

Background: GLIC, a pentameric ligand-gated ion channel (pLGIC), is inhibited by general anesthetics, but in anesthetic-bound crystal structures the channel appears open.
Results: Propofol induces structural rearrangements in the GLIC transmembrane domain that increase intrasubunit and decrease intersubunit cavity accessibilities.
Conclusion: Propofol inhibits GLIC by stabilizing a distinct closed channel state.
Significance: This study increases our understanding of how anesthetics modulate pLGIC function.
Keywords: Allosteric Regulation, Anesthetics, Cys-loop Receptors, Electrophysiology, Ligand-binding Protein, Membrane Proteins, Protein Conformation, Protein Structure, Substituted Cysteine Accessibility Method
Abstract
General anesthetics exert many of their CNS actions by binding to and modulating membrane-embedded pentameric ligand-gated ion channels (pLGICs). The structural mechanisms underlying how anesthetics modulate pLGIC function remain largely unknown. GLIC, a prokaryotic pLGIC homologue, is inhibited by general anesthetics, suggesting anesthetics stabilize a closed channel state, but in anesthetic-bound GLIC crystal structures the channel appears open. Here, using functional GLIC channels expressed in oocytes, we examined whether propofol induces structural rearrangements in the GLIC transmembrane domain (TMD). Residues in the GLIC TMD that frame intrasubunit and intersubunit water-accessible cavities were individually mutated to cysteine. We measured and compared the rates of modification of the introduced cysteines by sulfhydryl-reactive reagents in the absence and presence of propofol. Propofol slowed the rate of modification of L240C (intersubunit) and increased the rate of modification of T254C (intrasubunit), indicating that propofol binding induces structural rearrangements in these cavities that alter the local environment near these residues. Propofol acceleration of T254C modification suggests that in the resting state propofol does not bind in the TMD intrasubunit cavity as observed in the crystal structure of GLIC with bound propofol (Nury, H., Van Renterghem, C., Weng, Y., Tran, A., Baaden, M., Dufresne, V., Changeux, J. P., Sonner, J. M., Delarue, M., and Corringer, P. J. (2011) Nature 469, 428–431). In silico docking using a GLIC closed channel homology model suggests propofol binds to intersubunit sites in the TMD in the resting state. Propofol-induced motions in the intersubunit cavity were distinct from motions associated with channel activation, indicating propofol stabilizes a novel closed state.
Introduction
General anesthesia, a routine and indispensable tool in modern surgery, remains a poorly understood phenomenon. General anesthetics (GAs)2 are believed to exert many of their actions by binding to and modulating membrane-embedded pentameric ligand gated ion channels (pLGICs) (2). Heterologous expression studies and experiments using transgenic mice have shown that GAs potentiate anion-selective pLGICs like GABAARs and glycine receptors (3) and inhibit cation-selective pLGICs like nicotinic acetylcholine receptors (4) and GLIC, a prokaryotic pLGIC homolog (1, 5). The molecular details underlying the various actions of GAs on pLGICs, e.g. location of the GA binding site(s), protein movements triggered by GA binding, and structural elements that mediate their positive versus negative allosteric modulation, are not well understood.
pLGICs are composed of five identical or homologous subunits arranged pseudo-symmetrically around a central ion-conducting channel (Fig. 1). Structural knowledge of these proteins comes from a 4 Å-resolution cryo-EM structure of the Torpedo nicotinic acetylcholine receptor in a presumed unliganded closed state (6), crystal structures of full-length prokaryotic pLGIC homologs from Erwinia chrysanthemi (ELIC) and Gloeobacter violaceus (GLIC) solved in presumed closed and open channel conformations (7–9), and a recent crystal structure of an invertebrate glutamate-activated chloride channel (GluCl) in an apparent open channel conformation (10). In general, each subunit can be divided into two parts: an extracellular binding domain made of β-strands and a transmembrane channel domain (TMD) consisting of four α-helical membrane-spanning segments (M1-M4) (Fig. 1). The M2 helices of each of the subunits form the ion-conducting channel. The M1, M3, and M4 segments form an outer ring of helices that are partly exposed to lipid (for review, see Ref. 11). Neurotransmitter binds in the extracellular binding domain at interfaces between subunits (for review, see Ref.12).
FIGURE 1.

GLIC residues substituted to cysteine in the inter- and intrasubunit TMD cavities and channel pore. A, a propofol-bound structure of GLIC (PDB 3P50, (1)) shows the location of propofol (dark blue space-fill) in an intrasubunit cavity. B, the top view of the TMDs of two adjacent subunits (salmon and green) shows mutated residues as sticks. The intersubunit residues Asn-238, Leu-240, and Glu-242 are orange, intrasubunit residues Ile-201, Val-241, and Thr-254 are blue, and T243C facing the channel pore is yellow. The M2-M3 loop has been removed for clarity. C, partial sequences of the extracellular ends of TMD helices M1, M2, and M3 are shown. Residues mutated to cysteine are colored as in B. Residues that line the binding pocket in the propofol-bound crystal structure of GLIC are underlined (1).
Recent data suggest that GAs bind to pLGICs in water-accessible cavities in the TMD located between adjacent subunits (inter-), within a subunit (intra-), and in the channel pore. A photoreactive GA, azi-etomidate, labels residues in the β-α intersubunit cavity of the anion-selective GABAAR at Met-236 in αM1 and Met-286 in βM3 (13), and mutating residues in the intersubunit TMD cavity in GABAARs and glycine receptors alters GA actions (14–16). In cation-selective nicotinic acetylcholine receptors, photoreactive analogs of propofol or etomidate label residues in three locations: an intrasubunit site, an intersubunit site, and the channel pore (17, 18). In recently solved GA-bound crystal structures of the prokaryotic pLGIC homolog GLIC, propofol and desflurane occupy an intrasubunit cavity in the TMD (1). GAs inhibit GLIC current responses (1, 5), suggesting GAs stabilize a closed channel state, but in the propofol- and desflurane-bound structures the channel is in an apparent open conformation. When apo- and GA-bound GLIC structures are compared, GAs induce little change in protein conformation. Whether the static crystal structures of GLIC solved in detergent micelles accurately reflect GA actions on pLGICs embedded in membrane lipids remains largely untested and how GAs inhibit GLIC currents is still unclear. Molecular simulations of GLIC indicate that GAs can bind to multiple sites in the TMD of GLIC and induce motions that close the channel (19–24). So far GA-induced motions have not been demonstrated in functional GLIC channels embedded in a lipid membrane, especially in the resting state.
Here, we used the substituted cysteine accessibility method to test the hypothesis that propofol binds within the intrasubunit cavity of GLIC when the channel is in a resting, non-conducting state and to examine if propofol binding alone induces structural rearrangements in the TMD. Understanding the action of GAs on pLGICs requires not only the identification of their binding site(s) but also knowledge of the structural rearrangements that mediate their allosteric action on pLGIC gating. Using the propofol-bound GLIC crystal structure as a guide, we individually mutated residues in the intrasubunit cavity, intersubunit cavity, and channel pore to cysteine. We measured the rate at which the introduced cysteines were modified by sulfhydryl-specific reagents in the absence and presence of propofol. This approach has been used successfully in a variety of channels to identify binding sites and to detect drug-induced structural movements (25). Propofol increased the rate of modification of T254C in the intrasubunit cavity, indicating that this region moves in response to propofol binding and that in a resting, closed channel state propofol does not bind in the intrasubunit cavity. In silico docking using a GLIC closed channel homology model suggests propofol binds to an intersubunit cavity in the resting state.
MATERIALS AND METHODS
Site-directed Mutagenesis and Expression in Xenopus laevis Oocytes
The DNA sequence encoding GLIC (residues 44–359) was extracted by PCR amplification from G. violaceus cells (ATCC) and subcloned in vectors pUNIV (26) for two-electrode voltage clamp experiments. GLIC DNA sequence was preceded in pUNIV by the DNA sequence encoding the signal peptide of the GABAA receptor β2 subunit to promote cell surface expression. GLIC mutants were created using the QuikChange site-directed mutagenesis kit (Stratagene). Mutations were confirmed by DNA sequencing. Capped cRNAs encoding WT and mutant GLIC were transcribed in vitro using the mMessage mMachine T7 kit (Ambion). Single X. laevis oocytes were injected with 27 nl of cRNA (50–100 ng/μl). Injected oocytes were incubated at 16 °C in ND96 (5 mm HEPES, pH 7.4, 96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2) supplemented with 100 μg/ml gentamycin and 100 μg/ml bovine serum albumin for 2–5 days before use for electrophysiological recordings.
