

Background: The role of autophagy in osteocytes is unclear.
Results: Suppression of autophagy in osteocytes caused decreases in bone mass and bone remodeling similar to those caused by aging.
Conclusion: Autophagy in osteocytes maintains the rate of remodeling and bone mass.
Significance: A decline in autophagy in osteocytes may contribute to skeletal aging.
Keywords: Aging, Autophagy, Bone, Mouse, Osteocyte, Remodeling
Abstract
Bone mass declines with age but the mechanisms responsible remain unclear. Here we demonstrate that deletion of a conditional allele for Atg7, a gene essential for autophagy, from osteocytes caused low bone mass in 6-month-old male and female mice. Cancellous bone volume and cortical thickness were decreased, and cortical porosity increased, in conditional knock-out mice compared with control littermates. These changes were associated with low osteoclast number, osteoblast number, bone formation rate, and wall width in the cancellous bone of conditional knock-out mice. In addition, oxidative stress was higher in the bones of conditional knock-out mice as measured by reactive oxygen species levels in the bone marrow and by p66shc phosphorylation in L6 vertebra. Each of these changes has been previously demonstrated in the bones of old versus young adult mice. Thus, these results demonstrate that suppression of autophagy in osteocytes mimics, in many aspects, the impact of aging on the skeleton and suggest that a decline in autophagy with age may contribute to the low bone mass associated with aging.
Introduction
Aging causes bone loss but the underlying mechanisms are only partially understood. The adult skeleton is continuously remodeled by teams of osteoclasts, which resorb bone, and teams of osteoblasts, which form new bone (1). These teams function together within an anatomical structure known as the basic multicellular unit (BMU).2 Because bone mass declines with age, it is clear that the balance between resorption and formation within the BMU becomes negative with aging. Reduced production of sex steroids causes a rise in the rate of bone remodeling that subsides with time, but age-associated bone loss occurs even in individuals with normal levels of sex steroids (2). Importantly, rodents do not experience a significant decline in sex steroids with age but bone mass declines nonetheless, suggesting that mechanisms intrinsic to bone cells contribute to skeletal involution with age (3). However, these intrinsic mechanisms remain unclear.
Autophagy is an intracellular recycling pathway in which cellular components, including protein aggregates and organelles, are targeted to the lysosome for degradation (4). There are multiple forms of autophagy, which are distinguished from one another by the mechanisms used to target cellular components to the lysosome (5). Macroautophagy, hereafter referred to simply as autophagy, accomplishes this by engulfing the components to be degraded in a double-membrane structure known as the autophagosome, which eventually fuses with lysosomes (6). One of the major functions of autophagy is to transform cellular components into substrates for energy production when nutrients are limiting. A second important function is to recycle damaged or malfunctioning proteins and organelles. Indeed, suppression of autophagy leads to the accumulation of damaged mitochondria which produce elevated levels of reactive oxygen species (ROS) (7–9). Because this recycling function is important for the health and viability of long-lived cell types, reduced efficiency of the autophagic process has been proposed as an underlying cause of cellular and organismal aging (10–12). Consistent with this idea, genetic suppression of autophagy in cell types such as neurons and myocytes mimics the effects of aging on tissues containing these cell types (13, 14).
Osteocytes are former osteoblasts that are buried within the bone matrix and remain connected to one another and cells on the bone surface via a network of cell projections (15). In contrast to osteoclasts and osteoblasts, which reside on the bone surface and are short-lived cells, osteocytes may live many months or years, depending on the location within the skeleton and the rate of bone remodeling (16). Moreover, osteocytes are an important source of receptor activator of NFκB ligand (RANKL), which controls osteoclast formation, and sclerostin, which controls osteoblast formation (17–20). Thus changes in osteocyte function with age may alter remodeling and thereby contribute to the low bone mass associated with aging (16).
Herein we present evidence that genetic suppression of autophagy in murine osteocytes causes skeletal changes in young adult mice that are similar to those present in old wild type mice. Thus a decline in osteocyte autophagy may contribute to the skeletal changes that occur during old age.
EXPERIMENTAL PROCEDURES
Animal Studies
The generation of dentin matrix protein 1 (Dmp1)-Cre transgenic mice and mice harboring an Atg7 conditional allele has been described previously (21, 22). The experimental animals used in most of the studies described here were obtained using a two-step breeding strategy. Hemizygous Dmp1-Cre transgenic mice were crossed with heterozygous Atg7-flox mice to generate heterozygous Atg7-flox offspring with and without a Dmp1-Cre allele. These offspring were then intercrossed to generate the following offspring: wild type (WT) mice, mice hemizygous for the Dmp1-Cre allele, mice homozygous for the Atg7-flox allele, hereafter referred to as Atg7-f/f, and Atg7-f/f mice that were also hemizygous for the Dmp1-Cre allele. For studies requiring larger numbers of mice, Atg7-f/f mice were crossed with Atg7-f/f mice that were also hemizygous for the Dmp1-Cre allele. Offspring were genotyped by PCR using the following primer sequences: Cre-for, 5′-GCGGTCTGGCAGTAAAAACTATC-3′, Cre-rev, 5′-GTGAAACAGCATTGCTGTCACTT-3′, product size 102 bp; Hind-Fw, 5′-TGGCTGCTACTTCTGCAATGATGT-3′, Atg7-ex14-F, 5′-TCTCCCAAGACAAGACAGGGTGAA-3′, Pst-Rv, 5′-CAGGACAGAGACCATCAGCTCCAC-3′, product size 216 bp (WT) and 500 bp (floxed allele). Both the Dmp1-Cre and Atg7-flox mice were crossed into the C57BL/6 genetic background for more than 10 generations prior to initiating generation of mice for this study. Bone mineral density (BMD), microcomputed tomography (μCT), histomorphometry, and strength-testing were performed as previously described (17, 23, 24). CTX and PNIP in blood plasma were measured using kits according to the manufacturer's directions (Immunodiagnostic Systems, Scottsdale, AZ). Intracellular ROS levels were quantified in freshly isolated bone marrow cells from femurs using dichlorodihydrofluorescein diacetate dye (25). All studies involving mice were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and the Central Arkansas Veterans Healthcare System.
