

Background: AXL is an established therapeutic target in various cancers. HSP90 chaperoning is critical in maintaining the stability of several oncogenic kinases.
Results: HSP90 blockade by 17-AAG induced cytosolic mature AXL degradation via ubiquitin/proteasome pathway and impeded its membrane localization.
Conclusion: AXL depends on HSP90 for its stability and membrane translocation.
Significance: Targeting HSP90 would avail a strategy to counteract AXL.
Keywords: E3 Ubiquitin Ligase, Hsp90, Proteasome, Receptor Tyrosine Kinase, Ubiquitination, 17-AAG/Geldanamycin, AXL Receptor Tyrosine Kinase, CHIP E3 Ligase, HSP90 Inhibition
Abstract
The receptor tyrosine kinase AXL is overexpressed in many cancer types including thyroid carcinomas and has well established roles in tumor formation and progression. Proper folding, maturation, and activity of several oncogenic receptor tyrosine kinases require HSP90 chaperoning. HSP90 inhibition by the antibiotic geldanamycin or its derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG) causes destabilization of its client proteins. Here we show that AXL is a novel client protein of HSP90. 17-AAG induced a time- and dose-dependent down-regulation of endogenous or ectopically expressed AXL protein, thereby inhibiting AXL-mediated signaling and biological activity. 17-AAG-induced AXL down-regulation specifically affected fully glycosylated mature receptor present on cell membrane. By using biotin and [35S]methionine labeling, we showed that 17-AAG caused depletion of membrane-localized AXL by mediating its degradation in the intracellular compartment, thus restricting its exposure on the cell surface. 17-AAG induced AXL polyubiquitination and subsequent proteasomal degradation; under basal conditions, AXL co-immunoprecipitated with HSP90. Upon 17-AAG treatment, AXL associated with the co-chaperone HSP70 and the ubiquitin E3 ligase carboxyl terminus of HSC70-interacting protein (CHIP). Overexpression of CHIP, but not of the inactive mutant CHIP K30A, induced accumulation of AXL polyubiquitinated species upon 17-AAG treatment. The sensitivity of AXL to 17-AAG required its intracellular domain because an AXL intracellular domain-deleted mutant was insensitive to the compound. Active AXL and kinase-dead AXL were similarly sensitive to 17-AAG, implying that 17-AAG sensitivity does not require receptor phosphorylation. Overall our data elucidate the molecular basis of AXL down-regulation by HSP90 inhibitors and suggest that HSP90 inhibition in anticancer therapy can exert its effect through inhibition of multiple kinases including AXL.
Introduction
AXL belongs to the TAM (Tyro3, AXL, Mer) family of receptor tyrosine kinases, which includes Tyro3, AXL, and Mer, and it was initially isolated from primary human myeloid leukemia cells (1). Growth arrest-specific protein 6 (Gas6)3 is the ligand for AXL (2). AXL is overexpressed in several cancer types and has been associated to cancer aggressive phenotype, migration, invasion, and progression through the activation of multiple downstream pathways (3–6). Moreover, increased expression of AXL is often associated with resistance to anticancer conventional chemotherapy and targeted therapy (7–11). Several studies have shown the oncogenic potential of AXL, and its down-regulation through different strategies has shown antineoplastic activity both in in vitro and in vivo cancer models. We have shown previously that AXL and its ligand Gas6 are overexpressed in several thyroid cancer cell lines and human thyroid cancer samples, and inhibiting either or both proteins significantly impaired thyroid cancer cell growth, survival, invasiveness, and tumorigenicity in nude mice (6). Little is known on the mechanism regulating AXL protein maturation and stabilization. Stability and activity of several cancer-related, mutated, chimeric, and overexpressed signaling kinases are often maintained by the cytosolic heat shock protein 90 (HSP90), a member of the HSP chaperone family (12). Hence, targeting HSP90 would allow a combinatorial depletion of multiple oncogenic proteins, leading to the simultaneous disruption of most of the hallmarks of cancer (13). HSP90 promotes correct folding of client proteins in an ATP-dependent manner (14–16). HSP90 is a component of a multichaperone complex that also includes the co-chaperone HSP70 and the ubiquitin ligase carboxyl terminus of HSC70-interacting protein (CHIP) (17). CHIP binds to HSP70 and is responsible for ubiquitination and degradation of many misfolded signaling kinases. Under normal conditions, the probability of a client protein to be properly folded is higher than being ubiquitinated and degraded because the concentration of the HSP70-HSP90 complex is significantly higher than that of the destabilizing HSP70-CHIP complex. Conversely, this degradative mode of chaperone complex is attained under stress conditions or when the functional activity of HSP90 is curtailed (18).
Geldanamycin, a benzoquinone ansamycin antibiotic, is able to compete with ADP/ATP in the nucleotide binding pocket of HSP90, thereby inhibiting its ATP-dependent functional activity and inducing the degradative chaperone complex. Geldanamycin is unsuitable for clinical use due to its poor solubility and significant hepatotoxicity in mammals (19). However, geldanamycin analogues such as 17-allylamino-17-demethoxygeldanamycin (17-AAG) possess similar anticancer activity, much less hepatotoxicity, and better bioavailability (20, 21). Recently, a phase II clinical trial in HER2-positive metastatic breast cancer patients has shown a significant anticancer activity using 17-AAG in combination with the anti-HER2 antibody trastuzumab (22). Other similar HSP90 inhibitors are under clinical evaluation (23).
By using 17-AAG as a tool to inhibit HSP90, here we report that AXL is a novel HSP90 client protein that depends on this chaperone for its stability and maturation. AXL overexpression and constitutive activation are frequently found in cancer, and hence, 17-AAG-induced AXL down-regulation would be an effective therapeutic strategy to block AXL-driven oncogenic effects.
MATERIALS AND METHODS
Reagents and Antibodies
17-AAG, radicicol, and MG132 were purchased from Calbiochem. Lactacystin, ammonium chloride, and chloroquine were purchased from Sigma. Endoglycosidase H (Endo H) and peptide:N-glycosidase F (PNGase F) were from New England Biolabs (Ipswich, MA). Anti-HSP90 (catalogue number SPA-835) and anti-HSP70 (catalogue number SPA-810) were purchased from Stressgen Biotechnologies (Victoria, British Columbia, Canada). Anti-AXL, anti-myc, anti-HA, anti-c-KIT, and anti-PDGF receptor antibodies used in Western blot and immunoprecipitation experiments were from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody used for RET in Western blotting was described previously (24). The anti-AXL antibody directed to AXL extracellular domain and anti-phospho-AXL specific to tyrosine 779 were from R&D Systems (Abingdon, UK). Anti-DYKDDDDK tag (FLAG epitope) and the anti-poly(ADP-ribose) polymerase antibody were from Cell Signaling Technology (Beverly, MA). Secondary antibodies coupled to horseradish peroxidase were from Bio-Rad.
