

Background: Single domain variable regions of shark antibodies (V-NARs) are promising biotherapeutic candidates.
Results: A V-NAR specific for human serum albumin was humanized, and its crystal structure in complex with the antigen was solved, revealing an unusual recognition mode.
Conclusion: Humanization preserved antigen binding properties and activity of the parental shark antibody.
Significance: A structural framework for humanization of shark antibodies was established.
Keywords: Albumin, Antibodies, Antibody Engineering, Antigen, Crystal Structure, Humanization, IgNAR, V-NAR
Abstract
The immunoglobulin new antigen receptors (IgNARs) are a class of Ig-like molecules of the shark immune system that exist as heavy chain-only homodimers and bind antigens by their single domain variable regions (V-NARs). Following shark immunization and/or in vitro selection, V-NARs can be generated as soluble, stable, and specific high affinity monomeric binding proteins of ∼12 kDa. We have previously isolated a V-NAR from an immunized spiny dogfish shark, named E06, that binds specifically and with high affinity to human, mouse, and rat serum albumins. Humanization of E06 was carried out by converting over 60% of non-complementarity-determining region residues to those of a human germ line Vκ1 sequence, DPK9. The resulting huE06 molecules have largely retained the specificity and affinity of antigen binding of the parental V-NAR. Crystal structures of the shark E06 and its humanized variant (huE06 v1.1) in complex with human serum albumin (HSA) were determined at 3- and 2.3-Å resolution, respectively. The huE06 v1.1 molecule retained all but one amino acid residues involved in the binding site for HSA. Structural analysis of these V-NARs has revealed an unusual variable domain-antigen interaction. E06 interacts with HSA in an atypical mode that utilizes extensive framework contacts in addition to complementarity-determining regions that has not been seen previously in V-NARs. On the basis of the structure, the roles of various elements of the molecule are described with respect to antigen binding and V-NAR stability. This information broadens the general understanding of antigen recognition and provides a framework for further design and humanization of shark IgNARs.
Introduction
Antibody-based targeting has become an established paradigm of biologic drug development. High affinity, excellent specificity, generally good stability, and Fc-associated effector functions all make antibodies the molecules of choice for many diagnostic and therapeutic applications. At the same time, novel non-antibody scaffolds are constantly being sought by industry to allow for development of new therapeutic agents offering advantages over classical antibody platforms (1, 2). In particular, smaller size for better tissue penetration, reduced complexity for easier production, and enhanced biological and biophysical stability are some of the properties desired for the new generation of biologics.
Multiple formats and optimization strategies that try to incorporate these properties have been described. Some of the resulting molecules, such as scFv,3 DVD-IgTM, diabody, scFv-Fc, and others, represent novel designs or effector function variants based on traditional antibody scaffolds (3, 4). In addition, naturally occurring single variable domain antibodies from cartilaginous fish (IgNARs) and camelids (VHH antibodies; also known as nanobodies) provide an attractive alternative (5, 6). The variable domains of these antibodies can be linked in tandem to provide multispecificity and increase the size and thus the in vivo half-life of the molecules. They can also be linked to Fc domains of traditional antibodies to provide them with desired effector functions.
IgNARs were discovered in sharks in the 1990s (7, 8). Their variable regions (V-NARs) are small (12–13-kDa), independently folding domains that demonstrate high biophysical stability, solubility, and ability to bind to a variety of antigens including epitopes located in clefts on protein surfaces (e.g. enzyme active sites) that are non-accessible by traditional antibody variable domains (9, 10). A similar preference for cleft recognition was demonstrated for camelid VHH antibodies (11–14). In both cases, the key to such recognition is the structural organization of the CDR loops, in particular CDR3, which is often long (15–18 residues) and protruding from the V-NAR or VHH surface.
V-NARs are distinct from typical Ig VH and VL domains as well as camelid VHH domains, sharing higher structural homology to immunoglobulin VL and T-cell receptor V domains than with immunoglobulin VH. The most unique feature of V-NARs is the absence of a CDR2 loop and of two β-strands, C′ and C″, associated with it. Instead, a distinct “belt” is formed around the middle of the β-sandwich structure (10, 15). This region shows an elevated rate of somatic mutations and has thus been termed hypervariable region 2 (HV2) (16). Another region of increased mutation frequency is located between HV2 and CDR3, comprising a loop that links β-strands D and E similar to that in T-cell receptor V chains; thus, this region was termed HV4. Structurally, HV2 is most proximal to CDR3, whereas HV4 is in proximity to CDR1.
Several structural types of IgNAR variable domains have been classified based on the number and position of extra cysteine residues in CDRs and frameworks (FWs) in addition to the canonical cysteine pair (Cys23/Cys88 for VL; Kabat nomenclature) of the Ig fold (5). Type I V-NAR, found in nurse sharks, has 2 cysteines in CDR3 and 2 more cysteines in frameworks (FW2 and FW4). The more common type II has one extra cysteine pair, which links CDR1 and CDR3. Type III, detected primarily in neonatal shark development, is similar to type II but has a conserved Trp residue in CDR1 and limited CDR3 diversity. Another structural type of V-NAR, which we have termed type IV, has only two canonical cysteine residues. So far, this type has been found primarily in dogfish sharks (Ref. 17 and this study) and was also isolated from semisynthetic V-NAR libraries derived from wobbegong sharks (18).
The single domain nature and the lack of CDR2 in V-NARs heighten the requirement for CDR1 and CDR3 to provide specific and high affinity binding to prospective antigens. CDR3, which is more variable in terms of sequence, length, and conformation, plays the key role in antigen recognition. The placing of cysteine residues in different V-NAR types is important for determining the conformation of CDRs. For example, CDR3 is long and extended (and tethered to CDR1) in PBLA8, a type II V-NAR, which enables it to access the active site cavity of its target, hen egg white lysozyme (HEL; Ref. 10). In contrast, 5A7, a type I V-NAR also directed against lysozyme and targeting a similar surface epitope, has a long CDR3 that adopts a bent conformation and forms a rather flat binding surface that does not enter deep into the HEL active site (10, 15). Nevertheless, both HEL binders form comparable buried surface area with their target (∼700 Å2) and bind with low nanomolar affinity. The extent of the surface area is similar to values observed for the complexes of heavy chains of classical antibodies with their targets (19, 20).
Besides CDR1 and CDR3, a few other elements are involved in the HEL interaction by PBLA8 and 5A7. Of note is the residue Arg61 in the HV4 loop of PBLA8 that forms a hydrogen bond with Asp101 in HEL (10). Detailed mutational analysis of 5A7 by Fennell et al. (21) revealed a high degree of mutational plasticity within the V-NAR domain and suggested that residues outside of the CDR1 and CDR3 loops may form additional contacts with the antigen. For example, mutation of Ser61 to Arg in HV4 of 5A7 increases HEL binding ∼5-fold likely due to the formation of a contact with Asp101 similar to Arg61 in PBLA8. Likewise, mutation of Ala1 to Asp in 5A7 results in increased HEL binding due to a putative ionic interaction (21).