RNA and Genomic DNA Isolation
Osteocyte-enriched cortical bone was prepared as previously described (17). For genomic DNA isolation, bone pieces were decalcified in 14% EDTA for 1 week after collagenase digestion. Decalcified osteocyte-enriched bone was then digested with proteinase K (0.5 mg/ml in 10 mm Tris, pH 8.0, 100 mm NaCl, 20 mm EDTA, and 1% SDS) at 55 °C overnight. Genomic DNA was then isolated by phenol/chloroform extraction and ethanol precipitation. For soft tissues, ∼30 mg of tissue was digested with proteinase K and processed in the same way as osteocyte-enriched bone. RNA was purified from bone and soft tissues using Ultraspec reagent (Biotecx Laboratories, Houston, TX), according to the manufacturer's directions. RNA was purified from cultured cells using the RNeasy kit (Qiagen, Valencia, CA).
Taqman Assays
To quantify Atg7 gene deletion, the following custom Taqman assay for exon 14 was used: forward, 5′-ACCAGCAGTGCACAGTGA-3′, reverse, 5′-GCTGCAGGACAGAGACCAT-3′, probe, 5′-FAM-CTGGCCGTGATTGCAGNFQ-3′. All Taqman assays used in this study were obtained from Invitrogen (Foster City, CA). The custom Atg7 assay was used in combination with a Taqman copy number reference assay, Tfrc (catalogue number 4458367), and the relative amount of Atg7 exon 14 genomic DNA was calculated using the ΔCt method (26). To quantify mRNA levels, cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Invitrogen) according to manufacturer's directions. Taqman quantitative reverse transcription-PCR (RT-PCR) was performed as previously described (24) using the following primer probe sets: Sost, Mm00470479_m1; Mepe, Mm02525159_s1; RANKL, Mm00441908_m1; osteoprotegerin, Mm00435452_m1, tartrate-resistant acid phosphatase, Mm00475698_m1; cathepsin K, Mm00484036_m1; osterix-1, Mm00504574_m1; collagen 1a1, Mm00801666_g1; osteocalcin, (forward, 5′-GCTGCGCTCTGTCTCTCTGA-3′, reverse, 5′-TGCTTGGACATGAAGGCTTTG-3′, probe, 5′-FAM-AAGCCCAGCGGCC-NFQ-3′); and the housekeeping gene ribosomal protein S2, (forward, 5′-CCCAGGATGGCGACGAT-3′, reverse, 5′-CCGAATGCTGTAATGGCGTAT-3′, probe, 5′-FAM-TCCAGAGCAGGATCC-NFQ-3′. Relative mRNA levels were calculated using the ΔCt method (26). To quantify mitochondrial DNA, the following custom Taqman assay for the mitochondrial gene ND2 was utilized: forward, 5′-CATGACAAAAAATTGCTCCCCTATCAA-3′, reverse, 5′-ATGCCCCTATGAAAATAGAAGTAATTGCT-3′, probe, 5′-FAM-CCCGCTACTCAACTCT-NFQ-3′. The amount of mitochondrial DNA was calculated using results from the ND2 and Tfrc copy number reference assays and the ΔCt method (26).
Immunoblots
Protein was extracted from osteocyte-enriched cortical bone by freezing in liquid nitrogen followed by pulverization in liquid nitrogen. The pulverized bone powder was then incubated in RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1.0% Igepal CA-630 (Nonidet P-40), 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS)) containing 1× SDS-PAGE loading buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 6% glycerol, 1% β-mercaptolethanol, and 0.004% bromphenol blue) for 10 min at 100 °C. Proteins were then resolved in SDS-polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes. Membranes were subsequently blocked with 5% nonfat dry milk in TBS and were then incubated with primary antibodies and an appropriate horseradish peroxidase-linked secondary antibody. The following antibodies were used: anti-LC3 (Cell Signaling Technology, Danvers, MA), anti-p62 (Progen Biotechnik, Heidelberg, Germany), and anti-tubulin (Sigma-Aldrich). Blots were developed using enhanced chemiluminescence and the intensity of the bands was quantified using a ChemDoc XRS-plus system (Bio-Rad). Protein extraction from L6 vertebra and quantification of p66shc by immunoblotting was performed as previously described (3).
Cell Culture
Bone marrow cells were harvested from long bones and used to quantify colony forming units (CFU)-fibroblast (CFU-F) and CFU-osteoblasts (CFU-OB) as previously described (27). Osteoblast differentiation of bone marrow precursors was evaluated by plating bone marrow cells in 12-well plates at 5 × 106 cells/well and culturing in α-MEM containing 15% fetal bovine serum, 1% penicillin/streptomycin/glutamine, 1% ascorbic acid, and 10 mm β-glycerolphosphate. One-half of the culture medium was changed every 3 days. After 21 days, the cultures were fixed with 10% Millonig's modified phosphate buffered formalin and then stained with an aqueous solution of 40 mm alizarin red. After photography, the alizarin red was extracted with 10% acetic acid and quantified as previously described (28). Osteoblast-specific gene expression was evaluated in parallel cultures lacking β-glycerolphosphate and harvested after 12 days. Osteoclast differentiation of bone marrow progenitors was evaluated by culturing bone marrow cells in the presence of M-CSF (30 ng/ml) and RANKL (30 ng/ml) for 5 days followed by TRAP staining as previously described (29). The ability of bone marrow cells to support osteoclast differentiation was evaluated by plating bone marrow cells as above and adding vehicle or 10−7 m parathyroid hormone (PTH) for 12 days followed by RNA extraction and quantification of osteoclast-specific genes.
