELISA using anti-KLK5 and anti-KLK12 antibodies (R&D Systems) coupled with anti-mouse secondary antibody (Pierce).
RESULTS
Detection of KLK5 and KLK12 in Nasal Wash Samples
To determine whether KLK5 and KLK12 are present in the respiratory tract, the protease concentration from individual human nasal wash samples was assessed by ELISA. The presence of KLK5 and KLK12 was detected in two of the four and two of the eight samples examined, respectively. In cases in which kallikrein was detected, the average concentration was found to be 19 ng/ml for KLK5 and 12 ng/ml for KLK12 (Table 1). Interestingly, the presence of each peptidase in the nasal wash samples differed between individuals, suggesting that the spectrum of proteases able to activate influenza HA might vary from person to person or might be regulated under certain conditions. Ultimately, each peptidase was determined to be present in the nasal wash samples and therefore present in the human respiratory tract.
TABLE 1.
Peptidase concentration in nasal wash samples that were positive for KLK5 and KLK12
| Nasal wash subject | KLK5 | KLK12 |
|---|---|---|
| ng/ml | ng/ml | |
| 1 | 12.74 ± 0.37 | |
| 2 | 11.61 ± 3.65 | |
| 5 | 20.2 ± 7.2 | |
| 7 | 17.6 ± 8.9 |
HA Cleavage Activation by KLK5 and KLK12
To determine whether kallikrein 5 and kallikrein 12 are able to cleave HA, HAs from six strains of the influenza virus H1 subtype, one strain of the H2 subtype, and three strains of the H3 subtype were expressed in mammalian cells and treated with either peptidase. Although not relevant in vivo, trypsin is commonly used as a control in HA cleavage studies because trypsin cleaves most, if not all, HA subtypes and strains efficiently and therefore was used as a control in these experiments. Both KLK5 and KLK12 were able to cleave the HA precursor, forming the HA1 (55 kDa) and HA2 (25 kDa) products, as determined by a similar migration rate as in the trypsin control (Fig. 1). Of note, the migration rates of some HA1 products differed due to differences in the abundance of glycosylation. Cleavage of the H1 subtype strains by KLK5 resulted in a relatively high cleavage efficiency for A/California/04/09 HA and A/WSN/33 HA and a low cleavage efficiency for A/New Caledonia/99 HA, A/South Carolina/18 HA, A/PR/8/34 HA, and A/WS/33 HA (Fig. 1A and Fig. 2). In the case of KLK12, cleavage of the H1 subtype strains was relatively efficient for A/PR/8/34 HA and A/California/09 HA but inefficient for the A/New Caledonia/99 HA, A/South Carolina/18 HA, A/WS/33 HA, and A/WSN/33 HA (Fig. 1B and Fig. 2). For the H2 subtype, a relatively high cleavage efficiency was observed with KLK12, but virtually no cleavage was observed with KLK5 treatment (Fig. 1, C and E, and Fig. 2). Conversely, in the case of the H3 subtype, KLK5 displayed relatively high cleavability for the HA of each virus strain, but essentially no cleavage was observed with KLK12 treatment (Fig. 1, D and F) (Fig. 2). Thus, each peptidase appears to have a preference for particular subtypes and, in addition, a preference for particular strains within the H1 subtype.
FIGURE 1.

Cleavage of the human-adapted HA subtypes by KLK5 and KLK12. A and B, Western blot analysis of HA cleavage by KLK5 (A) and KLK12 (B) of the A/PR/8/34, A/New Caledonia/99 (A/NC/99), A/California/04/09 (A/CA/09), A/South Carolina/18 (A/SC/18), A/WS/33, and A/WSN/33 H1 subtype strains. A/WSN/33 HA in the absence of protease (Mock) or in the presence of trypsin was used as a control. C and E, Western blot analysis of HA cleavage of the A/Japan/305/57 (A/Japan/57) H2 subtype strain by KLK5 (C) and KLK12 (E). D and F, Western blot analysis of HA cleavage by KLK5 (D) and KLK12 (F) of the A/Aichi/2/68 (A/Aichi/68), A/Wyoming/3/03 (A/WY/03), and A/Wisconsin/67/05 (A/WI/05) (H3N2) H3 subtype strains. A/Aichi/2/68 HA in the absence of protease (Mock) or in the presence of trypsin were used as a control.