In contrast to typical antibodies, the structure described in this study utilizes CDR1 only minimally for antigen binding. An atypical “sideways” binding mode is observed that relies heavily on framework residues to achieve antigen binding in addition to CDR3. This binding mode has been described previously for other single domain antibodies (14) but has not been seen previously in V-NARs.
It is assumed that to be useful in therapeutic applications all novel non-human scaffolds, such as V-NARs or camelid VHH single domains, need to be humanized to reduce immunogenicity and/or improve thermodynamic stability, folding, and expression properties. Considerable expertise has been accumulated in this subject area, particularly with rodent mAbs (22–24). Typically, CDRs of a murine antibody of interest are grafted onto an appropriate human germ line framework (selected for sequence similarity, expression properties, or both), and then back-mutations are introduced at key positions responsible for particular CDR conformation and thus antigen binding. This approach has yielded many humanized antibodies with a number of them making it into the clinic.
With camelid VHH domains, humanization has been relatively straightforward because of the overall structural similarity and high sequence homology (∼80%) between human and camel or llama sequences. In most instances, only ∼10 mutations of “non-human” surface residues toward the human germ line of the closest VH3 type need to be introduced into VHH scaffolds; in addition, two of four VHH hallmark residues in FW2 (positions 42, 49, 50, and 52) can be changed (25, 26). All those changes can result in biophysically stable, well expressed, and biologically active VHH domains with nearly 100% framework identity to human germ line sequences.
Shark V-NARs represent an obvious challenge for humanization because of the structural differences (e.g. lack of CDR2) and low overall sequence identity (generally ∼30%) to human VH/VL sequences. However, available crystal structures of V-NAR domains demonstrate organization of key framework regions similar to that of human Ig variable domains, thus making an attempt at humanization possible. In this study, we describe the generation of humanized versions of type I and type IV V-NARs based on the human germ line VL scaffold, DPK9. We also provide detailed structural analysis of the type IV V-NAR clone, E06, in complex with its target, human serum albumin. Our analysis provides the foundation for further improvement and humanization of shark V-NARs.
EXPERIMENTAL PROCEDURES
Design and Cloning of Humanized V-NAR Variants
The sequences encoding humanized E06 variants were codon-optimized for expression in mammalian cells and synthesized by GeneArt AG (Germany). The sequences were cloned into a mammalian expression vector under the control of a murine CMV promoter. A signal peptide for extracellular secretion was added at the N terminus. C-terminal tags included His6, AAA-His6, or human IgG1 Fc (hFc). For hFc fusions, a Gly4-Ser-Gly4 linker was incorporated between the V-NAR and Fc.
Expression and Purification of V-NAR Proteins
V-NAR-hFc fusion proteins were expressed in COS-1 cells grown in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, penicillin, streptomycin, and glutamine. Cells were transfected using TransIT reagent (Mirus) following the manufacturer's recommendations, and serum-free conditioned medium (CM) was typically collected at days 3 and 7 post-transfection. hFc fusion proteins were purified on HiTrap protein A columns (GE Healthcare) followed by size exclusion chromatography on a Superdex 200 16/60 column run in PBS, pH 7.2.
Monomeric V-NARs tagged with AAA-His6 were similarly expressed in COS-1 cells and purified by chromatography on a HisTrap FF column (GE Healthcare). CM was loaded onto a column pre-equilibrated with 50 mm sodium phosphate, 300 mm NaCl, pH 8.0, and after extensive washing with the same buffer containing 20 mm imidazole, the bound proteins were eluted with a linear 10-column volume gradient to 250 mm imidazole. Fractions containing V-NAR proteins were dialyzed against PBS. Protein concentration was determined by A280 nm.
To obtain E06-His6 and huE06 v1.1-His6 proteins for crystallography, transient expression in HEK293-EBNA cells was performed in 1-liter spinners. Cells grown in serum-free FreeStyle293 expression medium (GE Healthcare) were transfected using polyethylenimine and harvested 120 h later. The expression of His6-tagged proteins in CM was confirmed by immunoblotting with anti-His4 antibody (Qiagen).
Isolation of E06-Human Serum Albumin Complexes
Clarified CM containing parental or humanized E06 with C-terminal His6 tag was applied to Ni2+-nitrilotriacetic acid Superflow resin (Qiagen). Following capture, the column was rigorously washed in PBS supplemented with 20 mm imidazole and subsequently eluted in PBS supplemented with 250 mm imidazole. E06 was dialyzed against PBS to remove excess imidazole in preparation for complex formation. To remove oligomeric species from commercial human serum albumin, lipid-free HSA (Sigma, catalog number A3782) was dissolved in PBS at 50 mg/ml and applied to a Superdex 200 16/60 size exclusion column. Fractions corresponding to the monomeric species were pooled and incubated at a 1:2 molar ratio with E06. After incubation for 1 h, the complex was concentrated and applied to a Superdex 200 16/60 column pre-equilibrated in Tris-buffered saline to remove excess E06. Consistent with complex formation, HSA and E06 co-eluted from the column at earlier elution volumes than both free species alone. Appropriate fractions were pooled and concentrated to 10 mg/ml for crystallization.
ELISA
Serum albumin proteins used for binding experiments were from Sigma. For direct ELISA to detect V-NAR binding to antigens, Costar assay plates were coated with 1 μg/ml antigen in PBS and then blocked with 1% nonfat milk in PBS. V-NAR-hFc proteins were diluted in assay buffer (0.5% nonfat milk, 0.05% Tween 20 in PBS) and incubated with antigen for 1 h followed by incubation with goat anti-hFc-HRP for 1 h. For sandwich ELISA, plates were coated with goat anti-hFc polyclonal antibody (Jackson ImmunoResearch Laboratories) at 2 μg/ml in PBS. V-NAR-hFc proteins were titrated starting from 1 μg/ml in assay buffer and incubated in the plate for 1 h followed by addition of 1 μg/ml antigen (e.g. HSA) in assay buffer for 1 h. Bound HSA was detected using goat anti-HSA-HRP conjugate (Bethyl Laboratories). Concentrations of expressed hFc fusion proteins in CM were determined by sandwich ELISA using goat anti-hFc polyclonal antibody for capture and HRP-conjugated goat anti-hFc polyclonal antibody for detection.