Apoptosis Quantification
Femurs from 6-month-old female mice were fixed in 10% Millonigs modified phosphate-buffered formalin for 24 h, decalcified in 5% formic acid and dehydrated in 100% ethylene glycol monoethyl ether, prior to paraffin-embedding and sectioning. Apoptotic osteocytes were detected using TACS® 2 TdT DAB (diaminobenzidine) Kit (Trevigen, Gaithersburg, MD).
Transmission Electron Microscopy
Decalcified bones were placed into fixative (2.5% glutaraldehyde, 2.0% paraformaldehyde, 0.025% calcium chloride in a 0.1 m sodium cacodylate buffer, pH 7.4) and allowed to fix for 3 h at room temperature. The bones were post fixed with osmium tetroxide, en bloc stained with 2.0% uranyl acetate, dehydrated in a graded ethanol series, and embedded in pure epoxy resin. Thin sections were cut using a diamond knife and an LKB 2088 ultramicrotome and placed on copper grids. Sections were stained with lead citrate and examined in a FEI Morgagni transmission electron microscope. Images were captured with an AMT 2K digital CCD camera (Advanced Microscopy Techniques, Danvers MA).
Statistics
Data were analyzed using SigmaStat (SPSS Science, Chicago, IL). All data that passed Levene's test for constant variance and the Shapiro-Wilk test for normality, either as the data were, or after log or reciprocal transformation, were evaluated for differences between group means using two-way ANOVA or Student's t test. For comparisons using ANOVA, post-hoc analysis was performed using the Holm-Sidak method. Data that did not pass the normality test were evaluated using the Mann-Whitney Rank Sum Test. All values are reported as the mean ± S.D.
RESULTS
Suppression of Autophagy in Osteocytes Decreases Bone Mass
To directly address the role of autophagy in osteocytes, we utilized mice harboring a conditional allele for Atg7, which is an E1-like enzyme that activates a ubiquitin-like protein known as LC3 (6). LC3 becomes conjugated to phosphatidyl ethanolamine and is required for autophagosome production (6). Importantly, Atg7 is essential for autophagy (22). Atg7-flox mice were crossed with transgenic mice expressing the Cre recombinase under the control of Dmp1 regulatory elements, hereafter designated Dmp1-Cre mice (21). The Dmp1-Cre transgene results in recombination in osteocytes and some mature osteoblasts (17, 21). Mice lacking Atg7 in all tissues die within 1 day after birth (22), but Dmp1-Cre;Atg7-f/f mice were obtained at the expected Mendelian frequency when analyzed at 21 days of age.
Analysis of genomic DNA extracted from osteocyte-enriched cortical bone revealed a 75% reduction of the Atg7 conditional allele that did not occur in soft tissues (Fig. 1A). Direct analysis of LC3 expression in protein extracted from bone shafts demonstrated reduced conversion from form I (unlipidated) to form II (lipidated) in the conditional knock-out mice (Fig. 1B) as well as accumulation of p62/sqstm1, a protein frequently elevated in cells with reduced autophagy (Fig. 1C). Taken together, these results confirm that autophagy was effectively suppressed in osteocytes by deletion of Atg7 using the Dmp1-Cre transgene.
FIGURE 1.
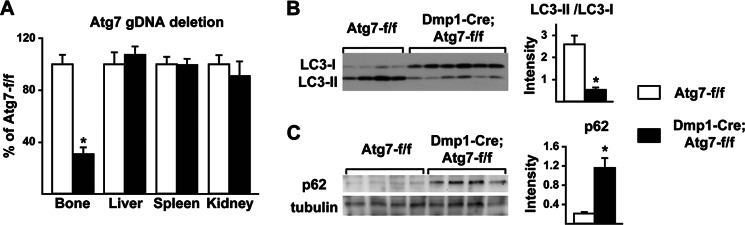
Atg7 deletion suppresses autophagy in osteocytes. A, quantitative PCR of loxP-flanked genomic DNA, normalized to a control locus, using genomic DNA isolated from osteocyte-enriched femoral cortical bone or the indicated soft tissues (n = 7–9 male 6-month-old mice per group). B, immunoblot of LC3 in protein extracted from osteocyte-enriched femoral cortical bone. The ratio LC3-II to LC3-I based on quantification of the bands in the immunoblot is shown on the right (n = 4–6 male 2-month-old mice per group). C, immunoblot of p62 in protein extracted from osteocyte-enriched femoral cortical bone. The intensity of the p62 band normalized to tubulin is shown on the right (n = 4 male 2-month-old mice per group). *, p < 0.05 using Student's t test.
BMD measurement in 6-week-old mice by dual energy x-ray absorptiometry (DEXA) revealed a small decrease in femoral BMD in male conditional knock-out mice, but no change in spinal or total BMD (supplemental Fig. S1). No change in BMD at any site was detected in female mice at this age, and body weight was not different from controls in either sex (supplemental Fig. S1). In contrast, at 6 months of age conditional knock-out mice had low bone mass at all skeletal sites compared with control littermates (Fig. 2A). The low bone mass at this age occurred in conditional knock-out mice of both sexes and was associated with normal body weight (supplemental Fig. S1). Littermates homozygous for the Atg7-conditional allele (Atg7-f/f) or harboring only the Dmp1-Cre transgene had bone mass indistinguishable from wild-type littermates demonstrating the specificity of the bone mass phenotype (Fig. 2A). Based on the latter observation, all further analysis was confined to Atg7-f/f and Dmp1-Cre;Atg7-f/f littermates.
FIGURE 2.