FIGURE 2.
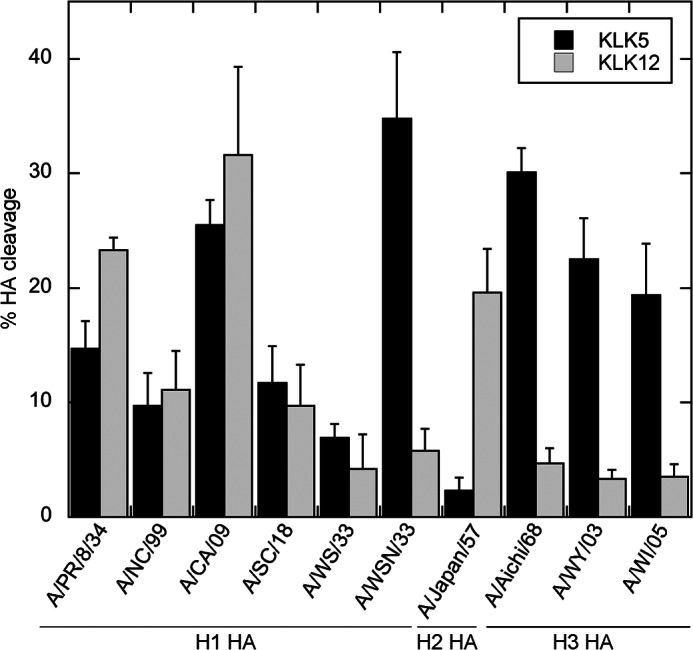
HA cleavage efficiency of KLK5 and KLK12. The bar graph depicts the HA cleavage efficiency of KLK5 (black bars) and KLK12 (gray bars) for all subtypes and strains examined. The percentage HA cleavage was determined by densitometry of the Western blot analysis shown in Fig. 1. A/NC/99, A/New Caledonia/99; A/CA/09, A/California/04/09; A/SC/18, A/South Carolina/18; A/Japan/57, A/Japan/305/57; A/Aichi/68, A/Aichi/2/68; A/WY/03, A/Wyoming/3/03; A/WI/05, A/Wisconsin/67/05.
To ensure that each peptidase was cleaving HA at the correct position, an R343V (H1 numbering) mutant of A/WSN/33 HA was generated and incubated with each protease. As expected, in the case of trypsin, no cleavage was observed with mutation of the Arg cleavage site (Fig. 3). Similarly, HA cleavage by each peptidase was abolished following mutation of the Arg cleavage site (Fig. 3). A relatively large excess (750 nm) of KLK12 was needed to cleave A/WSN/33 HA, consistent with data in Figs. 1 and 2. Although such an excess of KLK12 may not be relevant in vivo, these data confirm that both KLK5 and KLK12 cleave at the expected position on HA, i.e. Arg-343. However, a nonspecific cleavage product was observed for A/South Carolina/18 HA (Fig. 1, A and B), which is consistent with other proteases examined in our laboratory (23). We demonstrated that A/South Carolina/18 HA retains its fusogenic properties despite this nonspecific cleavage product. However, in this case, we were unable to determine whether cleavage by each kallikrein produced fusogenic A/South Carolina/18 HA due to the low cleavage efficiency.
FIGURE 3.