Crystallization
E06-HSA crystals were grown by hanging drop vapor diffusion at 18 °C in drops containing 1.0 μl of protein stock solution (11.0 mg/ml protein complex, 25 mm Tris, pH 7.4, 150 mm NaCl) mixed with 1.0 μl of well solution (16% PEG 2000 monomethyl ether, 100 mm sodium acetate, pH 4.6) and equilibrated against 0.5 ml of well solution. Chunky crystals grew in approximately 1 week, measuring ∼50 μm across.
huE06 v1.1-HSA crystals were grown by hanging drop vapor diffusion at 18 °C in drops containing 1.0 μl of protein stock solution (10.7 mg/ml protein complex, 25 mm Tris, pH 7.6, 150 mm NaCl) mixed with 1.0 μl of well solution (25% PEG 2000 monomethyl ether, 100 mm sodium acetate, pH 4.6) and 0.2 μl of the additive 30% 1,6-hexanediol and equilibrated against 0.5 ml of well solution. Diamond-shaped crystals appeared overnight and grew in approximately 1 week, measuring ∼50 μm in the longest dimension.
Data Collection and Processing
E06 complex crystals belong to the space group P3221 (Number 154) with unit cell parameters 127.98 × 127.98 × 151.76 Å3 and contain two molecules of E06 and two molecules of HSA in the asymmetric unit, implying a solvent content of 54.2%. Crystals were drawn through a solution of 20% DMSO and 80% well solution and cooled rapidly in liquid nitrogen. Diffraction data were recorded at Advanced Photon Source beamline 22-ID on a MAR-300 detector. Intensities were integrated and scaled using the program xia2 (27). huE06 v1.1 complex crystals belong to the space group P3121 (Number 152) with unit cell parameters 131.34 × 131.34 × 74.52 Å3 and contain one molecule of E06 v1.1 and one molecule of HSA in the asymmetric unit, implying a solvent content of 57.0%. Crystals were drawn through a solution of 25% ethylene glycol and 75% well solution and cooled rapidly in liquid nitrogen. Diffraction data were recorded at Advanced Photon Source beamline 22-ID on a MAR-300 detector. Intensities were integrated and scaled using the program autoProc (28).
Phasing, Model Building, and Refinement
The structure of E06 in complex with HSA was determined by molecular replacement with Phaser (29, 30) using the crystal structure of apo-HSA (Protein Data Bank code 1AO6) as a starting search model. A few rounds of refinement with PHENIX (31) were performed after which clear density for the β-sheet regions of E06 was obtained. After subsequent placement of a polyalanine model of E06 and several iterative cycles of model rebuilding with Coot (32) and refinement with autoBuster (28), final Rwork and Rfree values of 23.7 and 26.6%, respectively, were obtained (Table 1).
TABLE 1.
Statistics for data collection and refinement for E06-HSA and huE06 v1.1-HSA complexes
Values in parentheses represent the corresponding values for the highest resolution shells. r.m.s., root mean square.
| E06-HSA | huE06 v1.1-HSA | |
|---|---|---|
| Wavelength (Å) | 1.000 | 1.000 |
| Resolution range of data (Å) | 30.0–3.04 | 113.7–2.34 |
| Rmergea (%) | 22.2 | 8.6 |
| Completeness (%) | 93.2 | 99.7 |
| Redundancy | 6.4 | 4.6 |
| Total reflections | 170,816 | 144,138 |
| Unique reflections | 25,936 | 31,469 |
| I/σ(I)b | 13.9 (2.3) | 10.7 (1.1) |
| Resolution range of refinement (Å) | 30.0–3.04 | 38.0–2.34 |
| Rworkc (%) | 23.7 (26.2) | 22.1 (27.9) |
| Rfreed (%) | 26.6 (27.0) | 25.9 (31.4) |
| r.m.s. deviations from ideal geometry | ||
| Bonds (Å) | 0.008 | 0.004 |
| Angles (°) | 1.100 | 0.771 |
| Wilson B-value (Å2) | 74.7 | 45.1 |
| Average B-values (Å2) | 89.4 (HSA) | 71.2 (HSA) |
| 68.1 (E06) | 47.8 (huE06 v1.1) | |
| Protein atoms | 9,296 | 4,505 |
| Solvent atoms | 37 | 72 |
a Rmerge = |Ih − Ih|/Ih where Ih is the average intensity over symmetry equivalents.
b I/σ(I) = average I/average σ(I).
c Rwork = ‖Fobs| − |Fcalc‖/bar]Fobs|.
d Rfree is equivalent to Rwork but calculated for a randomly chosen 5% of reflections omitted from the refinement process.
The huE06 v1.1 structure was determined by molecular replacement using E06 as the starting molecular replacement model. After iterative cycles of model improvement and refinement using Coot and PHENIX, final Rwork and Rfree values of 22.1 and 25.9%, respectively, were obtained for huE06 v1.1 (Table 1).
Kinetic and Affinity Measurements of the E06-Serum Albumin Interaction
The kinetic constants of the E06-serum albumin interactions were determined by surface plasmon resonance (Biacore T100, GE Healthcare). Flow cells of a CM5 chip were immobilized with ∼600 resonance units of HSA, mouse serum albumin, and rat serum albumin in 10 mm glycine, pH 4.0 at 5 μl/min. Association of five concentrations of E06 proteins (from 1.23 to 100 nm at pH 7.4 and 0.062 to 30 nm at pH 6.0 depending on affinity) and a zero concentration (running buffer) was recorded for 2 min in 20 mm HEPES buffer with 150 mm NaCl, 3 mm EDTA, 0.05% P20, pH 7.4 or 20 mm MES buffer with 150 mm NaCl, 3 mm EDTA, 0.05% P20, pH 6.0. Dissociation of the complexes was measured for 3 min. Two 10-s pulses of 10 mm glycine, pH 1.5 regenerated the surface. Curves obtained after subtraction of the reference and buffer signals were fitted to a 1:1 Langmuir binding model with Biacore T100 Evaluation software.
Accession Numbers
Structure factors and coordinates have been deposited in the Worldwide Protein Data Bank under codes 4HGK (E06) and 4HGM (huE06 v1.1).
RESULTS
Isolation of Human Serum Albumin-binding Shark IgNARs
Spiny dogfish sharks (Squalus acanthias) were immunized with human serum albumin, and V-NAR sequences from a seropositive animal were isolated and made into a phage display library (33). Solid-phase panning of the library on HSA was carried out and led to the isolation of a number of positive clones, many of which also reacted to mouse and rat serum albumins. Binding at both pH 7.0 and pH 6.0 (to facilitate lysosomal recycling) was incorporated as part of the screening strategy. One of the best binders, which reacted to HSA, mouse serum albumin, and rat serum albumin at pH 7.0 and pH 6.0, was called E06.