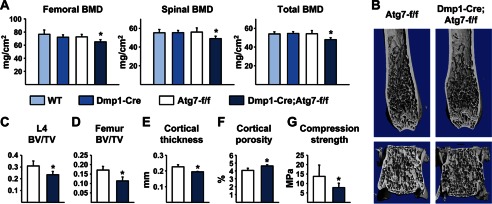
Low bone mass in Dmp1-Cre;Atg7-f/f mice. A, BMD measured by DEXA in the femur, spine, and whole body of wild type (WT), Dmp1-Cre, Atg7-f/f, and Dmp1-Cre;Atg7-f/f littermates. B, high resolution μCT images of the distal femur and L4 vertebra. C, bone volume per tissue volume (BV/TV) of cancellous bone in L4 vertebra. D–F, cancellous BV/TV in the distal femur, cortical thickness at the femoral diaphysis, and cortical porosity at the femoral diaphysis. G, compression strength (stress) of L4 vertebra. All values in Fig. 2 were determined in 6-month-old male mice (n = 6–9 mice per group). *, p < 0.05 using two-way ANOVA (A) or Student's t test (C–G).
Analysis of the skeleton by μCT revealed decreased cancellous bone volume in the spine and femur (Fig. 2, B–D). In addition, cortical thickness was reduced and cortical porosity was increased in the femurs of conditional knock-out mice (Fig. 2, E and F). Consistent with these changes in bone mass, biomechanical testing revealed that compression strength was reduced in lumbar vertebrae of the conditional knock-out mice (Fig. 2G).
Bone Turnover Is Reduced in Mice Lacking Autophagy in Osteocytes
To determine whether changes in bone remodeling might explain the low bone mass of the mice lacking autophagy in osteocytes, we performed histomorphometric analysis of lumbar vertebrae. Osteoclast number and the extent of bone surface covered by osteoclasts were decreased by ∼50% in the conditional knock-out mice (Fig. 3, A and B), as were osteoblast number and surface (Fig. 3, C and D). In line with reduced osteoblast number, the bone formation rate was reduced in the conditional knock-out mice due to a reduced amount of mineralizing surface but no change in the mineral apposition rate (Fig. 3, E–H). Reduced wall width is a consistent histological finding in aged human and murine bone and reflects the reduced amount of work (new bone matrix) performed by teams of osteoblasts (3, 30). Importantly, wall width was significantly lower in the conditional knock-out mice compared with control littermates (Fig. 3I). A circulating marker of bone resorption, C-terminal telopeptide of type I collagen (CTX), was reduced in the blood plasma of the conditional knock-out mice, although there was no significant change in the bone formation marker amino-terminal propeptide of type I collagen (P1NP) (Fig. 3, J–K). Taken together, these results demonstrate that 6-month-old mice lacking autophagy in osteocytes exhibit a low rate of bone remodeling similar to that observed in aged wild type mice.
FIGURE 3.
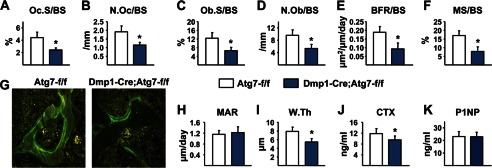
Dmp1-Cre;Atg7-f/f mice have low bone turnover. A–I, histomorphometric analysis of cancellous bone in lumbar vertebra 1–3. A and B, osteoclast surface per bone surface (Oc.S/BS) and osteoclast number per bone surface (N.Oc/BS). C and D, osteoblast surface per bone surface (Ob.S/BS) and osteoblast number per bone surface (N.Ob/BS). E and F, bone formation rate per bone surface (BFR/BS) and mineralizing surface per bone surface (MS/BS). G, photomicrographs of calcein-labeled surfaces in vertebral cancellous bone. H and I, mineral apposition rate (MAR) and wall thickness (W.Th). J and K, CTX and P1NP measured in blood plasma. All values in Fig. 3 were determined in 6-month-old male mice (n = 6–9 mice per group). *, p < 0.05 using Student's t test.
Autophagy plays an important role in the terminal differentiation of some cell types. For example, mice lacking autophagy in erythrocyte progenitors develop severe anemia and die by 14 weeks of age (31). To determine whether osteocyte differentiation from osteoblasts was altered by suppression of autophagy, we quantified osteocyte density and found that it was unchanged in either the cancellous or cortical bone of conditional knock-out mice (Fig. 4A). Similarly, the percentage of apoptotic osteocytes was not different between genotypes (Fig. 4B). Consistent with these findings, examination of osteocyte morphology in newly embedded and mature osteocytes by electron microscopy showed no obvious changes (Fig. 4C). Lastly, the expression of osteocyte-specific genes such as Sost and Mepe was not altered in the conditional knock-out mice (Fig. 4, D and E).
FIGURE 4.
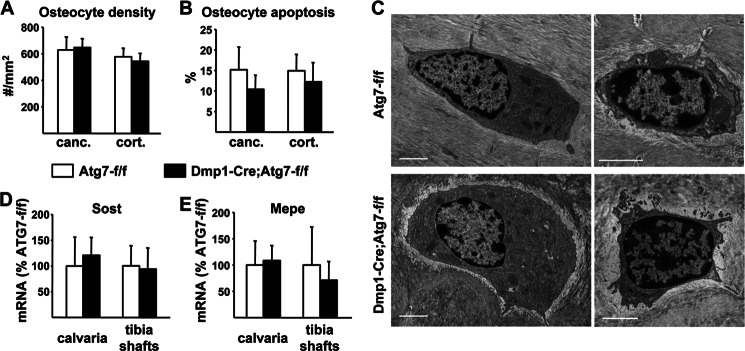
Osteocyte formation in Dmp1-Cre;Atg7-f/f mice. A, osteocyte density measured in cancellous (canc.) and cortical (cort.) bone of lumbar vertebra 1–3 of 6-month-old male mice (n = 6 per group). B, osteocyte apoptosis measured in cancellous (canc.) and cortical (cort.) bone of the femur of 6-month-old female mice (6 per group). C, TEM images of newly-embedded osteocytes (left) or mature osteoctyes (right) in femoral cortical bone from 2-month-old male mice; bar, 2 μm. D and E, quantitative RT-PCR of Sost and Mepe mRNA in calvaria and tibia shafts from 3-month-old male mice (n = 6–11 per group).