Cell Lines and Transfection Procedures
HeLa cells were from American Type Culture Collection (ATCC, Manassas, VA). The human thyroid papillary cancer cell line TPC1 and anaplastic thyroid cancer cell lines 8505C and CAL62 have been described previously (6) and were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (Invitrogen). Transient transfections were carried out by premixing each plasmid (1–1.5 μg) with FuGENE 6 (Roche Applied Science), and the mixture was added to cells at 70% confluence. Cells were cultured in the same medium for 48 h until lysis or further treatments.
Western Blotting and Immunoprecipitation
Cells were lysed at 4 °C in a buffer containing 50 mm HEPES, pH 7.5, 1% (v/v) Triton X-100, 150 mm NaCl, 5 mm EGTA, 50 mm NaF, 20 mm sodium pyrophosphate, 1 mm sodium vanadate, 2 mm phenylmethylsulfonyl fluoride (PMSF), and 1 μg/ml aprotinin. Lysates were clarified by centrifugation at 10,000 × g for 20 min. Lysates containing comparable amounts of proteins as estimated by a modified Bradford assay (Bio-Rad) were subjected to Western blot. For immunoprecipitation, 1 mg of protein-containing lysate was incubated with appropriate antibody for 3 h or overnight and then with protein A/G-Sepharose for 1 h at 4 °C. Antigen-antibody-bead complexes were centrifuged, washed using wash buffer (20 mm Tris-HCl, pH7.4, 150 mm NaCl, and 0.1% Triton X-100), resuspended in the sample buffer (60 mm Tris-Cl, pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, and 0.01% bromphenol blue), denatured, subjected to SDS-PAGE, transferred to nitrocellulose membrane, and probed with primary antibodies followed by secondary antibodies coupled to horseradish peroxidase. The proteins were detected with an enhanced chemiluminescence kit (Amersham Biosciences).
Plasmids and Constructs
We used pcDNA4/TO A His/myc (Invitrogen) and pFLAG5a (Sigma) vectors to subclone the full-length AXL (NM_001699, TrueORF cDNA clone, Origene) by PCR amplification. The truncated mutant AXL-EC and AXL kinase-dead (AXL K558R) mutant constructs were described previously (6). pcDNA3.1 CHIP-myc and CHIP-TPR-myc (K30A) vectors were a kind gift of L. Neckers. pcDNA-HA-ubiquitin (UbHA) vector was a kind gift from S. Giordano. HSP90-HA (25) expressing the Hsp90β wild type (WT) cDNA in pcDNA3 was obtained from the Addgene non-profit plasmid repository (Addgene plasmid 22487). RET, c-KIT, and PDGF receptor wild type expression constructs were described elsewhere (24, 26).
Biotinylation of Surface Proteins and Immunofluorescence
Cells grown around 70% confluence were washed twice with ice-cold PBS (with 10 mm Ca2+ and 1 mm Mg2+), and surface proteins were labeled for 30 min using 1 mg/ml EZ-Link Sulfo-NHS-SS-biotin (sulfo succinimidyl 2-(biotinamido)-ethyl-1,3′-dithiopropionate; Pierce). The unreacted biotin was removed by washing with 50 mm NH4Cl in PBS. All manipulations were carried out on ice to avoid the internalization at these steps. Cells were then lysed in the standard radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 1 mm EDTA). Comparable amounts of proteins were incubated overnight with streptavidin-agarose resin (Pierce), which was then thoroughly washed, denatured, and analyzed by Western blot with anti-AXL antibodies. The assessment of surface protein turnover by surface biotinylation and chase was performed as described previously (27). For immunofluorescence, cells plated on fibronectin-coated glass coverslips were subjected to 17-AAG treatment for 8 h, then fixed with 4% formaldehyde, blocked with PBS with 1% BSA, and incubated with a 1:50 dilution of 1 μg/μl anti-AXL antibody (R&D Systems) for 2 h at room temperature followed by a 1:1000 diluted rhodamine-conjugated anti-goat secondary antibody (Jackson ImmunoResearch Europe Ltd., Suffolk, UK) for 30 min. Analysis was performed using a Zeiss LSM 510 Meta confocal microscope.
[35S]Methionine-Cysteine Labeling and Pulse-Chase Analysis of AXL
CAL62 cells plated in a 60-mm dish at 70% confluence were starved for 1 h in methionine/cysteine-free DMEM (Sigma, catalogue number D0422) containing 2% dialyzed FBS. The cells were then metabolically labeled with 200 μCi of [35S]methionine-cysteine (Express35S protein labeling mixture, PerkinElmer Life Sciences) for 15 min in methionine/cysteine-free medium (pulse); unbound radioactive amino acids were washed and incubated with prewarmed complete medium (chase) in the presence of vehicle or 17-AAG (500 nm)/MG132 (10 μm). The cells were then disrupted in ice-cold lysis buffer, and comparable amounts of cell extracts were immunoprecipitated for AXL. Proteins were subjected to SDS-PAGE, the gel was dried, and labeled proteins were visualized by autoradiography.
Nickel Affinity His-tagged Protein Purification under Denaturing Conditions
HeLa cells transfected with His-tagged AXL (subcloned in pcDNA4/TO A His/myc) were harvested and lysed with Buffer A (6 m guanidine HCl, 100 mm sodium phosphate buffer, pH 8, 10 mm Tris-HCl, pH 8, 30 mm imidazole, and 10 mm β-mercaptoethanol). The lysate was rocked with nickel-nitrilotriacetic acid-agarose beads (Qiagen) for 4 h. The beads were collected and washed with Buffer B (8 m urea, 100 mm phosphate buffer, pH 6.3, 10 mm Tris-HCl, pH.6.3, 10 mm β-mercaptoethanol, and 0.2% Triton X-100), and nickel bound proteins were eluted by incubating the beads at 30 °C for 20 min in Buffer E (200 mm imidazole, 150 mm Tris-HCl, pH 6.7, 30% glycerol, 5% SDS, and 720 mm β-mercaptoethanol). Proteins were resuspended in sample buffer (2×) and subjected to SDS-PAGE and Western blotting.
Luciferase Activity Assay
Approximately 1 × 106 HeLa cells were transiently co-transfected with AXL and the AP1-driven luciferase reporter (AP1-Luc) vector (Stratagene, Garden Grove, CA) containing six AP1-binding sites upstream from the firefly luciferase cDNA. Twenty-four hours after transfection, cells were serum-starved, the indicated concentration of 17-AAG or vehicle was added, and cells continued to grow for the desired period. 10 ng of pRL-null (a plasmid expressing the enzyme Renilla luciferase from Renilla reniformis) served as an internal control. Cells were harvested, and firefly and Renilla luciferase activities were assayed using the Dual-Luciferase reporter system (Promega Corp., Madison, WI) and expressed as the percentage of residual activity compared with cells treated with vehicle. Light emission was measured by using a Berthold Technologies luminometer (Centro LB 960) (Bad Wildbad, Germany) and expressed as the ratio of firefly and Renilla luciferase activities. The analysis of variance multiple comparison test was used to assess the statistical significance of the luciferase assay, and InStat3 (GraphPad Software, La Jolla, CA) was used.