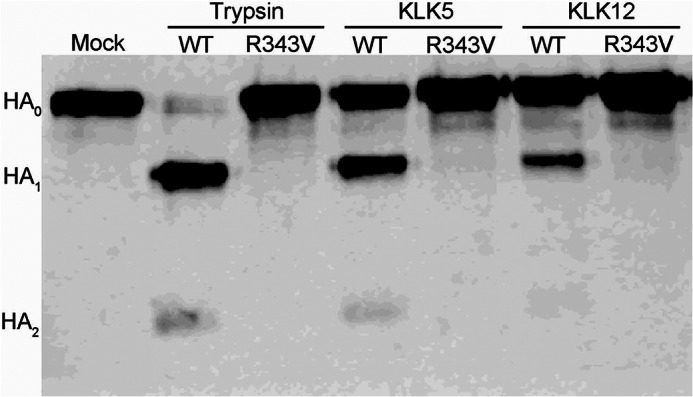
HA cleavage by kallikreins occurs at the proper arginine. Shown are the results from Western blot analysis of HA cleavage of A/WSN/33 HA and its R343V mutant by KLK5 and KLK12. Trypsin was included as a control.
To determine whether cleavage by KLK5 and KLK12 produces fusogenic HA, mammalian cells expressing HA of a representative strain from each subtype were treated with each peptidase and assessed for their fusogenic properties by monitoring syncytial formation (Fig. 4A). Cleavage by KLK5 induced syncytial formation for both the H1 and H3 subtypes, but not the H2 subtype, corresponding to the pattern of cleavage observed by Western blot analysis of HA (Fig. 1A). Likewise, KLK12 induced syncytial formation for the H1 and H2 subtypes, but not the H3 subtype, which also correlates with the Western blot analysis of HA cleavage (Fig. 1B). Quantification of the number of cells involved in syncytia revealed that KLK5 induced syncytial formation of the H3 subtype to a higher degree than the H1 and H2 subtypes, demonstrating a preference for the H3 subtype (Fig. 4B). In the case of KLK12, syncytial formation was the most abundant with treatment of the H1 subtype compared with the other subtypes, demonstrating a preference for the H1 subtype (Fig. 4B).
FIGURE 4.

Cleavage by kallikreins produces fusogenic HA. A, immunofluorescence microscopy of cells expressing A/PR/8/34 HA (upper panels), A/Japan/305/57 HA (middle panels), and A/Aichi/2/68 HA (lower panels) after trypsin treatment, mock treatment (without protease), or treatment with KLK5 or KLK12. The cell surface HA from each subtype was detected using HA-specific antisera, followed by Alexa Fluor 488-coupled secondary antibody (green), and the nucleus was stained with Hoechst 33258 (blue). B, quantification of the percentage syncytial formation in A.
As an additional means to determine that each kallikrein cleaves HA in a functional manner, a virus infection assay was employed. Non-cleaved influenza A/PR/8/34 HA (H1N1) and A/X-31 HA (H3N2) virions were generated by a single round of replication in 293T cells, which are incapable of cleaving HA. Each virus was then preincubated with KLK5 or KLK12 and subsequently incubated with mammalian cells. Both KLK5 and KLK12 activated influenza A/PR/8/34 HA, as indicated by immunofluorescence microscopy of the infected cells (Fig. 5). KLK5 treatment of A/X-31 HA virus resulted in a high abundance of infected cells (Fig. 5). As expected, no increase in infection was observed for KLK12 treatment of influenza A/X-31 HA compared with infection in the absence of protease (Fig. 5).
FIGURE 5.

Cleavage activation of A/PR/8/34 and A/X-31 HA influenza viruses by KLK5 and KLK12. Shown are the results from the immunofluorescence staining of influenza virus nucleoprotein of infected cells after trypsin treatment, mock treatment (no protease), or treatment with KLK5 or KLK12. The viral nucleoprotein was detected using nucleoprotein-specific antisera, followed by Alexa Fluor 488-coupled secondary antibody (green), and the nucleus was stained with Hoechst 33258 (blue).