We next sought molecular explanations for the reduced bone remodeling in the conditional knock-out mice. We and others have recently shown that mice lacking RANKL in osteocytes have reduced cancellous bone remodeling due to a reduction in osteoclast formation (17, 18). Thus it is possible that suppression of autophagy in this cell type altered production of RANKL or its soluble decoy receptor osteoprotegerin (OPG) to reduce osteoclast formation in Dmp1-Cre;Atg7-f/f mice. However, measurement of RANKL and OPG mRNAs in calvarial bone or osteocyte-enriched cortical bone did not reveal any changes in expression in conditional knock-out mice (Fig. 5A). We then examined the osteoclastogenic potential of bone marrow progenitors and found that, rather than being reduced, it was slightly elevated in conditional knock-out mice (Fig. 5B). It is also possible that the support of osteoclastogenesis by bone marrow stromal cells may have been altered by deletion of Atg7 in osteocytes. However, osteoclast differentiation in primary bone marrow cultures from conditional knock-out mice was similar to cultures from control mice (Fig. 5C).
FIGURE 5.

Osteoclast and osteoblast differentiation. A, quantitative RT-PCR of RANKL and OPG mRNA in calvaria and tibia shafts from 3-month-old male mice (n = 6–11 per group). B, osteoclast (oc) formation quantified in bone marrow from 12-month-old female mice (n = 3 wells per group). C, quantitative RT-PCR of TRAP and cathepsin K (CatK) in bone marrow cultures treated with vehicle or PTH for 11 days to induce osteoclast formation (n = 4 wells per group). D, CFU-F and CFU-OB in bone marrow cells from 12-month-old female mice (n = 3 wells/group). E, Alzarin Red staining and quantification of primary bone marrow cells cultured for 21 days in osteoblast differentiation medium (n = 3 wells/group). F, quantitative RT-PCR of osterix-1 (Osx1), collagen1a1 (Col1a1), and osteocalcin (Ocn) in 12-day primary bone marrow cultures (n = 3 wells/group). *, p < 0.05 using Student's t test.
Similar to the osteoclastogenic potential, we observed a small but significant increase in colony-forming units that give rise to fibroblasts (CFU-F) and osteoblasts (CFU-OB) in the bone marrow of conditional knock-out mice (Fig. 5D). In contrast, osteoblast differentiation, as measured by mineralizing nodule formation and osteoblast-specific gene expression in bone marrow cultures, was not affected by deletion of Atg7 in Dmp1-Cre-expressing cells (Fig. 5, E and F). Thus the reduced bone formation and lack of balance between bone resorption and bone formation in the conditional knock-out mice is not due to insufficient numbers of osteoblast progenitors or an inability of progenitors to differentiate into mature osteoblasts.
Suppression of Autophagy in Osteocytes Increases Oxidative Stress in Bone
We have shown previously that aging in wild type mice is associated with increased oxidative stress as revealed by elevated ROS production in bone marrow cells and increased phosphorylation of the p66shc adaptor protein in bone tissue (3). Strikingly, p66shc phosphorylation was increased in L6 vertebrae of Dmp1-Cre;Atg7-f/f mice, compared with control littermates (Fig. 6A). Moreover, ROS levels were significantly higher in the bone marrow of conditional knock-out mice (Fig. 6B). Previous studies have demonstrated that suppression of autophagy increases ROS in part by increasing the number of mitochondria (9, 32). Consistent with this, mitochondrial DNA content in osteocyte-enriched cortical bone was higher in conditional knock-out mice compared with control littermates (Fig. 6C). These results demonstrate that suppression of autophagy in osteocytes is sufficient to increase oxidative stress in the bones of young mice to levels normally seen in aged animals, possibly via suppression of mitochondrial turnover.
FIGURE 6.

Oxidative stress in bone from Dmp1-Cre;Atg7-f/f mice. A, immunoblot of phospho-p66shc in lumbar vertebra 6 from 3-month-old male mice (n = 5 per group). B, ROS in bone marrow isolated from tibiae of 3-month-old male mice (n = 5 per group). C, ratio of mitochondrial:nuclear DNA determined by Taqman PCR of DNA isolated from osteocyte-enriched femoral cortical bone of 6-month-old male mice (n = 7–9 per group). D, comparison of changes in old mice and mice lacking Atg7 in osteocytes. *, p < 0.05 using Student's t test (A and B) or Mann-Whitney Rank Sum test (C).
DISCUSSION
Compared with young adults, aged mice have low bone mass associated with low cancellous bone remodeling (3, 33). Because bone mass declines with age, the age-associated decrease in bone formation must exceed the decrease in bone resorption. Importantly, a different phenomenon occurs in cortical bone since cortical porosity, which depends on osteoclastic bone resorption, increases with age (34, 35). The molecular mechanisms responsible for altered bone remodeling in either compartment remain unclear but, as with many aging phenotypes, these changes are associated with increased oxidative stress (3, 36). Strikingly, all of these structural, cellular, and biochemical changes were also observed in Dmp1-Cre;Atg7-f/f mice at 6 months of age (Fig. 6D). Thus, suppression of autophagy in osteocytes is sufficient to mimic many of the skeletal changes associated with advanced age in young adult mice.