RESULTS
17-AAG Induced AXL Protein Down-regulation and Block of AXL-dependent Signaling
To test whether 17-AAG interfered with AXL expression, we used a panel of thyroid carcinoma cell lines (CAL62, 8505C, and TPC1) and HeLa cells, all endogenously expressing AXL. Cells were exposed to 17-AAG treatment in a dose- and time-dependent manner, and AXL protein levels were detected by Western blotting. In all the cell lines tested, 17-AAG induced a decrease of AXL protein levels (Fig. 1, A and B). AXL protein on Western blot is detected as a doublet of 140 and 120 kDa. Interestingly, we observed that 17-AAG induced a strong reduction of the slow migrating 140-kDa AXL isoform, whereas it caused an accumulation of the 120-kDa species. CAL62 cells were the most sensitive because 8-h treatment with 250 nm 17-AAG almost completely eliminated the 140-kDa AXL isoform (Fig. 1A). Therefore, we preferentially used this cell line for most of the experiments described in this work. In the time course experiment, the AXL protein level declined almost 50% within 4 h and was nearly undetectable after 8 h of 500 nm 17-AAG exposure (Fig. 1B). The same effect was observed on AXL using another HSP90 inhibitor, radicicol, that is structurally unrelated to 17-AAG (supplemental Fig. 1).
FIGURE 1.
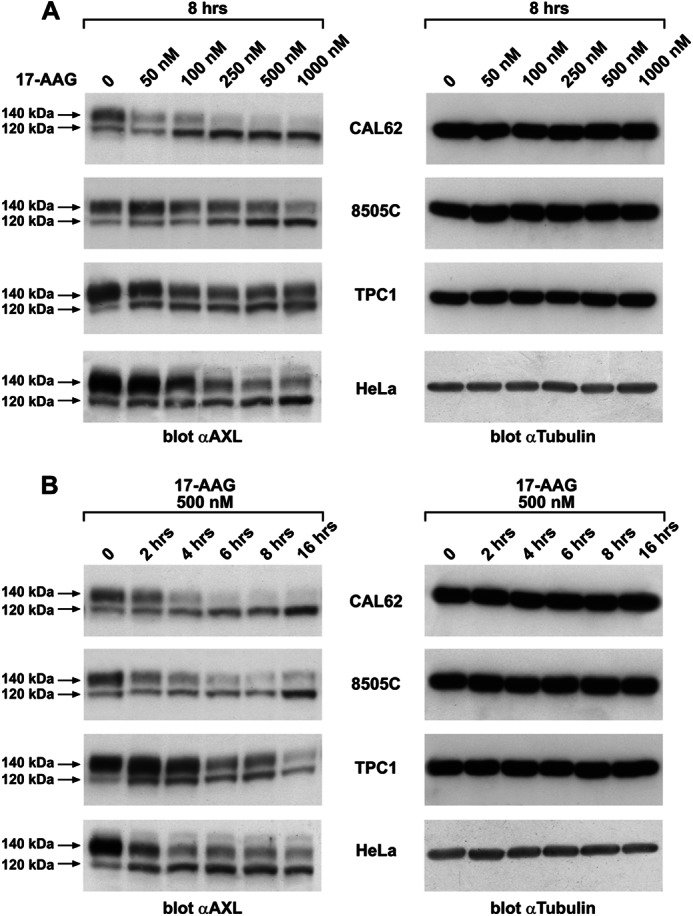
17-AAG induces AXL degradation. A, protein lysates from the indicated thyroid carcinoma cell lines (CAL62, 8505C, and TPC1) and HeLa cells that were subjected to vehicle (0) or 17-AAG treatment with the indicated doses for 8 h were immunoblotted for AXL. Tubulin immunoblotting were used as a loading control. B, protein lysates from the indicated thyroid carcinoma cell lines and HeLa cells treated with 500 nm 17-AAG and harvested at the indicated time points were immunoblotted using anti-AXL and anti-tubulin antibodies.
To address the impact of 17-AAG-induced AXL loss on AXL-mediated signaling and biological activity, we used two of the above tested ATC cell lines, CAL62 and 8505C, in which we demonstrated previously that AXL is overexpressed and active due to the autocrine production of ligand Gas6. The AXL-Gas6 axis in these cell lines is crucial in sustaining the survival advantage through activation of AKT and p70 S6 kinase (6). In both models, 17-AAG reduced AXL phosphorylation and inhibited its downstream signaling concomitantly with the loss of the 140-kDa AXL species (Fig. 2, A and B); we also showed similar effects by using the small molecule inhibitor bosutinib (Fig. 2, A and B), which was reported to abrogate AXL kinase activity (28). Consistently, 17-AAG treatment induced CAL62 cell apoptosis as assessed by TUNEL assay and poly(ADP-ribose) polymerase cleavage (supplemental Fig. 2, A and B).
FIGURE 2.

17-AAG inhibits AXL-mediated signaling and downstream activity. A, CAL62 and 8505C cells treated with the indicated doses of 17-AAG and bosutinib were harvested and lysed, and equal amounts of proteins were immunoprecipitated (IP) with anti-AXL and immunoblotted using anti-phospho-AXL (pAXL). NT, nontreated. B, CAL62 and 8505C cells treated with 17-AAG and bosutinib at varying doses were subjected to Western blotting. AKT and p70 S6 kinase immunoblotting was done with the respective phospho- and total antibodies. Tubulin immunoblotting was used as a loading control. NT, nontreated. C, HeLa cells were transiently transfected with AXL-FLAG-expressing vector and the AP1-Luc vector. pRL-null (a plasmid expressing the enzyme Renilla luciferase from R. reniformis) was used as an internal control. Firefly and Renilla luciferase activities are expressed as the percentage of residual activity of 17-AAG-treated cells with respect to untreated cells. Average results of three independent assays ± S.D. are indicated. The analysis of variance Bonferroni multiple comparison test was used to demonstrate statistical significance. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Although 17-AAG induced a significant reduction of AXL signaling, it is likely that reduction of cell survival and induction of apoptosis were due to the cumulative activity of 17-AAG on several HSP90 clients rather than AXL alone. To evaluate the effect of 17-AAG on AXL-specific downstream activity, we exploited the ability of AXL to activate an AP1-responsive promoter and show that HSP90 inhibition could interfere with such activity. To this aim, AXL-FLAG and AP1-Luc were co-transfected in HeLa cells. Despite displaying AXL expression, HeLa cells did not express the ligand Gas6, and AXL was not tyrosine-phosphorylated in these cells (data not shown). Consistently, the transfection of the AP1-Luc vector in HeLa cells did not result in significant Luc activity (Fig. 2C). Instead, AXL overexpression induced a strong ligand-independent phosphorylation of the receptor (data not shown) and consequently activation of the AP1-dependent luciferase activity. AXL-dependent activation of AP1 reporter was inhibited by 17-AAG in a dose-dependent fashion. The compound reduced AXL activity to ∼50% at 100 nm and completely abolished promoter activation at 200 nm.