Stock solutions of propofol (Sigma), methyl methanethiosulfonate (MMTS), and 4-(chloromercuri)benzenesulfonate (pCMBS−) (Toronto Research Chemicals, Toronto, Ontario, Canada) were prepared in DMSO, and that of 2-aminoethyl methanethiosulfonate (MTSEA) (Toronto Research Chemicals) was prepared in deionized water. Stocks were kept frozen at −20 °C and diluted to working concentration in ND96 just before use where final DMSO concentration (≤1%) did not affect GLIC function.
Electrophysiological Responses of GLIC to Acidic pH and Propofol
Oocytes were perfused continuously with ND96 at pH 7.6 at a flow rate of 5 ml/min while being held under a two-electrode voltage clamp at −60 mV in a bath volume of 200 μl. Borosilicate glass electrodes (Warner Instruments) used for recordings were filled with 3 m KCl and had resistances of 0.4–1.0 megaohms. Electrophysiological data were collected using GeneClamp 500 (Axon Instruments, Foster City, CA) interfaced to a computer with a Digidata 1200 A/D device (Axon Instruments) and recorded using the Whole Cell Program, Version 4.0.2 (kindly provided by J. Dempster, University of Strathclyde, Glasgow, UK). Proton-induced currents were measured by perfusing ND96 buffered at pH 6.5–3.8. For pH 5.0–3.8, HEPES was replaced with 5 mm sodium citrate as the buffering agent. For pH 6.5–6.0, 5 mm MES was used as the buffering agent.
Proton-induced currents were measured at pH 5.0 until peak current amplitudes varied by < 10%. pH-response curves were obtained by successive applications of 5–6 different pH pulses at room temperature separated by 3–7 min washes. pH dose-response data were fit to Equation 1,
where I is the peak response at a given pH, Imax is the maximum amplitude of current, pH50 is the pH inducing half-maximal response, and nH is the Hill coefficient. GraphPad Prism 4 was used for data analysis and curve fitting.
Propofol modulation experiments were performed at pH5–20. Modulation is defined as (I+propofol/I) where I+propofol is the current in the presence of propofol, and I is the control current elicited by pH5–20. Propofol was pre-applied for 15 s at pH 7.6 before co-application with pH5–20. Propofol alone did not elicit any current responses. To determine concentration dependence of propofol modulation of pH5–20 currents, 5–7 different concentrations of propofol were co-applied with pH5–20. Successive applications were separated by 5–10-min washes. (I+propofol/I) was determined for each propofol concentration and were fit to Equation 2,
where P is the propofol modulation (I+propofol/I) at a given propofol concentration, and Pmax is the peak propofol modulation. IC50 is the propofol concentration causing 50% of Pmax, and nH is the Hill coefficient. For N238C(15′), where propofol potentiated currents, the EC50 for potentiation was determined.
Covalent Modification of Substituted Cysteines
We used three sulfhydryl-specific reagents, MTSEA, MMTS, and pCMBS−, in this study. The functional effect of modifying substituted cysteines with these reagents was evaluated in oocytes using two-electrode voltage clamp. Currents were stabilized before the addition of sulfhydryl reagents by application of pH10–30 buffer at 5-min intervals until the proton-activated currents (I) varied by <10%. After the proton responses were stabilized, freshly diluted sulfhydryl-reactive reagent was applied for 3–5 min; the oocyte was washed for 5 min, and then pH10–30 responses were measured again. The effect of cysteine modification on proton current was calculated as (Imodified/I), where Imodified is the current elicited by protons after sulfhydryl reagent application and complete modification of all available cysteines, and I is the current elicited by protons before sulfhydryl reagent application. For each mutant, initial screening was performed to determine the concentration of sulfhydryl reagent that resulted in maximally altered current responses within 3–5 min (i.e. all available cysteines were modified).
The effect of covalent modification of substituted cysteines on propofol modulation was also determined. Modulation of pH5–20 currents by 100 μm propofol (I+propofol/I) was measured before and after sulfhydryl modification. After sulfhydryl modification, pH5–20 was redetermined by measuring the current responses to 3–4 different pH buffer pulses. This was done to ensure propofol modulation was measured at the same effective proton concentration before and after covalent modification of the cysteines. Propofol modulation of unmodified and modified receptors is plotted side by side for comparison (Figs. 4E and 5E). For T254C, modulation of pH5–20 currents by a lower concentration of propofol (10 μm) was also measured before and after sulfhydryl modification.
FIGURE 4.

Effects of cysteine mutations and MTSEA modification on propofol modulation. A, propofol concentration-inhibition curves from C26A and T254C GLIC channels are shown. B, shown is a propofol concentration-potentiation curve from N238C GLIC channels. Modulation is defined as the ratio of pH5–20 currents in the presence of propofol to those in its absence (I+propofol/I). Propofol IC50/EC50 and Hill coefficients were, respectively, 19 ± 3.6 μm and −0.8 ± 0.01 (C26A), 11 ± 0.5 μm and −1.4 ± 0.2 (T254C), and 14.6 ± 4.6 μm and 2.1 ± 0.4 (N238C). Data are the mean ± S.E. from ≥3 experiments. Mutant IC50/EC50 values were not significantly different compared with C26A value. Insets are representative currents from C26A and N238C showing propofol inhibition and propofol potentiation of proton-mediated currents, respectively. Solid line, pH5–20 application; open bars, 100 μm propofol application. C, MTSEA (5 mm, 2 min) modification of L240C inverts propofol modulation of pH 5.5 currents from inhibition to potentiation. D, MTSEA (5 mm, 2 min) modification of N238C inverts propofol modulation of pH 5.2 currents from potentiation to inhibition. Solid line, pH5–20 application; open bars, 100 μm propofol application. E, shown is a summary of the effects of cysteine mutation (hatched bars) and subsequent MTSEA modification (black bars) on 100 μm propofol modulation of pH5–20 currents (I+propofol/I). Data are the mean ± S.E. from ≥3 experiments. Locations of mutant residues are indicated on the left.
FIGURE 5.

Effect of different sulfhydryl reagent modifications of N238C and L240C on proton-mediated currents and propofol modulation. A, shown are chemical structures of MTSEA, MMTS, and PCMBS−. B, MMTS (5 mm, 2 min) modification of N238C inhibits GLIC currents and preserves propofol potentiation of pH 5.2 currents. C, PCMBS− (1 mm, 2min) modification of L240C potentiates GLIC currents and preserves propofol inhibition of pH 5.2 application currents. Solid line, pH5–20 application; open bars, 100 μm propofol. D, shown is a summary of the effects of MTSEA, MMTS, and PCMBS− modification of N238C and L240C on pH10–30 currents. Ratio of currents after modification to before modification are plotted (Imodified/I). Data are the mean ± S.E. from ≥3 experiments. E, modulation of pH5–20 currents by 100 μm propofol (I+propofol/I) before (unmodified) and after modification of N238C and L240C with MTSEA, MMTS, and PCMBS− are plotted. Data are the mean ± S.E. from ≥3 experiments.
Rate of Modification of Substituted Cysteines
The rate of reaction between cysteine and the sulfhydryl-specific reagents depends on the accessibility and ionization of the substituted cysteine (25). A water-accessible cysteine is more likely to be ionized and, hence, react faster. Steric block by ligands competing for the same site as the sulfhydryl-reactive reagent will reduce the rate of cysteine modification. We used MTSEA for all rate experiments except for N238C. For N238C, the reaction with MMTS was faster than MTSEA and allowed more accurate rate determination. For L240C, pCMBS− reaction rates were also determined to rule out any effect on reaction rates due to the switching of propofol modulation from inhibition to potentiation upon modification of L240C with MTSEA (Figs. 4 and 5).