V-NAR clone E06 is 103 residues in length and has low sequence similarity to human variable domain sequences (<30% identity) with the closest human germ line sequences being from VL6 and VH4 families. E06 is a typical shark V-NAR lacking the CDR2 region of mammalian antibody V domains and instead carrying an HV2 stretch in FW2 and an HV4 loop as part of the FW3 sequence. Similar to other Ig molecules, there is a 8-amino acid CDR1 sequence and relatively short 9-amino acid CDR3 sequence (Fig. 1). E06 belongs to a structural type IV family of shark V-NARs that is distinct from the better characterized type I (e.g. 5A7; Refs. 15 and 34) and type II domains (e.g. PBLA8, a phage display library clone from HEL-immunized nurse shark; Ref. 16). Type IV V-NARs have only 2 canonical Ig domain cysteine residues (positions 22 and 83 in E06) compared with 6 cysteines in type I and 4 cysteines in type II.
FIGURE 1.

Structural sequence alignment of V-NAR E06 and its humanized variants with human germ line Vκ light chain DPK9/Jκ1, shark V-NAR 5A7, and a humanized 5A7 variant, 5A7-IVabc. DPK9 framework residues and residues in V-NARs identical to DPK9 are shown in green. Mutations in E06 HV4 are shown in blue. The residue numbering refers to the E06 sequence.
Humanization Strategy for Type I and Type IV V-NARs
We first thought to humanize a type I V-NAR, 5A7. The humanization was done by resurfacing whereby multiple solvent-exposed and core framework residues of 5A7 were replaced by human residues from structurally related Ig domains. The selection was guided by structural superpositions of the closest human variable domains with the shark framework followed by modeling analysis of the humanized variants. Structures of human variable domains used for this study were found in the Protein Data Bank (codes 1DEE, 1DN0, 1E6J, and 2FBJ). Besides structural homology, one of the most important considerations was to preserve the favorable physicochemical properties of a shark V-NAR, i.e. solubility as a single domain, stability, and binding capability. For this reason, we considered the human antibody DP-47 germ line framework, a member of the variable heavy chain subgroup 3 (VH3), because this subgroup has been shown to have higher biophysical stability than other human frameworks (35). However, when humanized versions of 5A7 based on DP-47 were generated, the constructs had significantly compromised binding affinities and were not pursued further (data not shown). Thus, we selected the human antibody DPK9 germ line framework, a member of the variable κ subgroup 1 (Vκ1), that was more structurally similar to 5A7 when compared with many other human Ig variable domain sequences. The Vκ1 subgroup is also one of the most stable and well expressed human frameworks in IgG and scFv formats (35).
As shown in Fig. 1, the following structural elements were replaced in 5A7: FW1 (residues 6–21), FW2/part of HV2 (residues 38–47), FW3b (residues 67–82), and FW4 (residues 106–113). All 6 cysteine residues of the type I V-NAR scaffold were retained. The resulting molecule, which we call 5A7-IVabc, had 60 of 86 (69.8%) non-CDR residues and 60 of 112 (53.6%) total residues identical to DPK9. Importantly, 5A7-IVabc protein showed an excellent expression profile in mammalian cells with very little aggregation in either monomeric or dimeric (with human Fc) format and full retention of binding activity to HEL. The binding constant for monomeric V-NARs to HEL as determined by Biacore was 13.6 nm for parental 5A7 and 14.8 nm for humanized 5A7-IVabc (data not shown).
To further validate the humanization-by-resurfacing approach taken with 5A7, we created a series of humanized variants of E06 using the 5A7-IVabc molecule as a guide (Fig. 1). To make the humanized E06 variant 1.1 (huE06 v1.1), 30 residues of the 103 total residues in E06 were replaced with DPK9 residues. Specifically, the majority of the framework residues, FW1 (residues 6–21), FW2 (residues 38–40), FW3b (residues 66–82), and FW4 (residues 99–103), of E06 were made identical to DPK9. The majority of these changes equate to those used to make 5A7-IVabc. The regions left intact (shark) were the first 4 N-terminal residues, CDR1 (residues 28–33) and CDR3 (residues 86–94), HV2 (residues 43–52), and FW3a and HV4 (residues 53–65). In the huE06 v1.1 molecule, 54 of 85 (63.5%) non-CDR residues were identical to human DPK9.
To modify E06 further, mutations toward the 5A7-IVabc sequence were introduced into the HV4 region of v1.1 (K61S and T63S) to make huE06 v1.2. Further DPK9-like changes were made in the HV2 region (43SSNKE47 → 43KAPK46) to produce huE06 v1.7. These two sets of changes (in HV2 and HV4) were combined to make huE06 v1.3. A derivative of v1.3, which had its N terminus changed toward DPK9 (1TRVD4 to 1DIQMT5), was made and named huE06 v1.4. We also attempted to redesign E06 by shortening the FW3a/HV4 region; to do that, 5 shark residues were deleted, and 3 DPK9 residues were introduced in this region of the huE06 v1.3 molecule; in addition, a Y55F change was introduced. The resulting molecule was named huE06 v1.5. Finally, huE06 v1.10 was derived from v1.1 by restoring the 38RKN40 shark sequence in FW2 from DPK9-like 38QQK40; the specific rationale for this will be described below.
Functional Properties of Humanized E06 Variants
E06 and its humanized variants were first expressed as human Fc fusions. The expression levels of the humanized variants, such as 1.1, 1.2, 1.3, 1.5, and 1.7, were not dramatically different from the parental E06 molecule and were in the 10–40 μg/ml range when expressed transiently in COS-1 cells. However, when purified on protein A, there were progressively more high molecular weight species for humanized variants 1.1, 1.2, and 1.3 compared with parental E06 (data not shown). Therefore, the dimeric forms of parental and humanized E06 were purified further using size exclusion chromatography.
The binding of E06 and its humanized variants to HSA was first analyzed by ELISA using purified hFc fusions. For direct ELISA, plates were coated with human albumin, V-NAR-hFc samples were added, and the human Fc tag was detected with anti-hFc-HRP. For sandwich ELISA, V-NAR-hFc proteins were captured on the plate at different concentrations using goat anti-hFc, and bound HSA was detected using HRP-conjugated anti-HSA polyclonal antibody.