17-AAG Targeted the Cell Surface Isoform of AXL
In other receptor tyrosine kinases, like RET, the slow migrating isoform represents the fully glycosylated mature receptor that is localized at the plasma membrane, whereas the fast migrating isoform is a glycosylated mannose-rich immature isoform that remains confined to the intracellular compartments (29). We hypothesized that, as in the case of RET, the 17-AAG-sensitive 140-kDa slow migrating AXL isoform was the fully mature species of the receptor found on the cell surface. To confirm our hypothesis, we used the glycosylation inhibitor tunicamycin, Endo H, and PNGase F. Tunicamycin inhibits N-acetylglucosamine (GlcNAc) transfer, which catalyzes the first step of protein N-glycosylation in the endoplasmic reticulum. Endo H and PNGase F are two endoglycosidases that display selective cleavage of glycans exposed on a protein. Specifically, PNGase F removes any form of N-glycans from protein substrates, whereas Endo H cleaves only the high mannose type of N-glycans. CAL62 cells were treated with increasing doses of tunicamycin for 18 h, and AXL protein was analyzed by Western blot. As shown in Fig. 3A, upon 18 h of tunicamycin treatment, AXL protein accumulated as a 100-kDa protein, representing the core polypeptide, whereas the 140- and 120-kDa bands disappeared, indicating that both these species were N-glycosylated isoforms of AXL. Endo H and PNGase F treatment of CAL62 protein extracts clearly distinguished the fully glycosylated AXL isoform (140 kDa), which was resistant to Endo H and sensitive to PNGase F, from the glycosylated but immature AXL isoform (120 kDa), which was sensitive to both glycosidases (Fig. 3B). Thus, the 17-AAG-sensitive AXL species is the fully mature receptor. To confirm that 17-AAG induced a decrease of the AXL isoform exposed on the cell surface, we performed in vivo biotin labeling of cell surface proteins in CAL62 cells treated or not with 17-AAG. Biotinylated proteins were recovered with streptavidin-agarose and subsequently immunoblotted with anti-AXL antibodies. As shown in Fig. 3C, the 140-kDa AXL isoform was the only biotinylated product, indicating that only this AXL isoform was at the cell surface. No AXL biotinylated product was detected in 17-AAG-treated cells, indicating that the compound specifically affects cell surface-localized receptor. Consistently, immunofluorescence staining of unpermeabilized CAL62 cells treated or not with 17-AAG using an anti-AXL antibody directed to the AXL extracellular domain confirmed that 17-AAG treatment induced a strong reduction of surface-localized AXL (Fig. 3D).
FIGURE 3.

17-AAG targets the fully glycosylated isoform of AXL. A, lysates of CAL62 cells treated 18 h with the indicated doses of tunicamycin were immunoblotted for AXL and normalized with anti-tubulin. B, 50 μg of protein lysates of CAL62 cells preboiled for 5 min at 99 °C was subjected to overnight Endo H (0.01 unit) and PNGase F (1 unit) digestion at 37 °C. The enzyme-digested lysates were subjected to Western blotting for AXL and tubulin. C, CAL62 cells treated or not with 17-AAG (500 nm) for 8 h were subjected to surface protein biotin labeling. Cells were then lysed, immunoprecipitated (IP) using streptavidin-agarose resin, and immunoblotted for AXL. D, CAL62 cells grown on a coverslip were subjected to vehicle (0) or 17-AAG (500 nm) treatment for 8 h. Cells were then fixed and stained for immunofluorescence (IF) using AXL antibody targeting the extracellular domain. Representative microscopy images of untreated and 17-AAG-treated cells are presented.
Proteasomal Inhibitors Restored 17-AAG-induced Mature AXL Depletion
The 17-AAG effect is generally attributed to disruption of the HSP90 chaperoning activity and subsequent targeting of the misfolded HSP90 clients to the proteasomal degradation machinery through ubiquitination by E3 ligases (30, 31). Thus, we verified whether 17-AAG-induced AXL depletion was reverted by the proteasomal inhibitors lactacystin and MG132. As a control, we also used the lysosomal inhibitors chloroquine and ammonium chloride. Co-treatment with proteasomal inhibitors impaired 17-AAG-induced AXL depletion in CAL62 and HeLa cells. Conversely, when cells were treated with lysosomal inhibitors, 17-AAG retained its activity on AXL, indicating that the reduced levels of AXL protein induced by 17-AAG were mediated by proteasomal but not lysosomal degradation (Fig. 4).
FIGURE 4.

Proteasomal inhibitors restored 17-AAG-induced mature AXL depletion. Lysates of CAL62 (A) and HeLa (B) cells co-treated with 17-AAG (500 nm) and the proteasomal inhibitors MG132 (10 μm)/lactacystin (5 μm) or lysosomal inhibitors NH4Cl (20 mm)/chloroquine (100 μm) were subjected to Western blotting for AXL. Tubulin immunoblotting was used as a loading control. The 140-kDa AXL signals in the indicated treatments were quantified by densitometry, and the relative change compared with nontreated cells (NT) was plotted. The values are representative of three independent experiments. Error bars represent S.D. *, p < 0.05.
Effect of 17-AAG on Plasma Membrane-localized AXL
As we observed that 17-AAG specifically depleted AXL species on the cell surface, we asked whether this was due to an increased turnover or destabilization of the plasma membrane-associated receptor. We tested this hypothesis by biotin pulse-chase of surface proteins in the presence of vehicle or 17-AAG in CAL62 cells. Biotin-labeled surface proteins were isolated with streptavidin-agarose beads, separated by SDS-PAGE, and probed for AXL. This experiment showed that 17-AAG did not accelerate the removal of surface-bound receptor until 4 h of treatment (Fig. 5A). A very modest effect was only observed at 6 h (Fig. 5A, compare lane 5 with lane 8). However, AXL steady state levels were already affected at 2–4 h of 17-AAG treatment, indicating that the 17-AAG-induced loss of AXL was not due to destabilization or accelerated turnover of the membrane-localized AXL.
FIGURE 5.