Cleavage Efficiency of Peptide Mimics of the HA Cleavage Site Region by KLK5 and KLK12
To determine whether the HA cleavage site region affects the kallikrein cleavage efficiency, fluorogenic peptides were designed to mimic the consensus cleavage site region of the H1, H2, and H3 subtypes. In addition, a peptide was also designed to mimic the cleavage site region of the A/WSN/33 HA strain, which deviates from the H1 consensus cleavage site region. Treatment of each peptide with KLK5 resulted in a similar rate of cleavage, with a slightly higher rate for the peptide derived from A/WSN/33 HA (Fig. 6). Treatment of each peptide with KLK12 resulted in low cleavage efficiency for the H3 consensus- and A/WSN/33 HA-derived peptides but comparatively high cleavage efficiency for the H1 and H2 consensus-derived peptides (Fig. 6). The cleavage site region of HA appears to affect cleavage by KLK12 because HA and the peptide mimic of A/WSN/33 HA and the H3 subtype were cleaved with low efficiency. However, in the case of KLK5, the H2 peptide mimic was cleaved with relatively similar efficiency as the other peptide mimics but did not cleave H2 HA. Therefore, other factors such as glycosylation and secondary structure may affect the cleavage of the H2 subtype by KLK5.
FIGURE 6.
Cleavage of HA cleavage site region peptide mimics by KLK5 and KLK12. The bar graph depicts the cleavage rate of the fluorogenic peptides IPSIQSRGL (H1 consensus), IPSIQYRGL (A/WSN/33 cleavage site), VPQIESRGL (H2 consensus), and VPEKQTRGL (H3 consensus) after treatment with each peptidase. Black bars, KLK5; gray bars, KLK12. The rate of cleavage was determined by monitoring the increase in fluorescence at 390 nm.
Cleavage Activation of Thrombolytic Enzymes by KLK5 and KLK12
Plasmin, urokinase, and plasma kallikrein have all been shown to cleave and activate influenza virus HA. However, these enzymes circulate mainly in the inactive precursor form (33–35). To determine whether KLK5 and KLK12 have the ability to activate the thrombolytic zymogens, both peptidases were incubated with each zymogen and monitored for activation by cleavage of a fluorogenic substrate. In all cases, KLK5 and KLK12 cleaved and activated the thrombolytic zymogens (Fig. 7). Both KLK5 and KLK12 activated prekallikrein with similar efficiency, as determined by the rate of cleavage of the fluorogenic substrate by active plasma kallikrein. KLK12 activated prourokinase more efficiently compared with KLK5, with a 2-fold difference in cleavage of the fluorogenic substrate by active urokinase. Treatment of plasminogen with KLK5 and KLK12 resulted in the most striking difference in activation, where a 7.5-fold higher rate in cleavage by activated plasmin was observed with KLK12 treatment. A previous report demonstrated low plasminogen activation by KLK5 treatment (36), and our result is consistent with this.
FIGURE 7.

Activation of thrombolytic zymogens by KLK5 and KLK12. The increase in fluorescence (440 nm) of the GGGR-7-amino-4-methylcoumarin fluorogenic substrate was monitored upon treatment of plasminogen (A and B), prourokinase (C and D), and plasma prekallikrein (E and F) with kallikrein 5 (A, C, and E) and kallikrein 12 (B, D, and F). Each graph includes the change in fluorescence of zymogen alone (□), KLK5 and KLK12 alone (○), and zymogen with KLK5 and KLK12 (♢). RFU, relative fluorescence units.
DISCUSSION
Cleavage activation of the HA precursor is a critical step in the influenza virus life cycle. There is a short but growing list of possible host proteases that are involved in cleavage activation of influenza virus. In cell culture, exogenous trypsin is typically added to cleave and activate HA, and in embryonated chicken eggs, Factor Xa is thought to be the active protease (15). Recent work has demonstrated that certain membrane-bound serine proteases have the ability to cleave and activate viral HA (18, 20–23). Previous studies have also demonstrated that the thrombolytic enzymes plasmin, urokinase, and plasma kallikrein have the ability to cleave and activate HA (24). Moreover, proteases secreted by certain bacteria have been shown to cleave and activate viral HA (37). Finally, Kesic et al. (38) demonstrated that ozone-induced cleavage of HA is not cell-associated, where secreted proteases are sufficient for viral replication. A full understanding of the host proteases involved in cleavage activation of influenza virus would aid in the production of effective protease-based therapeutics.