Although the rate of cancellous bone remodeling is low in the conditional knock-out mice, this alone cannot explain their low bone mass. For example, some other genetic models with low bone remodeling, such as mice lacking a RANKL transcriptional enhancer (37) or mice lacking parathyroid hormone (38), have high bone mass as adults. Therefore, similar to aged mice, the low bone mass must be due to an imbalance between resorption and formation such that bone is incompletely replaced in each remodeling cycle. The decrease in wall width that occurs in the conditional knock-out mice is consistent with this idea.
Because the Dmp1-Cre transgene is active in at least some matrix-synthesizing osteoblasts, it is possible that the skeletal phenotype of the conditional knock-out mice is due in part to loss of autophagy in these cells. However, deletion of Atg7 in Dmp1-Cre-expressing cells did not alter osteoblast differentiation in vitro as measured by mineralized nodule formation and osteoblast gene expression in primary bone marrow cultures. This suggests that the skeletal phenotype is most likely due to suppression of autophagy in osteocytes rather than osteoblasts and that suppression of autophagy in osteocytes alters production of factors that control osteoclast and osteoblast number. Consistent with this idea, osteoclast numbers were reduced in the conditional knock-out mice even though osteoclasts or their progenitors do not express the Dmp1-Cre transgene (21).
The Dmp1-Cre transgene that we used has been shown to have activity in cell types other than osteoblasts and osteocytes, including odontoblasts and skeletal muscle cells (21, 39). It is possible that Atg7 deletion in odontoblasts altered tooth formation. However, altered dentition would affect the skeleton primarily via altered nutrition. The normal body weight of the Dmp1-Cre;Atg7-f/f mice confirms the normal nutritional status of these mice. Previous studies have shown that deletion of Atg7 using a muscle specific promoter resulted in significantly reduced body weight by 40 days of age (14). Therefore, the normal body weight of the Dmp1-Cre;Atg7-f/f mice also suggests that significant deletion of Atg7 did not occur in the muscle tissue of these mice. Taken together, these observations argue that the skeletal phenotype observed in Dmp1-Cre;Atg7-f/f mice is not the result of altered dentition or muscle mass.
There are several potential mechanisms by which suppression of autophagy in osteocytes may control bone remodeling. Suppression of autophagy in other long-lived cell types, such as neurons and myocytes, increases the basal rate of apoptosis. However, we did not detect any changes in osteocyte number or apoptosis in the conditional knock mice as measured by TUNEL staining, osteocyte density, or osteocyte-specific gene expression. Moreover, since osteocyte apoptosis has been associated with (40), or shown to cause (41–43), increased osteoclast formation and bone resorption, an increase in osteocyte apoptosis would not explain the reduced osteoclast formation in mice lacking autophagy in osteocytes.
Osteocytes have been shown to control osteoclast formation by producing RANKL and OPG and to suppress osteoblast formation by producing sclerostin (17, 18, 44, 45). However, we did not detect changes in the expression of mRNAs encoding these factors in osteocytes of the conditional knock-out mice. Nonetheless, it is possible that autophagy controls production of these factors via mechanisms other than transcriptional regulation. In support of this idea, autophagy has been shown to play an important role in the unconventional (not mediated by the classical endoplasmic reticulum/Golgi-dependent pathway) secretion of proteins such as IL-1β and IL-18 (46, 47). In addition, autophagy contributes to protein secretion in some cell types via a specialized structure known as the TOR-autophagy spatial coupling compartment (TASCC) (48). Thus it will be important to determine whether suppression of autophagy in osteocytes alters secretion or cell-surface expression of factors such as RANKL, OPG, and sclerostin. It is also possible that previously unrecognized factors produced by osteocytes contribute to the low remodeling and bone mass caused by deletion of Atg7 in these cells.
An increase in oxidative stress has been functionally associated with the bone loss caused by estrogen-deficiency and has been detected in the bones of aged wild type mice (3, 49). Thus, the increase in oxidative stress in the bones of mice lacking autophagy in osteocytes may contribute to the imbalance between bone resorption and formation. The elevated mitochondrial DNA content of the osteocyte-enriched bone suggests that the likely source of the elevated ROS is an accumulation of damaged mitochondria in the osteocytes of the conditional knock-out mice. Whether oxidative stress alters production of factors by osteocytes that control bone formation, or whether ROS produced by osteocytes act directly on osteoblasts, are important questions that will need to be addressed in future work.
We have shown previously that blockade of glucocorticoid action on osteoblasts and osteocytes blunts the loss of bone mass and strength caused by aging in mice (50). We also noted in those studies that corticosterone levels in the circulation increase with age. Together, these results suggested that an increase in endogenous glucocorticoids is partially responsible for the decrease in bone mass and strength caused by aging. The results of the present study suggest that a decline in osteocyte autophagy may also contribute to the adverse effects of age on the skeleton. Recent studies have provided evidence that glucocorticoids stimulate autophagy in the MLO-Y4 osteocytic cell line (51). Based on these studies, the increased endogenous glucocorticoid levels in aged mice might be expected to increase autophagy. However, it is important to note that autophagy is regulated by numerous signaling pathways and conditions such that the effects of glucocorticoids alone may not predominate in aging mice. Direct comparison of autophagy in osteocytes from young versus old mice will be required to determine whether this is the case. Our preliminary attempts to measure autophagy in old bone have been hampered by the increase in cortical porosity, and thus the presence of other cell types, in osteocyte-enriched bone from old mice (data not shown). Thus, development of novel approaches to directly measure autophagy in osteocytes will be required to address this question.