Effect of 17-AAG on plasma membrane-localized AXL. A, CAL62 cell surface proteins were biotinylated and chased in the presence of vehicle or 17-AAG (500 nm), and comparable amounts of lysates were immunoprecipitated (I.P.) with streptavidin-agarose and immunoblotted for AXL. AXL immunoblotting of total cell lysates was used to compare total AXL levels. The biotinylated AXL was analyzed by densitometry, and the relative change in the signal was plotted compared with no chase (0), which was considered equal to 1. The results are representative of three independent experiments. Error bars represent S.D. NT, non biotinylated. B, CAL62 cells biotinylated before and after treatments with 17-AAG (500 nm), MG132 (10 μm), and 17-AAG + MG132 for 5 h were subjected to streptavidin immunoprecipitation, and the recovered biotinylated proteins were subjected to Western blotting for AXL. AXL and tubulin immunoblotting was performed on total lysate. NT, non-treated cells.
To validate the above finding and to specifically evaluate the mechanism of membrane-associated AXL protein removal, we performed surface protein biotin labeling and chased in the presence of 17-AAG, MG132, and their combination for 5 h. Biotin-labeled surface proteins were isolated with streptavidin-agarose beads, separated by SDS-PAGE, and probed for AXL. As shown in Fig. 5B, no significant differences in AXL levels were observed between 17-AAG- and vehicle-treated cells (NT). Interestingly, MG132 increased the half-life of pre-existing membrane receptor species, indicating that the proteasome is involved in the degradation of membrane-localized AXL. However, this effect was not modified by 17-AAG co-treatment (Fig. 5B, Biotin labeling pre-treatment), confirming that HSP90 was not involved in membrane-localized AXL stability and turnover. Thus, we hypothesized that decreased levels of AXL protein on the cell membrane in the presence of 17-AAG (Fig. 3, C and D) could be due to misfolding of the protein in the intracellular compartment and its proteasome-dependent degradation, therefore impeding delivery to cell membrane. As the half-life of the AXL receptor is predicted to be around 2 h (Fig. 6A), treatment with 17-AAG with or without MG132 was carried out for 5 h, and surface proteins were biotinylated at the end of treatment as at this time point the pre-existing AXL species on the plasma membrane would have turned over, allowing specifically to monitor the receptor species that reached the surface during the treatment period. As expected, 17-AAG strongly depleted AXL surface levels. Surprisingly, when compared with untreated cells, MG132 treatment reduced the prevalence of surface receptor, an effect possibly due to endoplasmic reticulum stress caused by protein overload in conditions of proteasome blockade. Despite this, in 17-AAG-treated cells, a significant amount of surface AXL was rescued by MG132 co-treatment, strongly suggesting that HSP90 inhibition causes misfolding of intracellular AXL and targeting to the proteasome before it can reach the cell surface (Fig. 5B, Biotin labeling post-treatment).
FIGURE 6.

HSP90 inhibition blocked AXL transport to the cell surface. A, CAL62 cells pulsed with [35S]methionine for 15 min and chased with nonradioactive medium with or without 17-AAG (500 nm) for the indicated time points. Cells were lysed and subjected to AXL immunoprecipitation (IP), and the dried gel was subjected to standard autoradiography. The 140-kDa signal was analyzed by densitometry and graphically represented. The results are representative of three independent experiments. B, CAL62 cells pulsed with [35S]methionine for 15 min were harvested unchased (0 h) or were chased with nonradioactive medium in the presence of MG132 (10 μm), 17-AAG (500 nm), and their combination for 2 h. Cells lysates were immunoprecipitated for AXL followed by SDS-PAGE, and the dried gel was subjected to autoradiography and phosphorimaging. The quantification of 140-kDa AXL species was analyzed by densitometry. The non-treated cell (NT) signal was considered equal to 1. The results are representative of three independent experiments. Error bars represent S.D. *, p < 0.05.
17-AAG Blocked AXL Transport to the Cell Surface
To strengthen these data and to follow newly synthesized receptor biogenesis/trafficking in the presence of 17-AAG and MG132, we metabolically labeled CAL62 cells with [35S]cysteine-methionine for a pulse of 15 min and chased with nonradioactive medium for periods up to 4 h in the presence or absence of 17-AAG (Fig. 6A). In control CAL62 cells, AXL completed receptor glycosylation within 60 min as shown by the appearance of the 120- and 140-kDa species. Both isoforms progressively accumulated, and at 4 h of chase, the 140-kDa AXL isoform was the prevalent isoform; similar maturation kinetics were also observed in endogenous or ectopically expressed AXL in HeLa cells (supplemental Fig. 3). In contrast, when CAL62 cells were treated with 17-AAG, the 120-kDa isoform accumulated, and a significant lack of the fully mature receptor was observed. These observations are compatible with two possibilities. (i) HSP90 is by itself required for proper glycosylation of AXL protein so that its inhibition induces accumulation of the 120-kDa isoform and its targeting to the proteasome, or (ii) HSP90 is necessary for 140-kDa isoform stability after full glycosylation has occurred. To discriminate between these possibilities, we used [35S]cysteine-methionine pulse labeling and chasing in the presence of 17-AAG, MG132, and 17-AAG with MG132 for 2 h. Receptor levels were evaluated by AXL immunoprecipitation followed by autoradiography. As expected, 17-AAG significantly blocked the appearance of the 140-kDa AXL species. However, when MG132 was added to 17-AAG, it rescued the mature receptor from degradation, suggesting that HSP90 is not involved in the protein glycosylation process, whereas it is required for receptor stability upon full glycosylation (Fig. 6B).
17-AAG-induced AXL Degradation Requires Its Polyubiquitination
17-AAG by inhibiting HSP90 function alters the composition of the chaperone complex and induces recruitment of E3 ubiquitin ligases, thereby targeting the client protein to proteasome degradation. Proteasomal targeting is often mediated by protein polyubiquitination (32). To document whether AXL undergoes polyubiquitination upon 17-AAG treatment, HeLa cells were transfected with HA-tagged ubiquitin (UbHA) and treated with 17-AAG. The AXL immunocomplex displayed a time-dependent increase in total ubiquitination upon 17-AAG exposure (Fig. 7A) that peaked at 2–3 h of 17-AAG treatment and then decreased. The AXL ubiquitination peak preceded the loss of AXL protein at later time points as shown by the anti-HA immunoblot. AXL polyubiquitination upon 17-AAG treatment was also verified by immunoprecipitating ubiquitin (HA) and probing for AXL (supplemental Fig. 4). To confirm that the polyubiquitinated protein observed was AXL itself and not an associated protein pulled down by the AXL-ubiquitin immunocomplex, we used nickel pulldown of His/myc-tagged AXL under denaturing conditions. HeLa cells were co-transfected with UbHA and His/myc-tagged AXL and treated with 17-AAG. AXL protein was then purified by nickel pulldown and probed for HA to evaluate its ubiquitination level. This experiment confirmed that purified AXL protein was ubiquitinated upon HSP90 inhibition (Fig. 7B).
FIGURE 7.