As shown in Fig. 2A, in direct ELISA, all humanized variants tested apparently retained significant binding to human albumin compared with parental E06. However, the difference was very obvious by sandwich ELISA where the bivalent binding (avidity effect) is likely reduced. There, huE06 v1.1, 1.7, 1.2, and 1.3 demonstrated progressively reduced binding to HSA (Fig. 2B), which correlated with the extent of changes introduced into their HV2 and HV4 regions (Fig. 1). In particular, substitutions in HV4 region (K61S/T63S in v1.2 versus v1.1) caused a more significant reduction in activity compared with humanizing changes in HV2 (e.g. v1.7 versus v1.1). huE06 v1.5 demonstrated no binding to HSA when tested as conditioned medium (data not shown) and thus was not evaluated as purified protein. Based on these functional results, huE06 v1.1 was chosen as the humanized variant for crystallographic studies.
FIGURE 2.
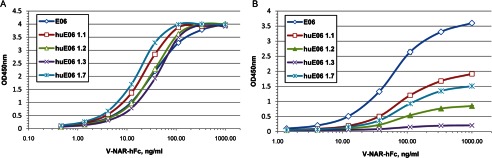
Albumin-binding ELISA with hFc-tagged E06 and its humanized variants 1.1, 1.2, 1.3, and 1.7. A, ELISA with HSA coated on the plate. B, sandwich ELISA with E06-hFc captured on the plate and HSA in solution.
Structure of E06 and huE06 v1.1 in Complex with Human Serum Albumin
E06 and E06 v1.1 were expressed as monomeric His6-tagged proteins and crystallized in complex with HSA (see “Experimental Procedures” for details). The structures were determined to 3.0- and 2.3-Å, respectively. The E06 structure contains two complexes in the crystallographic asymmetric unit with a root mean squared deviation calculated with Cα atoms between the two molecules of E06 of 0.157 Å. References to the E06 structure in this study refer to molecule C. huE06 v1.1 was crystallized in space group P3121 (Number 152) with one copy of the complex in the crystallographic asymmetric unit. The root mean squared deviation calculated with Cα atoms between E06 and huE06 v1.1 is 0.767 Å. The electron density for the V-NARs is good throughout, and the polypeptide chains are complete. All residues of huE06 v1.1 are present in the final model except for the C-terminal residues missing due to disorder, region 104–112. Regions of HSA that were omitted in the model due to lack of electron density include residues 1, 2, 79–85, 402, 403, 478, 479, and 493–585. The classical Ig-like fold is seen in both V-NAR structures with a short CDR3 and an internal disulfide bond between Cys22 and Cys83 with the absence of a CDR2 and the presence of an HV2 belt characteristic of V-NAR domains.
In contrast to the classical antigen-antibody recognition mode, we found that the most extensive interactions with HSA originate from the CDR3 residues and the framework residues on E06 (Fig. 3, A and B, and Table 2). Antigen binding results in a large buried surface area of 705Å2, which is 12.5% of the total surface area of E06. The structure of huE06 v1.1 in complex with HSA (Fig. 3B) shows that the humanizing changes in the V-NAR are at positions that are not directly involved in antigen binding with the exception of residues 38–40 (RKN in E06 and QQK in huE06 v1.1), which were not expected to participate in antigen recognition. However, as is clear from the binding assays, a number of additional humanizing changes present in other huE06 variants may affect antigen binding indirectly as detailed below.
FIGURE 3.

Ribbon diagrams of E06 and huE06 v1.1 in complexes with HSA. E06 and huE06 v1.1 are shown in color, and HSA is in gray. V-NARs are colored according to the following: residues with the original shark sequence are shown in green, residues in which the original shark sequence are identical to human germ line DPK9 are shown in cyan, and residues that have been mutated from the original shark sequence to correspond to DPK9 are colored in pink. A, the structure of the E06-HSA complex. B, A closer view of the huE06 v1.1 structure bound to HSA with HSA shown as a surface representation.
TABLE 2.
E06 residues within 5.0 Å of HSA
| Tyr-32, Tyr-35, Tyr-37, Lys-39, Ser-44, Asn-45,Lys-46, Gln-48, Arg-84, Met-86, Gly-87, Thr-88, Asn-89, Ile-90, Trp-91, Thr-92, Gly-93, Asp-94 |
In addition to the direct interactions at the interface of E06 and HSA in the complex structure, a number of residues appear to influence antigen binding through a set of second tier interactions as seen through analysis of variants of humanization with corresponding binding data and the complex structures. At the N terminus of E06, a network of hydrogen bonds is seen on the solvent-exposed side of the β-sheet including residues Thr1, Arg2, and Asp4 (Fig. 4A). This network likely contributes to the stability of this region. Val3 is buried in a hydrophobic pocket where it has contacts with Phe66 and Ala85 as well as Trp91 and other residues in CDR3 with little room for even a larger hydrophobic amino acid (e.g. Met). Disruption of either of these sets of interactions would disrupt the packing at this end of the V-NAR and likely result in a loss of stability and may impact the presentation of the directly neighboring CDR3.
FIGURE 4.

A, side chain interactions at the N terminus of huE06 v1.1 include a network of hydrogen bonds as well as hydrophobic interactions. Together these interactions may contribute to the presentation of the neighboring CDR3 and to the overall stability of the V-NAR. B, in the parental E06 shark sequence, the HV2 region features a network of hydrogen bonds that position Lys39, Ser44, and Lys46 for interaction with HSA. Disruption of these networks would negatively impact antigen binding. C, the HV4 region may influence the positioning of Tyr32, which is directly involved in HSA binding.
Similarly, residues in the HV2 region form a network of hydrogen bonds that orient residues Ser44, Asn45, and Lys46 for interaction with HSA (Fig. 4B). Also, Arg38 and Asn40 form an H-bond, which may help set up Lys39 for its interaction with HSA. Disruption of this network would impact the positions of these residues and adversely affect antigen binding.
In the HV4 region, Lys61 forms a hydrogen bond with the main chain of Tyr32, a key CDR1 residue at the interface with HSA. Thr63, through its contacts with Asn60 and possibly Ser85, contributes to a bend in HV4 loop, which positions Lys61 correctly for interaction with Tyr32 (Fig. 4C). Based on our observation that mutations at these residues are unfavorable for binding, it is possible that together these residues help position Tyr32 for interaction with HSA.
Albumin Binding by N-terminal Mutants of E06
The 5A7 molecule can tolerate well certain amino acid substitutions in the N terminus (21), but its activity drops significantly upon extension of the N terminus, e.g. in tandem constructs.4 Similarly, little or no antigen binding of C-terminal V-NAR in a tandem construct was observed by Simmons et al. (36). The N terminus of E06 as described above is in relative proximity to the CDRs and their antigen-binding sites and contains a network of intramolecular interactions. For this reason, we largely avoided humanization of the N terminus of E06 with the exception of v1.4 that had N-terminal residues from DPK9 in addition to other humanization changes equivalent to huE06 v1.3. To further probe the role of N-terminal residues of E06 in maintaining antigen binding, we generated a series of mutants of the parental E06 molecule (Fig. 5A). In particular, residues 2–4 (Gln-Met-Thr) were introduced from DPK9 combined with Thr (shark) or Ile (human) in position 1. In other variants, DPK9-like single amino acid (Asp) extension of the N terminus was introduced followed by either Thr or Ile.