The rate of covalent modification of introduced cysteines was determined by measuring the effect of sequential applications of sulfhydryl reagents on pH10–30 current responses. pH10–30 buffer was applied, and the cell was washed for 30 s; sulfhydryl reagent was applied for 5–20s in pH 7.6 buffer, and the cell was washed for 2.5min. The procedure was repeated until pH10–30 currents reached a plateau. The effect of propofol on the rate of sulfhydryl modification was tested by co-applying sulfhydryl reagent with propofol (100 μm) in pH 7.6 buffer. In all cases the wash times were adjusted to ensure that currents obtained from test pulses of pH10–30 buffer after brief exposure to high concentrations of propofol were stabilized. This ensured complete drug wash out and that any changes in current amplitudes were the result of sulfhydryl reagent application.
For all rate experiments, the peak currents elicited by the test pulses were plotted against the exposure (×) of sulfhydryl-specific reagent, defined as the cumulative time of MTSEA/MMTS/pCMBS− application multiplied by their respective concentrations,
The data were fit to a single exponential equation,
where I′ is the peak current elicited by the test pH10–30 pulse. I0 is the current elicited by pH10–30 buffer at time t = 0 when no sulfhydryl reactive reagent was applied, Iplateau is the current remaining after all available cysteines were modified (i.e. reaction had proceeded to apparent completion) and is defined by the curve fit, and k2 is the second order rate constant. To verify the accuracy of this protocol, second-order rate constants were determined using at least two different concentrations of sulfhydryl reagent. k2 values were similar, verifying pseudo-unimolecular kinetics. For easier visual comparison between control and experimental data sets, the data in Figs. 6 and 7 were normalized to the maximum current in each experiment.
FIGURE 6.

Propofol slows modification of L240C and has no effect on N238C and E242C modification. A, shown is an example of a rate experiment at L240C where modification by PCMBS− potentiates the proton-induced current; pH20-induced currents were measured before and after successive applications of 100 μm PCMBS− (top) or 200 μm PCMBS + 100 μm propofol (below) in pH 7.6 buffer. PCMBS− exposure time (s) is indicated on top of the arrows. B, current amplitudes were normalized to maximal potentiation by PCMBS− and plotted against cumulative PCMBS exposure time (s) weighted by concentration of PCMBS− (m) and fit with single exponential functions as described under “Materials and Methods.” B–E, shown are rates of modification of L240C (B and C), N238C (D), and E242C (E) in the absence (○) or presence (■) of 100 μm propofol. B and C, propofol significantly slowed modification of L240C by PCMBS− and MTSEA. D and E, propofol had no effects on modification of N238C and E242C by MMTS or MTSEA, respectively. Data points are the mean ± S.E. from ≥3 experiments. Single exponential fits of the data are shown as dotted (Control) and solid (+Propofol) lines. Second order rate constants are reported in Table 2.
FIGURE 7.

Propofol accelerates modification of T254C and has no effect on I201C and V241C modification A, shown is an example of a rate experiment at T254C where modification by MTSEA inhibits proton-induced currents. pH50 currents were measured before and after successive applications of 100 μm MTSEA (top) or 100 μm MTSEA + 100 μm propofol (below) in pH 7.6 buffer. Duration of MTSEA exposure (s) is indicated on top of the arrows. B, current amplitudes were normalized by the initial current and plotted against cumulative exposure time (s) weighted by concentration of MTSEA (m) and fit to single exponential equations. B–E, shown are rates of MTSEA modification of T254C (B and C), I210C (D), and V241C (E) in the absence (○) or presence (■) of 100 μm propofol. Propofol significantly accelerated modification of T254C when MTSEA was applied at pH7.6 (resting) (B) and pH 6.5 (activated/desensitized) (C). D and E, rates of modification of I201C and V241C were unaltered in the presence of propofol in pH 7.6 buffer. For V241C, the data were best fit with an equation describing two sequential pseudo-first order reactions (see “Materials and Methods”; inset, sample currents from a V241C rate experiment). Data are the mean ± S.E. from ≥3 experiments. Exponential fits of the data are shown as dotted (Control) and solid (+Propofol) lines. Second order rate constants are reported in Table 2.
For V241C, there was a delay in the appearance of a functional effect of MTSEA modification, and the rate data for V241C could not be fit using a single pseudo-unimolecular reaction model. Because GLIC is a homopentamer, mutation at a single site introduces five engineered cysteines in the protein. For V241C, modification of multiple cysteines may be required before a functional effect is observed. A similar effect has been reported when measuring the PCMBS− rates of modification for several GLIC cysteine mutants (27). Rate data for V241C were fit using two sequential pseudo unimolecular reactions as described in (27).
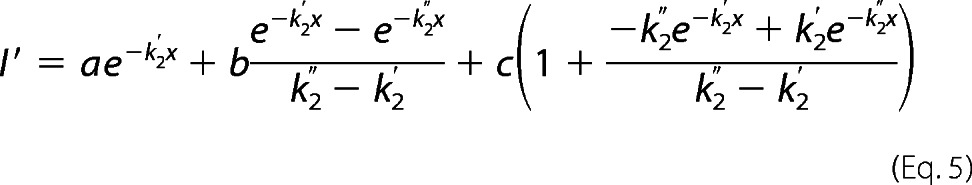 |
where I′ and x are as defined above, k2′ and k2″ are second-order rate constants for the first and the second reaction, respectively, and a, b, and c are constants determined from the fit whose initial values were chosen as a = I, b = 0.1 × I0, and c = 25 × I0.
For T254C, we also determined the rate of MTSEA modification at pH 6.5 in the presence and absence of propofol (100 μm). For this mutant, pH 6.5 elicited about 80% of Imax, and thus we probed the reaction rates of channels in a mixture of open, closed, and desensitized states. Because acidic pH reduces the ionization of cysteines and hence slows their rate of reaction, comparison of rates were performed only between control (pH 6.5 without propofol) and (pH 6.5 + propofol) but not between rates determined at pH 7.6 (closed, resting state) and pH 6.5 due to changes in the ionization state of the cysteine.
Data Analysis and Statistics
Fitting of data and statistical analysis was done using the Prism 4 or Prism 5 software (GraphPad Software Inc., San Diego, CA). All data sets are from ≥3 different oocytes from at least two different frogs. Significant differences in pH50, EC50, IC50, nH, (I+propofol/I), and (Imodified/I) between mutants and WT (C26A) were determined by one way analysis of variance followed by a post hoc Dunnett's test. Log values of EC50 and IC50 were used for statistical analysis. For (Imodified/I), variances among data sets were found to be significant by the Levene's test, so the data were transformed to log values before one-way analysis of variance. Statistical differences in second-order rate constants in the presence and absence of propofol were determined by Student's t test.
Homology Modeling and In-silico Docking
A closed state homology model of GLIC was built using Modeler (28) based on the crystal structure of ELIC (PDB 2VL0), which was solved in the putative closed state (7). GLIC and ELIC share a sequence similarity of only 26%. Therefore, structure-based alignment was performed on a single subunit of GLIC and ELIC using the Dali server (29). For creating multiple chain models the alignment file was modified to include all five chains separated by a slash (/). All the chains were constrained to have a similar conformation by use of pairwise symmetry restraints for C-α atoms in the modeler script used to generate the homology models. Five models returning the lowest molpdf, the standard modeler scoring function, were selected for further evaluation with DOPE (discrete optimized protein energy) score. DOPE is a statistical potential based on an improved reference state that corresponds to non-interacting atoms in a homogeneous sphere with the radius dependent on a sample native structure. The model with the best DOPE score was energy-minimized using the software suite Vega-ZZ (30). The steps for energy minimization were as follows; 1) the addition of hydrogens, 2) fixing charges and potential, and 3) minimization of potential energy. Charges and potential were fixed using the CHARMM22 char and CHARMM22 prot force fields, respectively. 1000 cycles of minimization was performed using conjugate gradient algorithm in the AMMP program included in the Vega-ZZ suite. This model was further energy-minimized using Sybyl (Tripos Corp.) and the Tripos force field until the deviation in positions was 0.05 Å or less (convergence). The final model was used for docking propofol.