Kallikrein-related peptidases 5 and 12 were examined for their ability to cleave viral HA from six strains of the H1 subtype, one strain of the H2 subtype, and three strains of the H3 subtype. Interestingly, cleavage by these peptidases revealed both HA strain and HA subtype specificity. For the H1 subtype, KLK5 preferentially cleaved A/California/09 HA and A/WSN/33 HA but displayed only limited cleavage of HA from the other strains examined. One intriguing aspect to this is that the amino acid sequences of the A/WS/33 HA and A/WSN/33 HA cleavage site regions differ only by the P2 amino acid (serine for A/WS/33 HA and tyrosine for A/WSN/33 HA), but a marked difference in cleavage by KLK5 was observed. KLK12 cleaved A/PR/8/34 HA and A/California/09 HA efficiently but displayed inefficient HA cleavage of the other strains tested. The most plausible explanation for inefficient cleavage of A/WSN/33 HA by KLK12 is that the mutation at the P2 position (S328Y) in the cleavage site region is not suitable for binding to the KLK12 active site because KLK12 also displayed a low cleavage rate with the A/WSN/33 HA-derived peptide. KLK12 cleaved H2 HA with relatively high efficiently, but virtually no cleavage was observed with KLK5 treatment. This result is intriguing because KLK5 cleaved the H2 HA consensus-derived peptide with relatively high efficiency but did not translate to cleavage of the HA protein. Therefore, other factors such as secondary structure and glycosylation may play a role in H2 HA cleavage. In the case of the H3 subtype, KLK5 cleaved HA with high efficiency, whereas only slight cleavage was observed with KLK12 treatment. Inefficient cleavage of the H3 HA consensus-derived peptide was also observed with KLK12 treatment, demonstrating that the cleavage site-flanking region may not be suitable for KLK12 binding and providing a plausible explanation for the lack of HA protein cleavage. Notably, the H3 HA cleavage site region contains a lysine residue at the P4 position that is unique to the subtypes tested and may be the determinant for the reduced cleavage efficiency observed for KLK12. To gain insight into the determinants of HA cleavage, attempts were made to cleave H3 HA by mutation of the KLK12 S4-binding pocket, but they have failed thus far (data not shown). An additional investigation of this nature will be conducted to determine the factors involved in the lack of H3 HA cleavage by KLK12.
There is still uncertainty regarding whether HA activation is dictated by secreted or membrane-bound proteases and, furthermore, whether activation occurs with HA in the virion, in the secretory pathway, or on the cell surface prior to incorporation into the virion. The recent characterization of HA cleavage by several members of the transmembrane serine protease family (principally TMPRSS2 and human airway trypsin-like protease (HAT) has clearly demonstrated trypsin-independent multicycle replication of influenza in vitro (18, 20, 22, 39). We attempted to compare the level of HA activation mediated by TMPRSS2 and HAT with that mediated by KLK5 and KLK12. However, in our hands, HA was highly sensitive to nonspecific cleavage by membrane-expressed TMPRSS2 and HAT, and complete degradation was observed upon coexpression of HA and each protease on the cell surface (data not shown). It is possible that the discrepancies in HA sensitivity to degradation by TMPRSS2 and HAT can be explained by expression levels or virus strain differences of the HAs examined. However, the role of TMPRSS family members in the context of severe acute respiratory syndrome (SARS) coronavirus infection is thought to be targeted via conspicuous degradation of the ACE2 receptor, with possible inactivation of the SARS coronavirus spike protein at high TMPRSS densities (40). Conversely, HA does not appear to be sensitive to degradation by KLK5 and KLK12, where little or no nonspecific degradation was observed with treatment of either peptidase for up to 3 h. Based on these observations, it would appear that the secreted KLK5 and KLK12 are highly specific for cleavage at the HA activation site. The known ability of members of the transmembrane TMPRSS family to cleave at multiple arginine or lysine residues (41) may in fact lead to down-regulation of influenza infection by nonspecific digestion of viral HA. As with KLK5 and KLK12, we did not observe nonspecific degradation of HA following the addition of exogenous trypsin (data not shown). It is possible that the presence of a secreted soluble protease is more compatible with specific cleavage of HA than is the presence of a transmembrane protease.