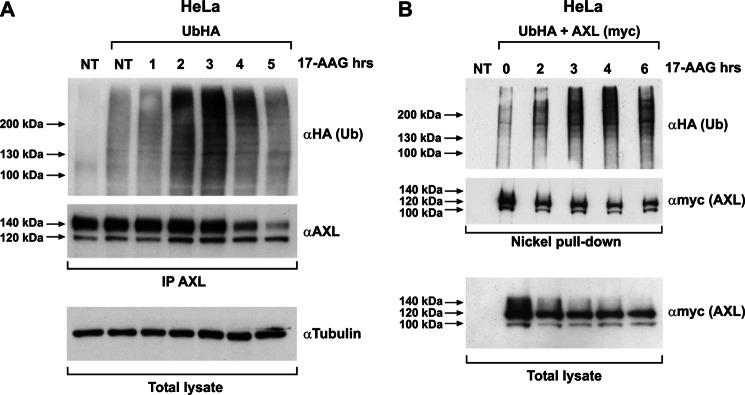
17-AAG induces AXL ubiquitination. A, HeLa cells transiently transfected with HA-tagged ubiquitin (UbHA) were subjected to vehicle (NT) or 500 nm 17-AAG treatment for the indicated time points. Cell lysates were immunoprecipitated (IP) using anti-AXL and immunoblotted using anti-HA and -AXL. Tubulin immunoblotting on total lysate is shown to normalize the input. NT, nontransfected/nontreated. B, HeLa cells co-transfected with UbHA and His-tagged AXL (subcloned in pcDNA4/TO A His/myc) were treated or not with 500 nm 17-AAG for the indicated times. Cells were harvested, and comparable amounts of cell suspension were subjected to nickel affinity protein purification under denaturing conditions following the protocol described under “Materials and Methods.” Purified proteins were subjected to Western blotting using anti-HA (Ub) and anti-myc (AXL). Comparable aliquot of cell suspension from the inputs were lysed using standard procedure and showed as total lysate, immunoblotted for myc (AXL). NT, nontransfected.
AXL Protein Interacted with HSP90 Chaperone Complex in a 17-AAG-sensitive Manner
We analyzed the association between AXL and the crucial components of the chaperone complex, like HSP90 and HSP70, and its modulation upon 17-AAG exposure. We immunoprecipitated endogenous AXL from HeLa cells and probed for HSP90 and HSP70 upon 17-AAG treatment. HSP90 was co-immunoprecipitated with AXL under basal conditions (Fig. 8A). Upon 17-AAG treatment, AXL-HSP90 interaction initially increased but then became undetectable. On the contrary, HSP70 co-immunoprecipitation with AXL was not detectable under basal conditions (Fig. 8A). Treatment of HeLa cells with 17-AAG, besides increasing the cellular concentration of HSP70, also promoted its interaction with AXL. These kinetics of association/dissociation of the chaperones suggest a shift in the chaperoning balance from folding (HSP90) to degradation mode (HSP70) due to the 17-AAG-dependent block of HSP90 activity (18).
FIGURE 8.

17-AAG modulates AXL interaction with HSP90 and HSP70. A, HeLa cells treated with 500 nm 17-AAG for the indicated times, and equal amounts of protein lysate were immunoprecipitated (I.P.) using AXL antibody followed by immunoblotting with anti-HSP90 and -HSP70. AXL, HSP90, HSP70, and tubulin levels were monitored using the respective antibody by Western blotting performed using total lysate. B, HeLa cells co-transfected with AXL-FLAG and HSP90-HA vectors were treated with vehicle (−) and 500 nm 17-AAG for the indicated times. Equal amounts of protein lysate were immunoprecipitated using anti-HA (HSP90) followed by immunoblotting with anti-FLAG (AXL). FLAG and HA immunoblotting was also performed on total lysates. NT, non-transfected cells. Arrows indicate the 100-, 120-, and 140-kDa isoforms of the receptor. C, HeLa cells co-transfected with AXL-FLAG and CHIP-myc were treated or not (−) with 500 nm 17-AAG for the indicated times. Comparable protein aliquots were immunoprecipitated using anti-myc (CHIP) followed by immunoblotting with anti-FLAG (AXL). FLAG and myc immunoblotting was performed on total lysate. NT, non-transfected cells.
To validate these interactions and to evaluate HSP90 interaction with AXL, we performed co-immunoprecipitation experiments by expressing AXL-FLAG and HSP90-HA in HeLa cells. As shown in Fig. 8B, HSP90 interacted with the AXL core polypeptide (100 kDa), the high mannose AXL isoform (120 kDa), and to a lesser extent the fully mature receptor (140 kDa) under basal conditions (Fig. 8B) even though this particular isoform seemed the most affected upon 17-AAG exposure. The HSP70-containing chaperone complex destabilizes misfolded proteins by recruiting many different ubiquitin ligases among which the ubiquitin E3 ligase CHIP is well described (33, 34). CHIP E3 ligase interacts with HSP70/90 via its amino-terminal tetratricopeptide repeat (TPR) domain (35). To test the involvement of CHIP in 17-AAG-induced AXL ubiquitination, we co-expressed CHIP-myc and AXL-FLAG in HeLa cells and assessed the recruitment of AXL in the CHIP-myc immunoprecipitate in the absence and presence of 17-AAG. As shown in Fig. 8C, although AXL interacted poorly with CHIP under normal conditions, it strongly associated with the E3 ligase upon 4 h of 17-AAG treatment, indicating a functional role for CHIP in mediating 17-AAG-dependent ubiquitination of the receptor. Surprisingly, CHIP also displayed a preferential association with the 120- and 100-kDa AXL isoforms over the 140-kDa isoform.
To test whether CHIP is involved in AXL polyubiquitination, we exogenously expressed CHIP together with UbHA in HeLa cells. AXL immunoprecipitation and probing for HA in the Western blot experiments showed an increased AXL polyubiquitination in CHIP-overexpressing samples. AXL ubiquitination increased in the presence of 17-AAG and was further enhanced by lactacystin treatment, which saved polyubiquitinated protein from proteasomal degradation (Fig. 9A). Unlike the wild type CHIP, CHIP K30A, a mutant in the CHIP TPR domain (35, 36), was unable to interact with AXL (Fig. 9B). Moreover, this mutant functioned as a dominant-negative protein, abrogating 17-AAG-induced AXL polyubiquitination (Fig. 9A). These data clearly demonstrated the role of CHIP E3 ligase in mediating 17-AAG-induced AXL polyubiquitination (37, 38).
FIGURE 9.

CHIP E3 ligase is responsible for AXL ubiquitination upon 17-AAG treatment. A, HeLa cells co-transfected with UbHA and CHIP WT or CHIP K30A were treated for 3 h with 500 nm 17-AAG with and without lactacystin (10 μm). Cells were lysed, and equal amounts of proteins were immunoprecipitated (I.P.) using AXL antibody and immunoblotted with anti-HA. AXL, myc, and tubulin immunoblotting was performed to monitor their levels in the total lysate. NT, non-transfected cells. “–”, nontreated. B, HeLa cells co-transfected with AXL-FLAG and myc-tagged CHIP WT or the mutant CHIP K30A were treated with 500 nm 17-AAG and subjected to myc (CHIP) immunoprecipitation. CHIP immunocomplexes on Western blotting were probed using anti-FLAG (AXL). FLAG and myc immunoblotting in total cell lysates was used to check AXL and CHIP levels, respectively.