Propofol was docked in the TMD of the closed state homology model of GLIC and the open state GLIC crystal structure as a control. Propofol was built using Sybyl Modeling software (Tripos Corp., St. Louis, MO) and energy-minimized using the Tripos force field, and then a random search was performed for the lowest energy conformation. Molecular docking was done using the Autodock4 suite of programs, including the Autodock Tools and the graphics interface ADT (Autodock) (31, 32). Propofol is found in the GLIC crystal structure (PDB 3P50) in an intrasubunit transmembrane cavity (1). As a control, we tested the Autodock parameters necessary to computationally dock propofol in the same site on PDB 3P50. All five monomers of the pentamer were prepared for docking using ADT, adding charges and desolvation to the receptor parameters. The receptor residues remained rigid during all docking experiments. The maximum allowed docking volume was selected using the box size parameters with a center of the box on the propofol binding site in the crystal structure (PDB 3P50). Docking was initiated with the Autodock Lamarkian genetic algorithm followed by a local search to minimize the fit of the bound molecule. Default parameters were modified to include 25 million energy searches, with 300 conformers and selection of 30 results to give a more thorough result. Our control docking placed the propofol in the crystal structure binding site as expected. However, we also found a near neighbor site under the M2-M3 loop in the interface between the TM and the extracellular region. The docking of propofol was then extended in a similar set of circumstances to the homology model of GLIC based on the ELIC crystal structure prepared using Modeler as described. The docking box was adjusted to include the whole TMD along with the channel pore. Autodock clustered the binding sites based on lowest energy and location. Within a cluster, the orientations were similar in the pockets, and the different binding pockets were equally probable. The PyMOL Molecular Graphics System (Version 1.5.0.4 Schrödinger, LLC) and the Autodock/Vina plugin (31–33) were used for visualization of all results.
RESULTS
Seven residues in the TMD of GLIC facing into the intrasubunit cavity, intersubunit cavity, and channel pore were individually mutated to cysteine (Fig. 1). Residues mutated in the intrasubunit cavity were Ile-201 in M1, Val-241 (18′) in M2, and Thr-254 in M3. In the propofol-bound GLIC crystal structure, these residues line the propofol binding pocket (1). In the intersubunit cavity, cysteines were introduced at Asn-238 (15′), Leu-240 (17′), and Glu-242 (19′) in M2. A cysteine was also engineered at Thr-243 (M2-20′), which faces into the ion channel pore. All of the introduced cysteines were made in a Cys-minus background by mutating the GLIC lone cysteine, Cys-26, to an alanine (C26A).
Effects of Mutations and MTSEA Modification on Proton-activated GLIC Currents
All of the mutant subunits assembled into functional GLIC channels. The C26A and E242C mutations had no effects on proton-induced current responses as compared with wild-type GLIC (pH50 ≈ 5.1 Fig. 2, Table 1). Except for T254C in M3, the other cysteine substitutions shifted the pH50 to slightly more acidic pH values. T254C shifted the pH50 to 6.8, a more basic pH (Fig. 2, Table 1). Substituting Thr-254 with alanine also shifts the pH50 to a more basic value (1), suggesting that threonine at this position helps stabilize a closed channel state.
FIGURE 2.

Effects of cysteine mutations on proton induced currents. A, pH-response curves for C26A (dotted line) and mutant GLIC channels are shown. Most mutations shift the pH50 to more acidic pH. T254C shifts the pH50 to a more basic pH. Data are the means ± S.E. from n > 3 experiments. B, shown are representative proton-induced currents from C26A GLIC. pH50 values are reported in Table 1.
TABLE 1.
Summary of pH responses from WT and mutant GLIC channels
Data are the mean ± S.E. from n experiments. pH50 is the pH value that elicited 50% of the maximal proton induced current (Imax). nH is the Hill coefficient.
| Channel | Location | pH50 | nH | Imax | n |
|---|---|---|---|---|---|
| μA | |||||
| WT | 5.1 ± 0.05 | 1.7 ± 0.07 | 13.0 ± 2.4 | 6 | |
| C26A | 5.2 ± 0.07 | 1.8 ± 0.12 | 12.8 ± 1.2 | 9 | |
| I201C (M1) | Intra | 4.6 ± 0.07a | 2.1 ± 0.25 | 5.0 ± 0.8a | 4 |
| N238C (M2-15′) | Inter | 4.4 ± 0.06a | 1.7 ± 0.13 | 4.3 ± 1.0a | 5 |
| L240C (M2-17′) | Inter | 4.6 ± 0.06a | 1.8 ± 0.04 | 9.1 ± 1.9 | 3 |
| V241C (M2-18′) | Intra | 4.3 ± 0.07a | 2.4 ± 0.28 | 1.7 ± 0.7a | 3 |
| E242C (M2-19′) | Inter | 5.0 ± 0.08 | 1.7 ± 0.14 | 10.9 ± 1.5 | 8 |
| T243C (M2-20′) | Pore | 4.6 ± 0.09a | 2.1 ± 0.09 | 4.5 ± 2.1a | 4 |
| T254C (M3) | Intra | 6.8 ± 0.03a | 3.1 ± 0.15a | 9.3 ± 1.2 | 4 |
a Values significantly different from WT; p < 0.01.
We measured currents elicited by pH10–30 concentrations before and after MTSEA application (Fig. 3) to examine how covalently modifying the introduced cysteines would affect proton-activated GLIC currents. MTSEA adds an ethylammonium group to a cysteine ionized sulfhydryl group via a disulfide bond. Treatment with 10 mm MTSEA for 2 min had no effects on the proton-activated currents elicited from C26A channels, indicating that any effects observed in the cysteine mutant channels are due to modification of the introduced cysteines. In the intrasubunit putative propofol binding site cavity, MTSEA modification of V241C and I201C significantly potentiated pH10–30 currents, whereas modification of T254C inhibited currents. In the intersubunit cavity and channel pore, MTSEA modification of N238C, L240C, E242C, and T243C significantly inhibited pH10–30 currents.
FIGURE 3.

MTSEA effects on proton induced currents from C26A and mutant GLIC. A, shown are currents before and after incubation with MTSEA from C26A (10 mm, 2 min), T254C (100 μm, 95s), and I201C (5 mm, 7 min). B, shown are ratios of pH10–30 currents after maximal reaction with MTSEA to currents before MTSEA exposure (Imodified/I). The locations of the introduced cysteines are indicated on the left. Note that x axis is in log scale. Data are the mean ± S.E. from ≥3 experiments. Significance from C26A was tested on log(Imodified/I). Black bars represent p < 0.01.
Effects of Cysteine Substitutions and Sulfhydryl Modification on Propofol Modulation of GLIC Currents
We also measured the effects of cysteine substitutions on propofol modulation of GLIC pH5–20 currents. Propofol inhibited C26A and T254C currents (Fig. 4A) with an IC50 of 19 ± 3.8 μm (n = 3) and 11 ± 0.5 μm (n = 4), respectively. The propofol IC50 for C26A is similar to values reported for wild-type channels (1, 5). Except for N238C (M2-15′, intersubunit cavity), the mutations had no effect on propofol maximal inhibition of GLIC currents (Fig. 4E). Interestingly, N238C switched propofol modulation of GLIC currents from inhibiting to potentiating. Propofol potentiated currents recorded from oocytes expressing N238C GLIC channels (Fig. 4B, E) with an EC50 = 14.6 ± 4.6 μm (n = 5), a value similar to the propofol IC50 for inhibiting C26A and T254C GLIC channels.
We next examined whether MTSEA modification of the introduced cysteines altered propofol modulation (Fig. 4). The intrasubunit cavity, lined by residues Ile-201, Val-241, and Thr-254, is hydrophobic. If propofol binds to this site, as suggested by the propofol-bound GLIC crystal structure (1), one might expect that propofol modulation of GLIC currents would be decreased by covalently attaching the -SCH2CH2NH3+ group into the site. Propofol maximal inhibition of GLIC pH5–20 currents was unaltered after MTSEA modification of these sites. For T254C, inhibition of GLIC pH5–20 current was also measured using a 10 μm concentration of propofol (IC50) before and after MTSEA modification. Propofol modulation was unaltered (data not shown). In summary, neither the cysteine substitutions nor MTSEA modifications at sites in the intrasubunit cavity significantly altered the ability of propofol to maximally inhibit GLIC currents.