FIGURE 5.

N-terminal mutants of E06 and their activity. A, sequences of N-terminal mutants of E06. DPK9-specific residues are underlined. B, direct ELISA on HSA with E06-hFc fusion proteins.
None of the tested N-terminal variants showed a significant difference in expression levels compared with the parental E06 (data not shown). However, most of them have strongly reduced binding to human albumin in direct ELISA, especially the variants with a 1-amino acid extension of the N terminus (Fig. 5B). This result is fully consistent with the structural analysis outlined above. huE06 v1.4 showed no HSA binding, which was likely a combination effect of changes in the N terminus, HV2, and HV4. These and other data indicate that the first position in IgNARs is relatively tolerant to changes, but subsequent positions and the overall length of the N terminus are quite important.
Humanized E06 Molecule That Retains All HSA Contact Sites
The structures of E06 and huE06 v1.1 in complex with HSA reveal that the only HSA contact residue lost in huE06 v1.1 is Lys39. This residue is part of the 38RKN40 sequence in E06 that is replaced with QQN sequence from DPK9 in huE06 v1.1 (Figs. 1 and 3). This finding prompted us to generate another humanized variant of E06, v1.10, with 38RKN40 sequence restored; otherwise, it is identical to huE06 v1.1 (Figs. 1 and 6A). When the HSA binding activity of huE06 v1.10-hFc was measured by ELISA, it showed an improvement over huE06 1.1, although it was still weaker than the parental E06 (Fig. 6, B and C). Neither parental E06 nor its humanized variants showed any appreciable binding to BSA in direct ELISA format (Fig. 6C).
FIGURE 6.
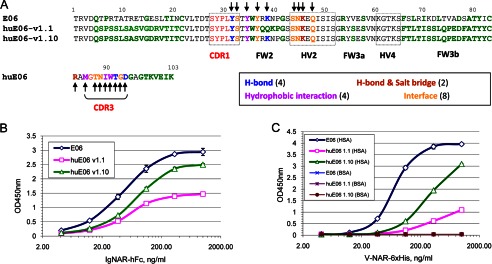
Retention of albumin binding by humanized E06 v1.1 and v1.10. A, sequence alignment of parental E06 with humanized E06 v1.1 and v1.10 with residues in E06 involved in the indicated modes of interactions with HSA marked by arrows. DPK9-like residues in E06 and huE06 are shown in bold. B, sandwich ELISA with purified E06 and huE06-hFc fusion proteins on human albumin. C, direct ELISA with purified monomeric E06 and huE06 on human and bovine albumin.
To assess the kinetic parameters of binding of E06, huE06 v1.1, and huE06 v1.10 to serum albumins, the monomeric (His6-tagged) V-NARs were tested in Biacore experiments. As shown in Fig. 7A and Table 3, the binding of huE06 v1.10 to mammalian serum albumins was reduced ∼25–35-fold at pH 7.4 compared with parental E06, whereas binding of v1.1 was reduced up to ∼85-fold. Parental E06 showed robust, generally subnanomolar binding to the albumins. The range of albumin affinities for both parental E06 and huE06 was human > mouse > rat (KD values of 0.19, 0.83, and 1.45 nm, respectively, for parental E06).
FIGURE 7.
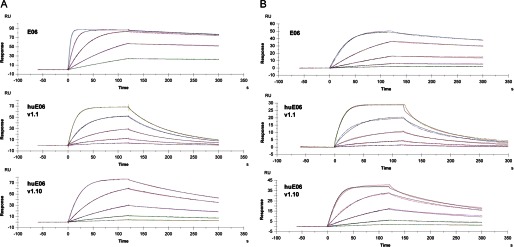
Biacore analysis of monomeric E06, huE06 v1.1, and huE06 v1.10 binding to human serum albumin. A, binding curves at pH 7.4. B, binding curves at pH 6.0. RU, resonance units.
TABLE 3.
Kinetic parameters of human, mouse, and rat albumin binding by E06 and its humanized variants at pH 7.4 and pH 6.0
n/a, not applicable.
| V-NAR | Albumin | pH | ka | kd | KD | -Fold change from E06 |
|---|---|---|---|---|---|---|
| 1/m s | 1/s | nm | ||||
| E06 | 3.48e+06 | 6.55e−04 | 0.19 | n/a | ||
| huE06 v1.1 | Human | 7.4 | 8.72e+05 | 1.40e−02 | 16.0 | 84.9 |
| huE06 v1.10 | 5.59e+05 | 3.57e−03 | 6.4 | 33.9 | ||
| E06 | 2.18e+06 | 1.80e−03 | 0.83 | n/a | ||
| huE06 v1.1 | Mouse | 7.4 | 8.41e+05 | 4.14e−02 | 49.2 | 59.4 |
| huE06 v1.10 | 4.12e+05 | 9.59e−03 | 23.2 | 28.1 | ||
| E06 | 2.21e+06 | 3.20e−03 | 1.45 | n/a | ||
| huE06 v1.1 | Rat | 7.4 | 4.36e+05 | 3.29e−02 | 75.3 | 52.0 |
| huE06 v1.10 | 4.49e+05 | 1.65e−02 | 36.8 | 25.4 | ||
| E06 | 1.12e+07 | 1.59e−03 | 0.14 | n/a | ||
| huE06 v1.1 | Human | 6.0 | 3.62e+06 | 1.84e−02 | 5.07 | 35.6 |
| huE06 v1.10 | 3.52e+06 | 6.37e−03 | 1.81 | 12.7 | ||
| E06 | 9.85e+06 | 3.23e−03 | 0.33 | n/a | ||
| huE06 v1.1 | Mouse | 6.0 | 3.43e+06 | 3.18e−02 | 9.38 | 28.3 |
| huE06 v1.10 | 3.14e+06 | 1.25e−02 | 3.98 | 12.2 |
At pH 6.0, the binding followed the same trend of species preferences and relative affinities between parental E06 and its humanized variants (Fig. 7B and Table 3). However, the humanized variants demonstrated lesser differences from parental E06 at this pH (∼10-fold drop in affinity for v1.10 and ∼30-fold drop for v1.1 compared with parental E06).