AXL Sensitivity to 17-AAG Was Dependent on AXL Kinase Domain but Not on Kinase Enzymatic Activity
We next determined the region of AXL conferring sensitivity to 17-AAG. The tyrosine kinase receptor HER2 has been shown to be an HSP90 client, and it is sensitive to 17-AAG. Moreover, it has been reported that the kinase domain of HER2 confers the receptor sensitivity to 17-AAG (39, 40). To verify whether AXL kinase domain was involved in 17-AAG sensitivity, we constructed an AXL mutant (AXL-EC) devoid of its intracellular domain but retaining the extracellular (EC) and transmembrane domains. We transiently transfected this mutant in HeLa cells and performed a treatment with tunicamycin or subjected protein to digestion with Endo H and PNGase F. The AXL-EC mutant was fully glycosylated and transported to the cell surface (supplemental Fig. 5 and data not shown). We then transfected the AXL-EC- and AXL WT-expressing vectors in HeLa cells and compared their sensitivity to 17-AAG. As shown in Fig. 10A, only full-length AXL was degraded upon 17-AAG, whereas its mutant AXL-EC failed to respond after up to 8 h of treatment, suggesting that the AXL tyrosine kinase domain is crucial for mediating 17-AAG sensitivity.
FIGURE 10.

The kinase domain but not the kinase activity sensitizes AXL to 17-AAG. A, FLAG-tagged AXL WT and AXL-EC mutant and myc-tagged AXL kinase-dead mutant (AXL K558R) constructs are schematically represented. TK, tyrosine kinase domain; TM, transmembrane domain. HeLa cells transiently transfected with FLAG-tagged AXL WT and AXL-EC expression vectors were treated with 500 nm 17-AAG (+) or vehicle (−) for 8 h, and the lysates were immunoblotted with anti-FLAG and anti-tubulin. B, CAL62 cells were treated with vehicle (−) or with 5 μm bosutinib in the presence or absence of 17-AAG (500 nm). Cell lysates were immunoblotted with anti-AXL, anti-phospho-AXL (pAXL), or anti-tubulin antibodies. C, HeLa cells transfected with myc-tagged AXL WT and AXL kinase-dead mutant (AXL K558R) were treated with 500 nm 17-AAG for the indicated time points. The lysates were subjected to Western blotting for myc and tubulin. NT, non-transfected cells.
We then asked whether AXL kinase activity was also required. To this aim, we treated CAL62 cells with bosutinib for 8 h in the presence and absence of 17-AAG and evaluated AXL degradation. As shown in Fig. 10B, no differences in 17-AAG-induced AXL degradation kinetics were detected between bosutinib-treated or untreated cells even though a significant reduction in AXL phosphorylation (Fig. 10B) was achieved by bosutinib pretreatment. Consistently, the AXL kinase-dead mutant (AXL K558R) overexpressed in HeLa cells retained its sensitivity to 17-AAG, confirming that kinase activity is not necessary for 17-AAG-induced AXL degradation (Fig. 10C).
DISCUSSION
This report sheds light on the molecular regulation of receptor tyrosine kinase AXL protein stability. We demonstrated that AXL protein is a substrate of the proteasome to which it is targeted in an HSP90-dependent and -independent manner. In particular, the plasma membrane-associated receptor is degraded by the proteasome but independently from the HSP90-HSP70 chaperone complex, whereas the intracellular mature fully glycosylated AXL isoform needs HSP90 for its stability. Thus, inhibition of the chaperone by 17-AAG promotes AXL mature isoform polyubiquitination and proteasome-dependent degradation.
HSP90 and its related co-chaperones play a regulatory role in maintaining conformational maturation and structural integrity of a variety of cellular proteins. Although the concept of client receptor tyrosine kinase destabilization upon HSP90 inhibition came to light decades ago, this is the first report dissecting the HSP90 inhibitory effect on differentially glycosylated species of a client protein. Such a different effect could be explained either by a direct involvement of HSP90 in the glycosylation process of AXL receptor or by its requirement for 140-kDa protein isoform folding and stability after glycosylation completion. We could confirm the latter hypothesis by showing that 17-AAG did not impede AXL protein full glycosylation because MG132 upon co-treatment could rescue newly synthesized mature AXL species from 17-AAG-induced degradation. By contrast to AXL, other HSP90 client receptor tyrosine kinases such as HER2 (41), RET (36), c-KIT (42), and PDGF receptor (43) require chaperone activity for the correct folding and stability of both mature and immature forms as shown by the proportionate degradation of both isoforms (supplemental Fig. 6). In this regard, AXL behaves differently from other receptor tyrosine kinases that also depend on HSP90 for their stability at the plasma membrane. However, TrkAI receptor is similar to AXL because 17-AAG causes the degradation of the fully mature receptor and the accumulation of the immature receptor (44).
17-AAG affected the interaction between HSP90 and AXL under normal conditions. Besides this, the AXL immunocomplex displayed an increasing association to HSP70 on time upon 17-AAG. This shift of AXL-associating partners of the chaperone complex with AXL not only demonstrates the alteration in the chaperone complex but also indicates the fate of AXL protein. The shift from the normal folding mode to the degradation mode of the chaperone complex mediates CHIP E3 ligase recruitment and links the chaperone-associated client to the proteasome degradation machinery (18). The co-immunoprecipitation analysis revealed that HSP90 and CHIP interacted with 120- and 100-kDa AXL isoforms and to a lesser extent with the 140-kDa isoform. This effect is probably due to tyrosine phosphorylation of the mature, membrane-localized AXL, which may render its interaction with the chaperone more labile and more difficult to detect. Indeed, the HSP90-interacting region of client kinases is characterized by neutral/positive charge; therefore, the negative charge introduced by tyrosine phosphorylation could be detrimental (45) to detection of the interaction in co-immunoprecipitation experiments. Accordingly, we observed that kinase-dead AXL displayed a better association of its 140-kDa species to both HSP90 and CHIP with respect to wild type receptor (supplemental Fig. 7).
The ability of CHIP WT, but not of the TPR CHIP mutant (CHIP K30A), to ubiquitinate AXL further supports the involvement of CHIP E3 ligase in AXL polyubiquitination at least in the context of HSP90 inhibition. Although CHIP K30A was capable of abolishing AXL ubiquitination, it could not completely stop the degradation of AXL induced by 17-AAG. These data were also confirmed by RNA interference of CHIP in HeLa and CAL62 cells (data not shown). We cannot exclude that our approaches could not achieve a complete inhibition of CHIP function, and the residual activity might still be sufficient to degrade client proteins. However, the possible involvement of other E3 ligases in 17-AAG-induced protein degradation of HSP90 clients should be considered as well. By using CHIP−/− mouse embryo fibroblasts, it has been shown that the role of CHIP in ubiquitination and turnover of HSP90 client proteins is not exclusive and that other E3 ligases may complement CHIP deficiency (35, 38).