In the intersubunit cavity, MTSEA modification of N238C (15′) switched propofol modulation from potentiating back to inhibiting (Fig. 4, D and E), whereas at L240C (17′), MTSEA modification switched propofol modulation from inhibiting to potentiating. At E242C (19′), MTSEA modification had no effect on propofol maximal inhibition. To further investigate how the local physicochemical environments near Asn-238 and Leu-240 influence propofol modulation of GLIC currents, we modified N238C and L240C with two additional sulfhydryl specific reagents: MMTS and pCMBS−.
MMTS covalently adds a relatively small and hydrophobic group, -SCH3, to a cysteine sulfhydryl (Fig. 5A). MMTS modification of N238C or L240C inhibited pH10–30 currents (Fig. 5, B and D), indicating that the introduced cysteines were modified. MMTS modification of these cysteines, however, had little effect on propofol modulation. After MMTS modification of N238C, propofol potentiated pH5–20 currents to a similar magnitude as propofol actions on non-covalently modified N238C channels (Fig. 5, B and E). Likewise, at L240C, propofol inhibited GLIC currents to a similar magnitude whether the cysteine was modified with MMTS or not.
pCMBS− is negatively charged at neutral pH (Fig. 5A) and covalently attaches a relatively large, -HgC6H4SO3−, group onto the sulfhydryl (Fig. 5A). pCMBS− modification of N238C and L240C potentiated GLIC currents (Fig. 5, C and D). Similar to MTSEA, pCMBS− modification of N238C switched propofol modulation from potentiating to inhibiting (Fig. 4E). At L240C, propofol inhibited GLIC currents to a similar magnitude whether the cysteine was modified with pCMBS− or not (Fig. 4, C and E). In summary, in the intersubunit cavity, propofol modulation of GLIC channel gating is strongly influenced by the charge and/or size of the functional groups at the M2-15′ and M2-17′ positions. In a similar manner, it has been reported that alcohol modulation of GLIC currents is influenced by side chain volume at M2-14′ and M2-17′ (34).
Effect of Propofol on Methanethiosulfonate Reaction Rates in the Closed State
To identify potential propofol binding sites and to probe for propofol-induced protein motions in the GLIC TMD, we measured the rates of modification of cysteines introduced in the intersubunit cavity (N238C, L240C, E242C), intrasubunit cavity (I201C, V241C, T254C), and channel pore (T243C) in the absence (control) and presence of 100 μm propofol (+propofol). The rate at which a sulfhydryl reagent reacts with a cysteine side chain depends mainly on the ionization of the thiol group and the access route to the engineered cysteine. Methanethiosulfonate reagents react 5 × 109 times faster with ionized thiolates (-S−) than with thiols (-SH) (25); thus, reaction is much more likely with water-accessible cysteines, which can ionize. Propofol-dependent movement is detected as a change in the rate of modification of the engineered cysteine by a sulfhydryl-specific reagent in the presence of propofol as compared with the rate in its absence. If a substituted cysteine faces into the core of a propofol binding site, we would expect that propofol would slow the rate of its modification by sterically blocking access to the sulfhydryl group. The reaction rates were measured at pH 7.6 to monitor the resting closed GLIC channel state. Based on our proton dose-response curves, the resting state should be predominant.
In the intersubunit cavity, propofol significantly decreased the rate of modification of L240C (17′) by pCMBS− and MTSEA but had no effect on the rates of modification of N238C (15′) or E242C (19′) (Fig. 6 and Table 2). The propofol-induced decrease in L240C modification could be due to propofol blocking access to the engineered cysteine (i.e. L240C lines part of a propofol binding site) or to propofol binding to another region of the protein and allosterically inducing conformational changes near L240C that decrease its accessibility and/or ionization. If Leu-240 lines part of a propofol binding site, one would expect that structural perturbations at the site would alter propofol modulation. Mutating Leu-240 to cysteine had no effect on propofol maximal inhibition of GLIC currents (Fig. 4). In addition, modification of L240C by MMTS and pCMBS− did not decrease the ability of propofol to bind and maximally inhibit GLIC currents (Fig. 5E). Taken together, the data suggest that propofol allosterically induces structural rearrangements in the TMD intersubunit cavity near L240C (M2-17′).
TABLE 2.
Second order rate constants in the absence and presence of propofol
Propofol slowed the rate of modification of L240C (intersubunit cavity) and increased the rate of modification of T254C. Rates were measured at pH 7.6 (resting state) except for T254C, for which rates were also measured at pH 6.5 (activated/desensitized). Data are the mean ± S.E. from n experiments.
| Channel | pH | Location | Reagent |
k2 |
p | |
|---|---|---|---|---|---|---|
| −Propofol | +Propofol | |||||
| m−1s−1 | ||||||
| I201C (M1) | 7.6 | Intra | MTSEA | 3.3 ± 0.4 (n = 4) | 3.1 ± 0.7 (n = 3) | 0.821 |
| N238C (M2 15′) | 7.6 | Inter | MMTS | 22.3 ± 3.0 (n = 6) | 20.7 ± 5.0 (n = 5) | 0.778 |
| L240C (M2 17′) | 7.6 | Inter | pCMBS | 907 ± 66.6 (n = 8) | 634 ± 71.5 (n = 9)a | 0.014 |
| MTSEA | 324 ± 31.0 (n = 5) | 161 ± 19.7 (n = 5)b | 0.002 | |||
| V241C (M2 18′) | 7.6 | Intra | MTSEA | k′ = 16.0 ± 2.8 (n = 5) | k′ = 14.7 ± 4.8 (n = 3) | 0.774 |
| k″ = 29.8 ± 6.15 (n = 5) | k″ = 22.6 ± 5.5 (n = 3) | 0.459 | ||||
| E242C (M2 19′) | 7.6 | Inter | MTSEA | 9.0 × 104 ± 1.0 × 104 (n = 4) | 8.9 × 104 ± 1.9 × 104 (n = 5) | 0.991 |
| T243C (M2 20′) | 7.6 | Pore | MTSEA | 1.2 × 105 ± 3.7 × 103 (n = 3) | 1.3 × 105 ± 7.3 × 103 (n = 3) | 0.513 |
| T254C (M3) | 7.6 | Intra | MTSEA | 374.1 ± 25.1 (n = 4) | 1267.0 ± 171.4 (n = 4)b | 0.002 |
| 6.5 | MTSEA | 70.0 ± 12.8 (n = 5) | 140.0 ± 16.6 (n = 5) a | 0.01 | ||
a Rates in the presence of propofol were significantly different than those in its absence; p < 0.05.
b Rates in the presence of propofol were significantly different than those in its absence; p < 0.01.
In the intrasubunit binding cavity, at pH 7.6, propofol significantly increased the rate of MTSEA modification of T254C and had no effect on the rates of modification of I201C and V241C, indicating that these residues are not lining the core of a propofol binding site in the resting, closed state of GLIC (Fig. 6, Table 2). The data suggest that propofol binding to GLIC in the resting state caused structural rearrangements near T254C that made it more accessible and/or ionized. These data are not consistent with the propofol-bound crystal structure of GLIC, where propofol occupies the intrasubunit cavity (1). However, the propofol bound GLIC crystal structure was solved at pH 4.0 with the channel in an apparent open state configuration (1, 8, 35–37). Therefore, to investigate if propofol binds in the intrasubunit cavity in an open/desensitized channel state, we compared the rates of modification of T254C at pH 6.5 in the presence and absence of propofol. For T254C channels, pH 6.5 elicits nearly 80% of the maximal currents (pH50 = 6.8). If T254C is part of a propofol binding site in open and/or desensitized states, then propofol should slow modification when co-applied with MTSEA at pH 6.5. Even at pH 6.5, propofol still significantly accelerated modification of T254C, suggesting that it does not line the core of a propofol binding site in an open/desensitized state (Fig. 7C; Table 2). Modification of T254C at pH 6.5 in the absence or presence of propofol was slower than at pH 7.6 (Table 2). This is expected as cysteine modification rate depends partly on its ionization, which is decreased at low pH. Interestingly, at pH 7.6, propofol increased the rate of modification of T254C 3-fold, whereas at pH 6.5 the rate increased 2-fold, suggesting that proton-induced conformational changes near T254C may offset some of the propofol-induced increase in its modification.