Previous reports have demonstrated that the thrombolytic enzymes plasmin, urokinase, and plasma kallikrein have the ability to cleave and activate certain subtypes of influenza and may play a role in infection. These blood proteases are thought to localize to the site of tissue injury, where influenza infection would presumably cause tissue damage of the respiratory tract. However, the majority of the circulating thrombolytic enzymes are found in the inactive precursor form. Both KLK5 and KLK12 were examined for their ability to activate these thrombolytic enzymes. Both peptidases activated each of the thrombolytic zymogens examined, with KLK12 being a more potent activator. Activation of the thrombolytic enzymes by KLK5 and KLK12 has the potential to produce a cascade effect of HA-cleaving proteases, with the thrombolytic proteases themselves cleaving HA and activating other proteases that may be involved in HA cleavage. Cleavage activation of the thrombolytic zymogens by KLK5 and KLK12 also sheds light on their biological role, as they may be involved in thrombolysis.
To conclude, with limited information on the host proteases involved in influenza activation, it is necessary to study a number of proteases for their ability to cleave HA to determine the critical ones involved in influenza infection. Determination of the key proteases involved in influenza activation will aid in the production of effective protease-based therapeutics. Investigation of the kallikrein-related peptidases 5 and 12 revealed HA subtype specificity, with KLK5 cleaving the H1 and H3 subtypes most efficiently and KLK12 cleaving the H1 and H2 subtypes most efficiently. In addition, each peptidase appears to have a preference for particular strains within the H1 subtype. The HA strain and subtype specificity observed with KLK5 and KLK12 raises the question of whether each human-adapted subtype has evolved to be activated by a set of host proteases that are distinct from the other human-adapted subtypes; this observation warrants further investigation.
Acknowledgments
We thank Theresa Fitzgerald for providing nasal wash samples, Nadia Chapman for technical assistance, and Jean Millet for critical reading of this manuscript. We also thank David Steinhauer for generous provision of reagents.
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI48678 and United States Department of Health and Human Services Contract HHSN266200700008C from the NIAID Centers of Excellence for Influenza Research and Surveillance.