Structural Analysis of Residues That May Influence Species Specificity of Albumin Binding by E06
In anticipation of the use of E06 or a humanized variant in future in vivo studies (such as described by Müller et al. in Ref. 33), a thorough understanding of the species specific differences in the E06-albumin interaction is of interest and could contribute to the comparison of studies in different models. As described above, a Biacore study of the binding kinetics of E06 and humanized E06 v1.1 and v1.10 to HSA, mouse serum albumin, and rat serum albumin showed measurable differences in their KD values (Table 3). Our analysis of the structural basis of the varying affinity of E06 for human, mouse, rat, or bovine serum albumin found that species-specific differences in affinity can be attributed to a very small number of albumin residues (Fig. 8A). In particular, Ser232, Asp269, and Ser270 appear to drive the higher affinity of E06 to human versus rat or mouse serum albumin. The side chain atoms of Ser232 are surrounded by close contacts, leaving little room to accommodate the extra methyl group of Thr (Fig. 8B). Asp269 may interact with E06, whereas an Ala in this position would not. The corresponding side chain for interaction in E06 is disordered in the x-ray structures. Ser270 participates in H2O-mediated contacts with E06 and possibly provides a direct H-bond as well. A Thr in this position would likely be less favorable as either contact to E06 would be lost or the additional methyl would be solvent-exposed.
FIGURE 8.
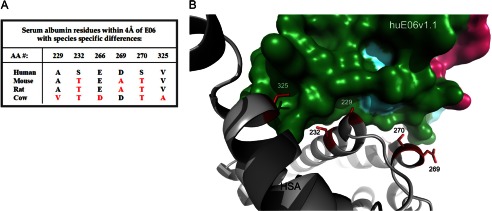
Structural determinants of species specificity of E06-albumin binding. A, E06-proximal residues that differ among human, mouse, rat, and bovine albumins. B, ribbon diagram with side chain sticks of HSA depicting residues that likely confer species specificity. The corresponding binding area of E06 is shown as a surface representation. AA, amino acid.
In addition to the contributions of residues 232, 269, and 270, Val229 and Ala325 are likely to contribute to the lower affinity of E06 specifically to bovine serum albumin. Ala229 of human albumin occupies a small pocket on the surface of E06. There does not appear to be enough space to accommodate Val at this position without steric hindrance (Fig. 8B). Val325 of human albumin fills a hydrophobic pocket on the surface of E06; Ala would be less effective, likely resulting in decreased affinity.
DISCUSSION
Shark IgNARs demonstrate a surprising ability for specific and high affinity binding to diverse antigens by using just two variable domains per molecule, each carrying only two CDRs. The structural basis for this property is a result of additional recombination events in the IgNAR V-D-J cluster and the introduction of junctional diversity, which results in significant heterogeneity of CDR3 sequences. The varying length of CDR3 and the unique positioning of non-canonical cysteine residues in several structural types of V-NARs all help create a remarkable structural plasticity for antigen binding.
Although CDR1 and CDR3 are considered the two major determinants for antigen binding by V-NAR domains, other regions, such as HV2 and HV4, show an increased frequency of somatic mutations, indicating their potential involvement in antigen recognition. However, such an involvement has not been demonstrated previously for shark single domains. In this study, we solved the crystal structure of a type IV V-NAR complexed with its target and demonstrated the remarkable role of V-NAR framework residues in antigen recognition. This structure provides an example of antigen recognition using essentially one single CDR, which is remarkable when viewed in contrast to the six CDRs available for antigen binding for classical antibodies. This is made possible by the extensive use of framework residues for antigen recognition. A significant fraction of those framework interactions come from the HV2 region, corresponding to the observation of increased frequency of somatic mutation. To achieve these contacts, antigen binding is sideways with respect to the antibody framework and the face typically contacting antigen. Although previously described in camelids (VHH in complex with pancreatic α-amylase; Protein Data Bank codes 1KXV and 1KXT; Ref. 14), our structures are the first example of such binding for V-NAR domains. Interestingly, in both cases, CDR1 essentially does not participate in the antigen binding interface, and the same alternative face of the antibody domain makes up the rest of the antigen recognition surface. This alternative face is the same face as would be buried in the dimerization interface of a conventional antibody. Thus, it would seem that the sideways binding phenomenon may be exclusive to single domain antibodies as the side that provides the necessary framework residues is normally buried and not available for antigen recognition. Furthermore, this face of an Ig domain seems to be primed for protein-protein interaction despite the absence of the hydrophobic patch present in conventional antibodies that drives complex formation between heavy and light chains. It remains to be seen whether this binding mode is a more universal phenomenon for type IV V-NARs or an antigen-specific case. A strikingly similar mode of binding is seen in the interaction between the fibronectin type III domain (Fn3 monobody) and estrogen receptor α (Protein Data Bank 2OCF) that features the binding of an α-helical structure (estrogen receptor α) by a surface of β-sheets and loops corresponding to CDR3 and HV2 (37).
The other key aspect of our study is the design of a humanized version of shark V-NAR domain. Such an approach becomes possible due to a high degree of structural homology between V-NAR and Ig VL framework regions despite a low degree of sequence identity. We have demonstrated that replacement of most framework elements in V-NAR with those shared with a human VL scaffold can result in a functional humanized V-NAR molecule with >50% overall human content. As expected, changes in the regions specific to V-NARs (such as HV2 and HV4) result in partial loss of activity. We have provided a structural explanation for the importance of these regions in maintaining overall V-NAR structures. Humanized E06 can serve as a universal scaffold for humanization of other V-NAR binders provided that key elements involved in antigen recognition and V-NAR stability are identified and preserved in humanized molecules.
Acknowledgments
We thank Mark Johnson for assistance in screening and Julia Bianco and Xiaotian Zhong for protein expression. We thank the Aberdeen team for the isolation of E06.
The atomic coordinates and structure factors (codes 4HGK and 4HGM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
O. V. Kovalenko and L. Tchistiakova, unpublished data.
- scFv
- single chain Fv
- IgNAR
- immunoglobulin new antigen receptor
- HSA
- human serum albumin
- HEL
- hen egg white lysozyme
- CDR
- complementarity-determining region
- FW
- framework
- HV
- hypervariable region
- V
- variable region
- VH
- variable heavy chain
- VL
- variable light chain
- VHH
- variable domain of the H chain of heavy chain antibodies
- hFc
- human IgG1 Fc
- CM
- conditioned medium.