In the extracellular end of the channel, MTSEA modification of T243C facing the channel pore was unaltered in the presence of propofol, suggesting that this region does not contribute to a propofol binding site. To summarize, comparison of cysteine modification rates measured at pH 7.6 in the presence and absence of propofol indicates that propofol binding to GLIC in the resting state of the channel induces motions in the extracellular end of the TMD and alters the accessibility of L240C and T254C.
Propofol Preferentially Docks in Intersubunit Sites in Closed-state Models of GLIC
To further investigate potential sites for propofol binding in GLIC, we built a closed state homology model of GLIC based on the crystal structure of ELIC (7) (PDB 2VL0). This closed-state homology model was then used for in silico docking of propofol. Propofol docked at two sites (Fig. 8). One site was nestled under the M2-M3 loop in an intersubunit cavity between adjacent subunits (Fig. 8B). The second propofol site was in an intersubunit hydrophobic pocket near the cytoplasmic end of the TMD facing the lipid (Fig. 8C). The second location is similar to a site recently identified on ELIC for bromoform (38). In our dockings, propofol never docked in the TMD intrasubunit cavity or channel lumen, consistent with our experimental data. In addition, when we used the recently solved locally closed GLIC structure (39) (PDB 3TLU), propofol docked in the intersubunit cavity under the M2-M3 loop and not in the intrasubunit cavity (data not shown). Although propofol decreased modification of L240C (Fig. 6, B and C), our docking results suggest that propofol does not bind near this residue. Thus, decreases in L240C modification rates are likely due to propofol-induced structural motions. We also docked propofol using the crystal structure of GLIC in the apparently open conformation (PDB 3P50) (Fig. 8D). Propofol docked in the intrasubunit cavity as previously observed crystallographically (1) and computationally (21, 24). In addition, propofol was found in the intersubunit site under the M2-M3 loop though less frequently.
FIGURE 8.

Propofol docks at intersubunit sites in a GLIC closed state homology model. A, propofol docking to a GLIC closed state homology model built using the crystal structure of ELIC (PDB 2VL0) is shown. Propofol docked at two locations in the intersubunit interface; one site is under the M2-M3 loop between adjacent subunits (salmon and green), and the second site is a hydrophobic pocket bordered by the M3 and M1 helices of adjacent subunits near the cytoplasmic end of the TMD facing the lipid. Propofol docking poses are shown as yellow sticks overlaid with the Van der Waal's surface of the ensemble. B, an expanded top view of TMD of two adjacent subunits shows the docked propofol poses under the M2-M3 loop. C, an upright expanded view of two adjacent subunits shows the docked propofol poses near the cytoplasmic end of the TMD. D, propofol docking to GLIC crystal structure (PDB 3P50) is shown. Propofol docks in both the intra- and intersubunit cavities. In all panels, two adjacent subunits in the foreground are colored salmon and green, the mutated intersubunit residues Asn-238, Leu-240, and Glu-242 are orange sticks, and intrasubunit residues Ile-201, Val-241, and Thr-254 are blue sticks.
DISCUSSION
General anesthetics exert many of their actions by binding to and modulating membrane-embedded pLGICs (2–4). Although the crystallization of GLIC with the GAs propofol and desflurane provide the first atomic resolution structures of a GA-bound pLGIC (1), it is unclear whether the intrasubunit TMD anesthetic binding pocket identified corresponds to the site, where anesthetics interact in functional membrane-embedded pLGICs. In addition, the anesthetic-bound GLIC structures are indistinguishable from the apoGLIC structure (1) and yield little information about the structural mechanisms underlying GA inhibition of GLIC currents. Here, using functional GLIC channels expressed in oocytes, we provide evidence that propofol does not bind in the TMD intrasubunit cavity in the resting state. In silico docking using GLIC closed channel models suggests propofol binds to intersubunit sites in the TMD in the resting state. In addition, using SCAM (substituted cysteine accessibility method), we detect propofol-induced motions in the TMD in both the intersubunit and intrasubunit cavities, indicating propofol binding causes structural rearrangements in these cavities that stabilize a distinct closed channel conformation.
Several lines of evidence support our conclusion that Ile-201, Val-241, and Thr-254 in the GLIC TMD intrasubunit cavity, which lie near propofol in the crystal structure (1), do not line the core of the propofol binding site in the resting state. First, neither their mutation to cysteine nor covalent addition of a charged ethyl ammonium group impaired propofol maximal inhibition of GLIC currents (Fig. 3E). For Thr-254, mutation to cysteine decreased propofol IC50 ∼2-fold, and cysteine modification had no effect on propofol IC50 for inhibition. Second, propofol did not slow covalent modification of I201C, V241C, and T254C (Fig. 7; Table 2). If propofol was binding in the intrasubunit cavity near these residues, we would expect it to sterically block access and slow their modification. Third, in silico docking using a GLIC closed state homology model, suggests propofol does not bind in the intrasubunit cavity (Fig. 8). In our putative resting state model, the size of the intrasubunit cavity is smaller than in the GLIC putative open state structure and likely prevents propofol from docking in this pocket. Recently, using a photoreactive propofol analog, residues in the intrasubunit cavity of nicotinic acetylcholine receptor and GLIC were only labeled in the presence of agonist (18, 40), suggesting that, in the absence of agonist (i.e. resting state), propofol does not bind in the TMD intrasubunit cavity.
The effects of mutating Ile-201, Val-241, and Thr-254 in GLIC have been reported in a previous study (1). Mutation of Ile-201 to alanine, tryptophan, and tyrosine had no effects on the IC50 for propofol inhibition of GLIC currents, consistent with our results that I201C had little effect on propofol maximal inhibition. Mutation of Val-241 to methionine and Thr-254 to alanine significantly decreased propofol IC50 ∼10-fold (i.e. increased propofol apparent affinity). For T254C, we measured only a 2-fold decrease in propofol IC50. The difference is likely due to substituting a cysteine versus an alanine at this position. Overall, the mutations had little adverse effect on propofol inhibition of GLIC currents. One might expect, if these residues line the propofol binding site and contact propofol, that mutating the residues would inhibit propofol modulation rather than enhance it. Alternatively, as suggested by Nury et al. (1), the decrease in propofol IC50 may result from the Val-241 and Thr-254 mutations decreasing propofol mobility in the binding site and promoting favorable contacts with propofol. Our data demonstrating that covalently attaching sulfhydryl-reactive reagents at these sites has little to no effect on propofol maximal modulation and that propofol increases the rate of modification of T254C rather than decreases the rate support the idea that these residues do not line the core of the propofol binding site in the resting state and the effects of the mutations are due to an allosteric mechanism.
Where does propofol bind to GLIC in the resting state? Because Ile-201, Val-241, and Thr-254 are oriented somewhat deep into the intrasubunit cavity, we cannot rule out the possibility that propofol binds along the lipid protein interface bordering the intrasubunit cavity. Ordered lipid molecules seen at the mouth of the intrasubunit cavity in the apoGLIC crystal structure (8) were displaced in the propofol-bound structure (1), hinting at a possible interaction of lipids and GAs. Alternatively, our in silico docking studies revealed two potential TMD intersubunit propofol binding pockets in a GLIC closed channel state homology model. One intersubunit cavity was positioned under the M2-M3 loop (Fig. 8B), which is a crucial structural element that couples ligand binding at the orthosteric site to channel gating (41–43). Binding of GAs near this loop may interfere with motions involved in channel gating (8) and thus stabilize a closed state. The other potential propofol binding site identified was positioned in the TMD between the M3 helix of one subunit and the M1 helix of the adjacent subunit near the cytoplasmic ends of the helices (Fig. 8C). A similar site was recently identified in ELIC, which binds bromoform (38). In eukaryotic GABAA receptors, an etomidate binding site is located at a TMD interface between the β and α subunits (13) at a position slightly above the site where we observed propofol docking. The potential intersubunit propofol binding sites we have identified by in silico docking are distant from the residues we probed. Further experimentation is needed to establish if these sites correspond to a native propofol binding site in GLIC.