REFERENCES
- 1. Neumann G., Noda T., Kawaoka Y. (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmed R., Oldstone M. B. A., Palese P. (2007) Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat. Immunol. 8, 1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson M. I., Holmes E. C. (2007) The evolution of epidemic influenza. Nat. Rev. Genet. 8, 196–205 [DOI] [PubMed] [Google Scholar]
- 4. Medina R. A., García-Sastre A. (2011) Influenza A viruses: new research developments. Nat. Rev. Microbiol. 9, 590–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schnitzler S. U., Schnitzler P. (2009) An update on swine-origin influenza virus A/H1N1: a review. Virus Genes 39, 279–292-292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pappas C., Aguilar P. V., Basler C. F., Solórzano A., Zeng H., Perrone L. A., Palese P., García-Sastre A., Katz J. M., Tumpey T. M. (2008) Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc. Natl. Acad. Sci. U.S.A. 105, 3064–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watanabe Y., Ibrahim M. S., Suzuki Y., Ikuta K. (2012) The changing nature of avian influenza A virus (H5N1). Trends Microbiol. 20, 11–20 [DOI] [PubMed] [Google Scholar]
- 8. Tumpey T. M., Belser J. A. (2009) Resurrected pandemic influenza viruses. Annu. Rev. Microbiol. 63, 79–98 [DOI] [PubMed] [Google Scholar]
- 9. Suzuki Y., Ito T., Suzuki T., Holland R. E., Jr., Chambers T. M., Kiso M., Ishida H., Kawaoka Y. (2000) Sialic acid species as a determinant of the host range of influenza A viruses. J. Virol. 74, 11825–11831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chutinimitkul S., Herfst S., Steel J., Lowen A. C., Ye J., van Riel D., Schrauwen E. J. A., Bestebroer T. M., Koel B., Burke D. F., Sutherland-Cash K. H., Whittleston C. S., Russell C. A., Wales D. J., Smith D. J., Jonges M., Meijer A., Koopmans M., Rimmelzwaan G. F., Kuiken T., Osterhaus A. D. M. E., García-Sastre A., Perez D. R., Fouchier R. A. M. (2010) Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A (H1N1) virus affects receptor binding. J. Virol. 84, 11802–11813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu V. C., Whittaker G. R. (2004) Influenza virus entry and infection require host cell N-linked glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 101, 18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oshansky C. M., Pickens J. A., Bradley K. C., Jones L. P., Saavedra-Ebner G. M., Barber J. P., Crabtree J. M., Steinhauer D. A., Tompkins S. M., Tripp R. A. (2011) Avian influenza viruses infect primary human bronchial epithelial cells unconstrained by sialic acid α2,3 residues. PLoS ONE 6, e21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wiley D. C., Skehel J. J. (1987) The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56, 365–394 [DOI] [PubMed] [Google Scholar]
- 14. Thoennes S., Li Z.-N., Lee B.-J., Langley W. A., Skehel J. J., Russell R. J., Steinhauer D. A. (2008) Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370, 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinhauer D. A. (1999) Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258, 1–20 [DOI] [PubMed] [Google Scholar]
- 16. Taubenberger J. K. (1998) Influenza virus hemagglutinin cleavage into HA1, HA2: no laughing matter. Proc. Natl. Acad. Sci. U.S.A. 95, 9713–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun X., Tse L. V., Ferguson A. D., Whittaker G. R. (2010) Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J. Virol. 84, 8683–8690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Böttcher E., Matrosovich T., Beyerle M., Klenk H.-D., Garten W., Matrosovich M. (2006) Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80, 9896–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kido H., Yokogoshi Y., Sakai K., Tashiro M., Kishino Y., Fukutomi A., Katunuma N. (1992) Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J. Biol. Chem. 267, 13573–13579 [PubMed] [Google Scholar]