REFERENCES
- 1. Binz H. K., Amstutz P., Plückthun A. (2005) Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 23, 1257–1268 [DOI] [PubMed] [Google Scholar]
- 2. Skerra A. (2007) Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 18, 295–304 [DOI] [PubMed] [Google Scholar]
- 3. Carter P. J. (2006) Potent antibody therapeutics by design. Nat. Rev. Immunol. 6, 343–357 [DOI] [PubMed] [Google Scholar]
- 4. Presta L. G. (2008) Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 20, 460–470 [DOI] [PubMed] [Google Scholar]
- 5. Barelle C., Gill D. S., Charlton K. (2009) Shark novel antigen receptors—the next generation of biologic therapeutics? Adv. Exp. Med. Biol. 655, 49–62 [DOI] [PubMed] [Google Scholar]
- 6. Wesolowski J., Alzogaray V., Reyelt J., Unger M., Juarez K., Urrutia M., Cauerhff A., Danquah W., Rissiek B., Scheuplein F., Schwarz N., Adriouch S., Boyer O., Seman M., Licea A., Serreze D. V., Goldbaum F. A., Haag F., Koch-Nolte F. (2009) Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 198, 157–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greenberg A. S., Avila D., Hughes M., Hughes A., McKinney E. C., Flajnik M. F. (1995) A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 374, 168–173 [DOI] [PubMed] [Google Scholar]
- 8. Roux K. H., Greenberg A. S., Greene L., Strelets L., Avila D., McKinney E. C., Flajnik M. F. (1998) Structural analysis of the nurse shark (new) antigen receptor (NAR): molecular convergence of NAR and unusual mammalian immunoglobulins. Proc. Natl. Acad. Sci. U.S.A. 95, 11804–11809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henderson K. A., Streltsov V. A., Coley A. M., Dolezal O., Hudson P. J., Batchelor A. H., Gupta A., Bai T., Murphy V. J., Anders R. F., Foley M., Nuttall S. D. (2007) Structure of an IgNAR-AMA1 complex: targeting a conserved hydrophobic cleft broadens malarial strain recognition. Structure 15, 1452–1466 [DOI] [PubMed] [Google Scholar]
- 10. Stanfield R. L., Dooley H., Verdino P., Flajnik M. F., Wilson I. A. (2007) Maturation of shark single-domain (IgNAR) antibodies: evidence for induced-fit binding. J. Mol. Biol. 367, 358–372 [DOI] [PubMed] [Google Scholar]
- 11. Transue T. R., De Genst E., Ghahroudi M. A., Wyns L., Muyldermans S. (1998) Camel single-domain antibody inhibits enzyme by mimicking carbohydrate substrate. Proteins 32, 515–522 [DOI] [PubMed] [Google Scholar]
- 12. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lauwereys M., Arbabi Ghahroudi M., Desmyter A., Kinne J., Hölzer W., De Genst E., Wyns L., Muyldermans S. (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17, 3512–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Desmyter A., Spinelli S., Payan F., Lauwereys M., Wyns L., Muyldermans S., Cambillau C. (2002) Three camelid VHH domains in complex with porcine pancreatic α-amylase. Inhibition and versatility of binding topology. J. Biol. Chem. 277, 23645–23650 [DOI] [PubMed] [Google Scholar]
- 15. Stanfield R. L., Dooley H., Flajnik M. F., Wilson I. A. (2004) Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 305, 1770–1773 [DOI] [PubMed] [Google Scholar]
- 16. Dooley H., Stanfield R. L., Brady R. A., Flajnik M. F. (2006) First molecular and biochemical analysis of in vivo affinity maturation in an ectothermic vertebrate. Proc. Natl. Acad. Sci. U.S.A. 103, 1846–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J. L., Anderson G. P., Delehanty J. B., Baumann R., Hayhurst A., Goldman E. R. (2007) Selection of cholera toxin specific IgNAR single-domain antibodies from a naive shark library. Mol. Immunol. 44, 1775–1783 [DOI] [PubMed] [Google Scholar]
- 18. Streltsov V. A., Varghese J. N., Carmichael J. A., Irving R. A., Hudson P. J., Nuttall S. D. (2004) Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 12444–12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janin J., Chothia C. (1990) The structure of protein-protein recognition sites. J. Biol. Chem. 265, 16027–16030 [PubMed] [Google Scholar]
- 20. Lo Conte L., Chothia C., Janin J. (1999) The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285, 2177–2198 [DOI] [PubMed] [Google Scholar]
- 21. Fennell B. J., Darmanin-Sheehan A., Hufton S. E., Calabro V., Wu L., Müller M. R., Cao W., Gill D., Cunningham O., Finlay W. J. (2010) Dissection of the IgNAR V domain: molecular scanning and orthologue database mining define novel IgNAR hallmarks and affinity maturation mechanisms. J. Mol. Biol. 400, 155–170 [DOI] [PubMed] [Google Scholar]
- 22. Ewert S., Honegger A., Plückthun A. (2004) Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 34, 184–199 [DOI] [PubMed] [Google Scholar]
- 23. Hwang W. Y., Almagro J. C., Buss T. N., Tan P., Foote J. (2005) Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods 36, 35–42 [DOI] [PubMed] [Google Scholar]
- 24. Tsurushita N., Hinton P. R., Kumar S. (2005) Design of humanized antibodies: from anti-Tac to Zenapax. Methods 36, 69–83 [DOI] [PubMed] [Google Scholar]
- 25. Conrath K., Vincke C., Stijlemans B., Schymkowitz J., Decanniere K., Wyns L., Muyldermans S., Loris R. (2005) Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J. Mol. Biol. 350, 112–125 [DOI] [PubMed] [Google Scholar]
- 26. Vincke C., Loris R., Saerens D., Martinez-Rodriguez S., Muyldermans S., Conrath K. (2009) General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 284, 3273–3284 [DOI] [PubMed] [Google Scholar]
- 27. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 [Google Scholar]
- 28. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Smart O. S., Vonrhein C., Womack T. O. (2009) BUSTER, Version 2.8.0, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Müller M. R., Saunders K., Grace C., Jin M., Piche-Nicholas N., Steven J., O'Dwyer R., Wu L., Khetemenee L., Vugmeyster Y., Hickling T. P., Tchistiakova L., Olland S., Gill D., Jensen A., Barelle C. J. (2012) Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. MAbs 4, 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dooley H., Flajnik M. F., Porter A. J. (2003) Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol. Immunol. 40, 25–33 [DOI] [PubMed] [Google Scholar]
- 35. Ewert S., Huber T., Honegger A., Plückthun A. (2003) Biophysical properties of human antibody variable domains. J. Mol. Biol. 325, 531–553 [DOI] [PubMed] [Google Scholar]
- 36. Simmons D. P., Abregu F. A., Krishnan U. V., Proll D. F., Streltsov V. A., Doughty L., Hattarki M. K., Nuttall S. D. (2006) Dimerisation strategies for shark IgNAR single domain antibody fragments. J. Immunol. Methods 315, 171–184 [DOI] [PubMed] [Google Scholar]
- 37. Koide A., Abbatiello S., Rothgery L., Koide S. (2002) Probing protein conformational changes in living cells by using designer binding proteins: application to the estrogen receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 1253–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]