Regardless of where propofol binds, in the absence of agonist, propofol triggered distinct structural rearrangements in the TMD of GLIC. Propofol decreased modification of L17′C (M2) and increased modification of T254C (M3) (Figs. 6 and 7). Interestingly, proton activation of GLIC speeds modification of L17′C, suggesting channel opening makes it more accessible (27). Thus, depending on the ligand and its functional effect on GLIC gating, structural changes near L17′C in the intersubunit cavity differ. Propofol inhibits GLIC current responses and stabilizes a closed channel conformation, which results in L17′C being more buried and less reactive and T254C being more exposed and reactive. We envision propofol binding triggers an inward motion of M2 toward the pore axis that stabilizes a novel closed channel conformation that is different from resting and open state conformations. Distinct propofol-induced conformational states have been observed in eukaryotic GABAARs (44).
Our data also demonstrate that the amino acid composition of the intersubunit cavity plays a crucial role in propofol allosteric modulation of GLIC. Altering the hydrophobicity/hydrophilicity of the M2-15′ and M2-17′ side chains by mutation or covalent modification controls whether propofol inhibits or potentiates GLIC currents (Figs. 4 and 5). Similar results have been reported for alcohol modulation of GLIC. Mutating Leu-17′ to methionine switches ethanol modulation of GLIC currents from potentiating to inhibiting and mutating Phe-14′ to cysteine or alanine switches butanol modulation from inhibiting to potentiating (34). Notably, in eukaryotic GABAARs and glycine receptors, side-chain volume (45, 46) and hydrophobicity (47) at M2-15′ are also important determinants of the extent and direction of alcohol and GA modulation, highlighting the evolutionarily conserved role of this position in pLGIC modulation. Statistical covariance analysis suggests that this residue is part of a cluster that co-evolved in the pLGIC family (48).
The marked effects that the mutations and modifications in the intersubunit cavity of GLIC have on propofol and alcohol modulation raises the possibility that these modifications uncover a separate potentiating site in the intersubunit cavity. In a recent communication, an ethanol-bound crystal structure of a mutant GLIC (F14′A) shows ethanol in the intersubunit cavity near N15′(49). Ethanol potentiates F14′A GLIC, suggesting that the intersubunit cavity may be a potentiating site for allosteric modulators. In our study we did not find evidence for propofol binding near Asn-15′ and Glu-19′ (Fig. 6, D and E); i.e. rates of modification of N15′C and E19′E were unchanged in the presence of propofol. Propofol, however, decreased the modification rate of L17′C. The decrease in modification could be due to propofol blocking access (i.e. L17′C lines part of a propofol binding site) or to propofol allosterically inducing conformational changes. Modification of L17′C had no effect on propofol maximal modulation, suggesting an allosteric mechanism. We cannot, however, rule out the possibility that propofol slows the rate of modification of L17′C by steric block, and further experiments are needed.
Whether potentiation and inhibition by GAs are mediated by binding to separate sites on pLGICs or a single site is still unclear (34, 50–53). In GABAARs, both positive and negative benzodiazepine modulators bind to the same site to mediate their actions (54–56) and point mutations in the benzodiazepine binding site can convert a BZD-positive modulator to a negative modulator (57). Likewise in GPCRs, structurally similar agonists binding to the same orthosteric site can activate various G-proteins via different structural mechanisms (58–60). More recently, a novel allosteric modulator of the ionotropic serotonin receptor 5HT3R was demonstrated to mediate both positive and negative allosteric modulation via binding to a single intersubunit site (28). Regardless of presence of single or multiple binding sites, our results underline the importance of the intersubunit cavity in allosteric modulation of pLGICs.
In summary, our data provide new insights into the mechanism of inhibition of cation-selective pLGICs by GAs and supports the use of GLIC as a simple and effective model for studying allosteric modulation of eukaryotic pLGICs. Although x-ray crystal structures have advanced our understanding of the structural basis underlying pLGIC function, detailed functional studies are required to test these mechanisms. We have shown that propofol binding induced structural rearrangements in GLIC that altered the intersubunit and intrasubunit cavities in the TMD. Propofol-induced motions in the intersubunit cavity were distinct from motions associated with channel activation, indicating that the intersubunit TMD cavity plays a critical role in mediating propofol actions. Moreover, our data provide structural evidence for a novel closed state stabilized by propofol binding. Anesthetic modulation of GLIC shares several features with GA modulation of eukaryotic pLGICs and indicates that GLIC is a good model for understanding of anesthetic actions in pLGICs.
Acknowledgments
We thank Dr. Cosma Dellisanti for cloning GLIC in pUNIV vector and James Raspanti and Gaoussou Diarra for oocyte extraction from X. laevis.
This work was supported, in whole or in part, by National Institutes of Health Grant 34727 (NINDS; to C. C.).
- GA
- general anesthetic
- pLGIC
- pentameric ligand-gated ion channel
- GLIC
- G. violaceus ligand gated ion channel
- TMD
- transmembrane domain
- MTSEA
- 2-aminoethyl methanethiosulfonate
- MMTS
- methyl methanethiosulfonate
- pCMBS−
- p-chloromercuribenzenesulfonate
- GABAAR
- GABAA receptor
- ELIC
- Erwinia chrysanthemi ion channel.
REFERENCES
- 1. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 2. Franks N. P., Lieb W. R. (1994) Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614 [DOI] [PubMed] [Google Scholar]
- 3. Zeller A., Jurd R., Lambert S., Arras M., Drexler B., Grashoff C., Antkowiak B., Rudolph U. (2008) Inhibitory ligand-gated ion channels as substrates for general anesthetic actions. Handb. Exp. Pharmacol. 182, 31–51 [DOI] [PubMed] [Google Scholar]
- 4. Akk G., Mennerick S., Steinbach J. H. (2008) Actions of anesthetics on excitatory transmitter-gated channels. Handb. Exp. Pharmacol. 182, 53–84 [DOI] [PubMed] [Google Scholar]
- 5. Weng Y., Yang L., Corringer P. J., Sonner J. M. (2010) Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth. Analg. 110, 59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 7. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 8. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 9. Hilf R. J., Dutzler R. (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 10. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baenziger J. E., Corringer P. J. (2011) 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 60, 116–125 [DOI] [PubMed] [Google Scholar]
- 12. Miller P. S., Smart T. G. (2010) Binding, activation, and modulation of Cys-loop receptors. Trends Pharmacol. Sci. 31, 161–174 [DOI] [PubMed] [Google Scholar]
- 13. Li G. D., Chiara D. C., Sawyer G. W., Husain S. S., Olsen R. W., Cohen J. B. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 26, 11599–11605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Belelli D., Lambert J. J., Peters J. A., Wafford K., Whiting P. J. (1997) The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 94, 11031–11036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mihic S. J., Ye Q., Wick M. J., Koltchine V. V., Krasowski M. D., Finn S. E., Mascia M. P., Valenzuela C. F., Hanson K. K., Greenblatt E. P., Harris R. A., Harrison N. L. (1997) Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 389, 385–389 [DOI] [PubMed] [Google Scholar]
- 16. Ahrens J., Leuwer M., Stachura S., Krampfl K., Belelli D., Lambert J. J., Haeseler G. (2008) A transmembrane residue influences the interaction of propofol with the strychnine-sensitive glycine α1 and α1β receptor. Anesth. Analg. 107, 1875–1883 [DOI] [PubMed] [Google Scholar]