It is somewhat surprising that the conversion of matrix-synthesizing osteoblasts to osteocytes was not altered by deletion of Atg7 in Dmp1-Cre-expressing cells. Osteoblasts and recently-embedded osteocytes, also known as osteoid osteocytes (15), contain abundant endoplasmic reticulum and mitochondria that are progressively reduced as the cells mature within mineralized bone. Because autophagy is required for similar reductions in cellular components during the maturation of other cell types (31), one might have anticipated altered osteocyte morphology or increased cell death when Atg7 was deleted using the Dmp1-Cre transgene. However, since this transgene does not become active until the matrix-synthesizing stage of osteoblast differentiation, it is possible that even after the Atg7 gene was deleted, sufficient Atg7 protein remained to allow autophagy to continue until osteocytes were fully formed. This would of course depend on the half-life of the Atg7 protein, which likely varies in different cell types and conditions. Nonetheless, it remains possible that the process of osteocyte formation may be affected when the Atg7 gene is deleted using Cre driver strains that become active at earlier stages of osteoblast differentiation.
In summary, the results presented herein demonstrate that suppression of autophagy in osteocytes causes skeletal changes very similar to those caused by aging and suggest the possibility that reduced autophagy may contribute to the detrimental effects of aging on bone mass and strength. It will now be important to identify the molecular mechanisms that account for the low and unbalanced bone remodeling caused by suppression of autophagy in osteocytes. In addition, recent studies in other organs, such as the brain and liver, have demonstrated that stimulation of autophagy can prevent or reverse age-associated damage (52, 53). Thus, it will also be important to complement our loss-of-function studies with gain-of-function studies to determine whether maintenance or stimulation of autophagy in osteocytes is sufficient to prevent any aspects of skeletal aging.
Acknowledgments
We thank P.E. Cazer, A. Deloose, S.B. Berryhill, and J.J. Goellner for technical support and H.W. Virgin for supplying the Atg7-flox mice. We also thank the staff of the UAMS Department of Laboratory Animal Medicine.
This work was supported by National Institutes of Health Grants AG13918 (to S. C. M.) and AR049794 (to C. A. O.) and the Central Arkansas Veteran's Healthcare System (Merit Review 1I01BX000294, to C. A. O.). Additional support was provided by the UAMS Translational Research Institute (UL1RR029884) and by UAMS tobacco settlement funds.

This article contains supplemental Fig. S1.
- BMU
- basic multicellular unit
- ROS
- reactive oxygen species
- RANKL
- receptor activator of NFκB ligand
- BMD
- bone mineral density
- CFU
- colony forming unit
- PTH
- parathyroid hormone
- DEXA
- dual energy x-ray absorptiometry
- OPG
- osteoprotegerin.
REFERENCES
- 1. Manolagas S. C. (2000) Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 21, 115–137 [DOI] [PubMed] [Google Scholar]
- 2. Riggs B. L., Melton L. J., Robb R. A., Camp J. J., Atkinson E. J., McDaniel L., Amin S., Rouleau P. A., Khosla S. (2008) A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J. Bone Miner. Res. 23, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almeida M., Han L., Martin-Millan M., Plotkin L. I., Stewart S. A., Roberson P. K., Kousteni S., O'Brien C. A., Bellido T., Parfitt A. M., Weinstein R. S., Jilka R. L., Manolagas S. C. (2007) Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 282, 27285–27297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang Z., Klionsky D. J. (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol. 12, 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He C., Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tal M. C., Sasai M., Lee H. K., Yordy B., Shadel G. S., Iwasaki A. (2009) Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scherz-Shouval R., Elazar Z. (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 36, 30–38 [DOI] [PubMed] [Google Scholar]
- 10. Vellai T., Takács-Vellai K., Sass M., Klionsky D. J. (2009) The regulation of aging: does autophagy underlie longevity? Trends Cell Biol. 19, 487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salminen A., Kaarniranta K. (2009) Regulation of the aging process by autophagy. Trends Mol. Med. 15, 217–224 [DOI] [PubMed] [Google Scholar]
- 12. Yen W. L., Klionsky D. J. (2008) How to live long and prosper: autophagy, mitochondria, and aging. Physiology 23, 248–262 [DOI] [PubMed] [Google Scholar]
- 13. Komatsu M., Wang Q. J., Holstein G. R., Friedrich V. L., Jr., Iwata J., Kominami E., Chait B. T., Tanaka K., Yue Z. (2007) Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. U.S.A. 104, 14489–14494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Masiero E., Agatea L., Mammucari C., Blaauw B., Loro E., Komatsu M., Metzger D., Reggiani C., Schiaffino S., Sandri M. (2009) Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515 [DOI] [PubMed] [Google Scholar]
- 15. Bonewald L. F. (2011) The amazing osteocyte. J. Bone Miner. Res. 26, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manolagas S. C., Parfitt A. M. (2010) What old means to bone. Trends Endocrinol. Metab. 21, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J.Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 19. Winkler D. G., Sutherland M. K., Geoghegan J. C., Yu C., Hayes T., Skonier J. E., Shpektor D., Jonas M., Kovacevich B. R., Staehling-Hampton K., Appleby M., Brunkow M. E., Latham J. A. (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D'Agostin D., Kurahara C., Gao Y., Cao J., Gong J., Asuncion F., Barrero M., Warmington K., Dwyer D., Stolina M., Morony S., Sarosi I., Kostenuik P. J., Lacey D. L., Simonet W. S., Ke H. Z., Paszty C. (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 23, 860–869 [DOI] [PubMed] [Google Scholar]
- 21. Lu Y., Xie Y., Zhang S., Dusevich V., Bonewald L. F., Feng J. Q. (2007) DMP1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res. 86, 320–325 [DOI] [PubMed] [Google Scholar]
- 22. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Brien C. A., Jia D., Plotkin L. I., Bellido T., Powers C. C., Stewart S. A., Manolagas S. C., Weinstein R. S. (2004) Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145, 1835–1841 [DOI] [PubMed] [Google Scholar]