- 20. Böttcher-Friebertshäuser E., Freuer C., Sielaff F., Schmidt S., Eickmann M., Uhlendorff J., Steinmetzer T., Klenk H.-D., Garten W. (2010) Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 84, 5605–5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chaipan C., Kobasa D., Bertram S., Glowacka I., Steffen I., Tsegaye T. S., Takeda M., Bugge T. H., Kim S., Park Y., Marzi A., Pöhlmann S. (2009) Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83, 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertram S., Glowacka I., Blazejewska P., Soilleux E., Allen P., Danisch S., Steffen I., Choi S.-Y., Park Y., Schneider H., Schughart K., Pöhlmann S. (2010) TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 Cells. J. Virol. 84, 10016–10025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamilton B. S., Gludish D. W. J., Whittaker G. R. (2012) Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J. Virol. 86, 10579–10586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheiblauer H., Reinacher M., Tashiro M., Rott R. (1992) Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J. Infect. Dis. 166, 783–791 [DOI] [PubMed] [Google Scholar]
- 25. Emami N., Diamandis E. P. (2007) New insights into the functional mechanisms and clinical applications of the kallikrein-related peptidase family. Mol. Oncol. 1, 269–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michael I. P., Pampalakis G., Mikolajczyk S. D., Malm J., Sotiropoulou G., Diamandis E. P. (2006) Human tissue kallikrein 5 is a member of a proteolytic cascade pathway involved in seminal clot liquefaction and potentially in prostate cancer progression. J. Biol. Chem. 281, 12743–12750 [DOI] [PubMed] [Google Scholar]
- 27. Korbakis D., Gregorakis A. K., Scorilas A. (2009) Quantitative analysis of human kallikrein 5 (KLK5) expression in prostate needle biopsies: an independent cancer biomarker. Clin. Chem. 55, 904–913 [DOI] [PubMed] [Google Scholar]
- 28. Shaw J. L. V., Diamandis E. P. (2007) Distribution of 15 human kallikreins in tissues and biological fluids. Clin. Chem. 53, 1423–1432 [DOI] [PubMed] [Google Scholar]
- 29. Yousef G. M., Magklara A., Diamandis E. P. (2000) KLK12 is a novel serine protease and a new member of the human kallikrein gene family–differential expression in breast cancer. Genomics 69, 331–341 [DOI] [PubMed] [Google Scholar]
- 30. Planque C., Li L., Zheng Y., Soosaipillai A., Reckamp K., Chia D., Diamandis E. P., Goodglick L. (2008) A multiparametric serum kallikrein panel for diagnosis of non-small cell Lung carcinoma. Clin. Cancer Res. 14, 1355–1362 [DOI] [PubMed] [Google Scholar]
- 31. Belin D., Vassalli J.-D., Combépine C., Godeau F., Nagamine Y., Reich E., Kocher H. P., Duvoisin R. M. (1985) Cloning, nucleotide sequencing and expression of cDNAs encoding mouse urokinase-type plasminogen activator. Eur. J. Biochem. 148, 225–232 [DOI] [PubMed] [Google Scholar]
- 32. Steinhauer D. A., Wharton S. A., Wiley D. C., Skehel J. J. (1991) Deacylation of the hemagglutinin of influenza A/Aichi/2/68 has no effect on membrane fusion properties. Virology 184, 445–448 [DOI] [PubMed] [Google Scholar]
- 33. Deryugina E. I., Quigley J. P. (2012) Cell surface remodeling by plasmin: a new function for an old enzyme. J. Biomed. Biotechnol. 2012, 564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Renné T., Dedio J., Meijers J. C. M., Chung D., Müller-Esterl W. (1999) Mapping of the discontinuous H-kininogen binding site of plasma prekallikrein. Evidence for a critical role of apple domain-2. J. Biol. Chem. 274, 25777–25784 [DOI] [PubMed] [Google Scholar]
- 35. Sharma J. N., Sharma J. (2002) Cardiovascular properties of the kallikrein-kinin system. Curr. Med. Res. Opin. 18, 10–17 [DOI] [PubMed] [Google Scholar]
- 36. Michael I. P., Sotiropoulou G., Pampalakis G., Magklara A., Ghosh M., Wasney G., Diamandis E. P. (2005) Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J. Biol. Chem. 280, 14628–14635 [DOI] [PubMed] [Google Scholar]
- 37. Tashiro M., Ciborowski P., Klenk H.-D., Pulverer G., Rott R. (1987) Role of Staphylococcus protease in the development of influenza pneumonia. Nature 325, 536–537 [DOI] [PubMed] [Google Scholar]
- 38. Kesic M. J., Meyer M., Bauer R., Jaspers I. (2012) Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza A infection. PLoS ONE 7, e35108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Böttcher E., Freuer C., Steinmetzer T., Klenk H.-D., Garten W. (2009) MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27, 6324–6329 [DOI] [PubMed] [Google Scholar]
- 40. Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S., Gallagher T. (2011) A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 85, 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hooper J. D., Clements J. A., Quigley J. P., Antalis T. M. (2001) Type II transmembrane serine proteases. J. Biol. Chem. 276, 857–860 [DOI] [PubMed] [Google Scholar]