- 17. Hamouda A. K., Stewart D. S., Husain S. S., Cohen J. B. (2011) Multiple transmembrane binding sites for p-trifluoromethyldiazirinyl-etomidate, a photoreactive Torpedo nicotinic acetylcholine receptor allosteric inhibitor. J. Biol. Chem. 286, 20466–20477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jayakar S. S., Dailey W. P., Eckenhoff R. G., Cohen J. B. (2013) Identification of propofol binding sites in a nicotinic acetylcholine receptor with a photoreactive propofol analog. J. Biol. Chem. 288, 6178–6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brannigan G., LeBard D. N., Hénin J., Eckenhoff R. G., Klein M. L. (2010) Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. U.S.A. 107, 14122–14127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Q., Cheng M. H., Xu Y., Tang P. (2010) Anesthetic binding in a pentameric ligand-gated ion channel. GLIC. Biophys. J. 99, 1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu R., Perez-Aguilar J. M., Liang D., Saven J. G. (2012) Binding site and affinity prediction of general anesthetics to protein targets using docking. Anesth. Analg. 114, 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willenbring D., Liu L. T., Mowrey D., Xu Y., Tang P. (2011) Isoflurane alters the structure and dynamics of GLIC. Biophys. J. 101, 1905–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lebard D. N., Hénin J., Eckenhoff R. G., Klein M. L., Brannigan G. (2012) General anesthetics predicted to block the GLIC pore with micromolar affinity. PLoS Comput. Biol. 8, e1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mowrey D., Cheng M. H., Liu L. T., Willenbring D., Lu X., Wymore T., Xu Y., Tang P. (2013) Asymmetric ligand binding facilitates conformational transitions in pentameric ligand-gated ion channels. J. Am. Chem. Soc. 135, 2172–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karlin A., Akabas M. H. (1998) Substituted-cysteine accessibility method. Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 26. Venkatachalan S. P., Bushman J. D., Mercado J. L., Sancar F., Christopherson K. R., Boileau A. J. (2007) Optimized expression vector for ion channel studies in Xenopus oocytes and mammalian cells using alfalfa mosaic virus. Pflugers Arch. 454, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parikh R. B., Bali M., Akabas M. H. (2011) Structure of the M2 transmembrane segment of GLIC, a prokaryotic Cys loop receptor homologue from Gloeobacter violaceus, probed by substituted cysteine accessibility. J. Biol. Chem. 286, 14098–140109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 30. Pedretti A., Villa L., Vistoli G. (2004) VEGA. An open platform to develop chemo-bioinformatics applications using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 18, 167–173 [DOI] [PubMed] [Google Scholar]
- 31. Huey R., Morris G. M., Olson A. J., Goodsell D. S. (2007) A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 28, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 32. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 [Google Scholar]
- 33. Trott O., Olson A. J. (2010) AutoDock Vina. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Howard R. J., Murail S., Ondricek K. E., Corringer P. J., Lindahl E., Trudell J. R., Harris R. A. (2011) Structural basis for alcohol modulation of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U.S.A. 108, 12149–12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nury H., Poitevin F., Van Renterghem C., Changeux J. P., Corringer P. J., Delarue M., Baaden M. (2010) One-microsecond molecular dynamics simulation of channel gating in a nicotinic receptor homologue. Proc. Natl. Acad. Sci. U.S.A. 107, 6275–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu F., Hummer G. (2010) Pore opening and closing of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U.S.A. 107, 19814–19819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu F., Hummer G. (2009) Gating transition of pentameric ligand-gated ion channels. Biophys. J. 97, 2456–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spurny R., Billen B., Howard R. J., Brams M., Debaveye S., Price K. L., Weston D. A., Strelkov S. V., Tytgat J., Bertrand S., Bertrand D., Lummis S. C., Ulens C. (2013) Multi-site binding of a general anesthetic to the prokaryotic pentameric ligand-gated ion channel ELIC. J. Biol. Chem. 288, 8355–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prevost M. S., Sauguet L., Nury H., Van Renterghem C., Huon C., Poitevin F., Baaden M., Delarue M., Corringer P. J. (2012) A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat. Struct. Mol. Biol. 19, 642–649 [DOI] [PubMed] [Google Scholar]
- 40. Chiara D. C., Chen Q., Tillman T., Gill J. F., Dailey W. P., Eckenhoff R. G., Xu Y., Tang P., Cohen J. B. (2012) Photoaffinity labeling the propofol binding site in GLIC. Biophys. J. 104, 636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kash T. L., Jenkins A., Kelley J. C., Trudell J. R., Harrison N. L. (2003) Coupling of agonist binding to channel gating in the GABA(A) receptor. Nature 421, 272–275 [DOI] [PubMed] [Google Scholar]
- 42. Lee W. Y., Sine S. M. (2005) Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 438, 243–247 [DOI] [PubMed] [Google Scholar]
- 43. Jha A., Cadugan D. J., Purohit P., Auerbach A. (2007) Acetylcholine receptor gating at extracellular transmembrane domain interface. The cys-loop and M2-M3 linker. J. Gen. Physiol. 130, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Williams D. B., Akabas M. H. (2002) Structural evidence that propofol stabilizes different GABA(A) receptor states at potentiating and activating concentrations. J. Neurosci. 22, 7417–7424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ye Q., Koltchine V. V., Mihic S. J., Mascia M. P., Wick M. J., Finn S. E., Harrison N. L., Harris R. A. (1998) Enhancement of glycine receptor function by ethanol is inversely correlated with molecular volume at position α267. J. Biol. Chem. 273, 3314–3319 [DOI] [PubMed] [Google Scholar]
- 46. Koltchine V. V., Finn S. E., Jenkins A., Nikolaeva N., Lin A., Harrison N. L. (1999) Agonist gating and isoflurane potentiation in the human γ-aminobutyric acid type A receptor determined by the volume of a second transmembrane domain residue. Mol. Pharmacol. 56, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 47. Morris K. D., Amin J. (2004) Insight into the mechanism of action of neuroactive steroids. Mol. Pharmacol. 66, 56–69 [DOI] [PubMed] [Google Scholar]
- 48. Chen Y., Reilly K., Chang Y. (2006) Evolutionarily conserved allosteric network in the Cys loop family of ligand-gated ion channels revealed by statistical covariance analyses. J. Biol. Chem. 281, 18184–18192 [DOI] [PubMed] [Google Scholar]
- 49. Sauguet L., Howard R. J., Malherbe L., Lee U. S., Corringer P. J., Adron Harris R., Delarue M. (2013) Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat. Commun. 4, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Borghese C. M., Henderson L. A., Bleck V., Trudell J. R., Harris R. A. (2003) Sites of excitatory and inhibitory actions of alcohols on neuronal α2β4 nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 307, 42–52 [DOI] [PubMed] [Google Scholar]
- 51. Collins T., Millar N. S. (2010) Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol. Pharmacol. 78, 198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trattnig S. M., Harpsøe K., Thygesen S. B., Rahr L. M., Ahring P. K., Balle T., Jensen A. A. (2012) Discovery of a novel allosteric modulator of 5-HT3 receptors. Inhibition and potentiation of Cys-loop receptor signaling through a conserved transmembrane intersubunit site. J. Biol. Chem. 287, 25241–25254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murail S., Howard R. J., Broemstrup T., Bertaccini E. J., Harris R. A., Trudell J. R., Lindahl E. (2012) Molecular mechanism for the dual alcohol modulation of Cys-loop receptors. PLoS Comput. Biol. 8, e1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boileau A. J., Czajkowski C. (1999) Identification of transduction elements for benzodiazepine modulation of the GABA(A) receptor. Three residues are required for allosteric coupling. J. Neurosci. 19, 10213–10220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hanson S. M., Czajkowski C. (2008) Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. J. Neurosci. 28, 3490–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sancar F., Czajkowski C. (2011) Allosteric modulators induce distinct movements at the GABA-binding site interface of the GABA-A receptor. Neuropharmacology 60, 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morlock E. V., Czajkowski C. (2011) Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Mol. Pharmacol. 80, 14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghanouni P., Gryczynski Z., Steenhuis J. J., Lee T. W., Farrens D. L., Lakowicz J. R., Kobilka B. K. (2001) Functionally different agonists induce distinct conformations in the G protein coupling domain of the β2 adrenergic receptor. J. Biol. Chem. 276, 24433–24436 [DOI] [PubMed] [Google Scholar]
- 59. Swaminath G., Xiang Y., Lee T. W., Steenhuis J., Parnot C., Kobilka B. K. (2004) Sequential binding of agonists to the β2 adrenoceptor. Kinetic evidence for intermediate conformational states. J. Biol. Chem. 279, 686–691 [DOI] [PubMed] [Google Scholar]
- 60. Swaminath G., Deupi X., Lee T. W., Zhu W., Thian F. S., Kobilka T. S., Kobilka B. (2005) Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J. Biol. Chem. 280, 22165–22171 [DOI] [PubMed] [Google Scholar]