- 24. O'Brien C. A., Jilka R. L., Fu Q., Stewart S., Weinstein R. S., Manolagas S. C. (2005) IL-6 is not required for parathyroid hormone stimulation of RANKL expression, osteoclast formation, and bone loss in mice. Am. J. Physiol. Endocrinol. Metab. 289, E784–E793 [DOI] [PubMed] [Google Scholar]
- 25. Huang X., Frenkel K., Klein C. B., Costa M. (1993) Nickel induces increased oxidants in intact cultured mammalian cells as detected by dichlorofluorescein fluorescence. Toxicol. Appl. Pharmacol. 120, 29–36 [DOI] [PubMed] [Google Scholar]
- 26. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 27. Di Gregorio G. B., Yamamoto M., Ali A. A., Abe E., Roberson P., Manolagas S. C., Jilka R. L. (2001) Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17β-estradiol. J. Clin. Invest. 107, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gregory C. A., Gunn W. G., Peister A., Prockop D. J. (2004) An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal. Biochem. 329, 77–84 [DOI] [PubMed] [Google Scholar]
- 29. Martin-Millan M., Almeida M., Ambrogini E., Han L., Zhao H., Weinstein R. S., Jilka R. L., O'Brien C. A., Manolagas S. C. (2010) The estrogen receptor-α in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol. Endocrinol. 24, 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parfitt A. M., Villanueva A. R., Foldes J., Rao D. S. (1995) Relations between histologic indices of bone formation: implications for the pathogenesis of spinal osteoporosis. J. Bone Miner. Res. 10, 466–473 [DOI] [PubMed] [Google Scholar]
- 31. Mortensen M., Ferguson D. J., Edelmann M., Kessler B., Morten K. J., Komatsu M., Simon A. K. (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Bosch-Marce M., Shimoda L. A., Tan Y. S., Baek J. H., Wesley J. B., Gonzalez F. J., Semenza G. L. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Shahnazari M., Dwyer D., Chu V., Asuncion F., Stolina M., Ominsky M., Kostenuik P., Halloran B. (2012) Bone turnover markers in peripheral blood and marrow plasma reflect trabecular bone loss but not endocortical expansion in aging mice. Bone 50, 628–637 [DOI] [PubMed] [Google Scholar]
- 34. Ferguson V. L., Ayers R. A., Bateman T. A., Simske S. J. (2003) Bone development and age-related bone loss in male C57BL/6J mice. Bone 33, 387–398 [DOI] [PubMed] [Google Scholar]
- 35. Silbermann M., Weiss A., Reznick A. Z., Eilam Y., Szydel N., Gershon D. (1987) Age-related trend for osteopenia in femurs of female C57BL/6 mice. Compr. Gerontol. A 1, 45–51 [PubMed] [Google Scholar]
- 36. Jilka R. L., Almeida M., Ambrogini E., Han L., Roberson P. K., Weinstein R. S., Manolagas S. C. (2010) Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 9, 851–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galli C., Zella L. A., Fretz J. A., Fu Q., Pike J. W., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2008) Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-κB ligand gene reduces bone remodeling and increases bone mass. Endocrinology 149, 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miao D., He B., Lanske B., Bai X. Y., Tong X. K., Hendy G. N., Goltzman D., Karaplis A. C. (2004) Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology 145, 2046–2053 [DOI] [PubMed] [Google Scholar]
- 39. Sheng M. H., Zhou X. D., Bonewald L. F., Baylink D. J., Lau K. H. (2013) Disruption of the insulin-like growth factor-1 gene in osteocytes impairs developmental bone growth in mice. Bone 52, 133–144 [DOI] [PubMed] [Google Scholar]
- 40. Aguirre J. I., Plotkin L. I., Stewart S. A., Weinstein R. S., Parfitt A. M., Manolagas S. C., Bellido T. (2006) Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 21, 605–615 [DOI] [PubMed] [Google Scholar]
- 41. Emerton K. B., Hu B., Woo A. A., Sinofsky A., Hernandez C., Majeska R. J., Jepsen K. J., Schaffler M. B. (2010) Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46, 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tatsumi S., Ishii K., Amizuka N., Li M., Kobayashi T., Kohno K., Ito M., Takeshita S., Ikeda K. (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464–475 [DOI] [PubMed] [Google Scholar]
- 43. Jilka R. L., Noble B., Weinstein R. S. (2013) Osteocyte Apoptosis. Bone 54, 264–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kramer I., Halleux C., Keller H., Pegurri M., Gooi J. H., Weber P. B., Feng J. Q., Bonewald L. F., Kneissel M. (2010) Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 30, 3071–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Bezooijen R. L., Roelen B. A., Visser A., van der Wee-Pals L., de Wilt E., Karperien M., Hamersma H., Papapoulos S. E., ten Dijke P., Löwik C. W. (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 199, 805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Deretic V., Jiang S., Dupont N. (2012) Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 22, 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narita M., Young A. R., Arakawa S., Samarajiwa S. A., Nakashima T., Yoshida S., Hong S., Berry L. S., Reichelt S., Ferreira M., Tavaré S., Inoki K., Shimizu S., Narita M. (2011) Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332, 966–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lean J. M., Davies J. T., Fuller K., Jagger C. J., Kirstein B., Partington G. A., Urry Z. L., Chambers T. J. (2003) A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J. Clin. Investig. 112, 915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weinstein R. S., Wan C., Liu Q., Wang Y., Almeida M., O'Brien C. A., Thostenson J., Roberson P. K., Boskey A. L., Clemens T. L., Manolagas S. C. (2010) Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell 9, 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia X., Kar R., Gluhak-Heinrich J., Yao W., Lane N. E., Bonewald L. F., Biswas S. K., Lo W. K., Jiang J. X. (2010) Glucocorticoid-induced autophagy in osteocytes. J. Bone Miner. Res. 25, 2479–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schaeffer V., Lavenir I., Ozcelik S., Tolnay M., Winkler D. T., Goedert M. (2012) Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 135, 2169–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang C., Cuervo A. M. (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]