Our data also showed that an AXL receptor carrying an intact intracellular domain retained sensitivity to 17-AAG, whereas the AXL-EC, an intracellular domain deletion mutant, completely lost the response. However, AXL kinase activity is not required for the 17-AAG effect. This favors the prevailing hypothesis that the HSP90 binding determinants are located in the kinase domain, and this association depends on the intrinsic structure and stability of the receptor tyrosine kinase domain (46, 47) rather than on its activity. However, the exact specific factors and molecular determinants favoring this interaction have yet to be addressed.
AXL overexpression and activation appear to be a feature of many different types of cancer, and there is enough evidence to support its oncogenic activity (48). We have demonstrated previously that AXL is overexpressed and active in thyroid cancer. We have shown that silencing AXL by RNA interference hampers thyroid cancer cell proliferation, survival, migration, and tumor formation in immunodeficient mice (6). Furthermore, AXL overexpression sustains resistance to different anticancer agents (7–11). By using 17-AAG, we showed that the functional inhibition of HSP90 induced AXL degradation, which efficiently interfered with its downstream signaling and biological activity. HSP90 inhibitors have already been used to down-regulate multiple oncogenic signal transducers, and their activity toward AXL widens the application of these drugs. Moreover, in the advent of optimized and clinically relevant HSP90 inhibitors (49–51), our data strongly suggest that AXL overexpression and activity could be targeted in AXL-addicted tumors or in AXL-mediated drug-resistant cancers.
Acknowledgments
We thank M. Billaud (Génétique moléculaire, signalisation et cancer UMR 5201 (CNRS/Université Lyon 1), Université Claude Bernard Lyon, 169373 Lyon Cedex 08, France) for the CHIP-myc and CHIP-TPR-myc (K30A) vectors, S. Giordano (Institute for Cancer Research and Treatment, University of Torino School of Medicine, Torino, Italy) for the pcDNA-HA-ubiquitin (UbHA) vector, William C. Sessa (Department of Pharmacology and Molecular Cardiobiology Program, Yale University School of Medicine, New Haven, CT) for the HSP90 plasmid.
This work was supported by the Associazione Italiana per la Ricerca sul Cancro, the Istituto Superiore di Oncologia, Project “Sviluppo di nuovi farmaci capaci alterare il microambiente tumorale e ripristinare la risposta immune anti-tumorale” (Alleanza Contro il Cancro), Project “Molecular diagnostic and prognostic markers of thyroid neoplasis” RF-CAM-353005 of the Health Ministry, Italian Ministero della Salute Grant PIO-2, and Ministero dell'Università e della Ricerca Grants E61J11000300001 and E61J10000210001.

This article contains supplemental Figs. 1–7.
- Gas6
- growth arrest-specific protein 6
- HSP
- heat shock protein
- CHIP
- carboxyl terminus of HSC70-interacting protein
- 17-AAG
- 17-allylamino-17-demethoxygeldanamycin
- Endo H
- endoglycosidase H
- PNGase F
- peptide:N-glycosidase F
- Luc
- luciferase
- Ub
- ubiquitin
- TPR
- tetratricopeptide repeat
- EC
- extracellular domain.
REFERENCES
- 1. O'Bryan J. P., Frye R. A., Cogswell P. C., Neubauer A., Kitch B., Prokop C., Espinosa R., 3rd, Le Beau M. M., Earp H. S., Liu E. T. (1991) Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 11, 5016–5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stitt T. N., Conn G., Gore M., Lai C., Bruno J., Radziejewski C., Mattsson K., Fisher J., Gies D. R., Jones P. F., Masiakowski P., Ryan T. E., Tobkes N. J., Chen D. H., DiStefano P. S., Long G. L., Basilico C., Goldfarb M. P., Lemke G., Glass D. J., Yancopoulos G. D. (1995) The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670 [DOI] [PubMed] [Google Scholar]
- 3. Holland S. J., Pan A., Franci C., Hu Y., Chang B., Li W., Duan M., Torneros A., Yu J., Heckrodt T. J., Zhang J., Ding P., Apatira A., Chua J., Brandt R., Pine P., Goff D., Singh R., Payan D. G., Hitoshi Y. (2010) R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 70, 1544–1554 [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y. X., Knyazev P. G., Cheburkin Y. V., Sharma K., Knyazev Y. P., Orfi L., Szabadkai I., Daub H., Kéri G., Ullrich A. (2008) AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 68, 1905–1915 [DOI] [PubMed] [Google Scholar]
- 5. Rankin E. B., Fuh K. C., Taylor T. E., Krieg A. J., Musser M., Yuan J., Wei K., Kuo C. J., Longacre T. A., Giaccia A. J. (2010) AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 70, 7570–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Avilla E., Guarino V., Visciano C., Liotti F., Svelto M., Krishnamoorthy G., Franco R., Melillo R. M. (2011) Activation of TYRO3/AXL tyrosine kinase receptors in thyroid cancer. Cancer Res. 71, 1792–1804 [DOI] [PubMed] [Google Scholar]
- 7. Huang F., Hurlburt W., Greer A., Reeves K. A., Hillerman S., Chang H., Fargnoli J., Graf Finckenstein F., Gottardis M. M., Carboni J. M. (2010) Differential mechanisms of acquired resistance to insulin-like growth factor-I receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 70, 7221–7231 [DOI] [PubMed] [Google Scholar]
- 8. Liu L., Greger J., Shi H., Liu Y., Greshock J., Annan R., Halsey W., Sathe G. M., Martin A. M., Gilmer T. M. (2009) Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 69, 6871–6878 [DOI] [PubMed] [Google Scholar]
- 9. Hong C. C., Lay J. D., Huang J. S., Cheng A. L., Tang J. L., Lin M. T., Lai G. M., Chuang S. E. (2008) Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 268, 314–324 [DOI] [PubMed] [Google Scholar]
- 10. Dufies M., Jacquel A., Belhacene N., Robert G., Cluzeau T., Luciano F., Cassuto J. P., Raynaud S., Auberger P. (2011) Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2, 874–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Z., Lee J. C., Lin L., Olivas V., Au V., LaFramboise T., Abdel-Rahman M., Wang X., Levine A. D., Rho J. K., Choi Y. J., Choi C. M., Kim S. W., Jang S. J., Park Y. S., Kim W. S., Lee D. H., Lee J. S., Miller V. A., Arcila M., Ladanyi M., Moonsamy P., Sawyers C., Boggon T. J., Ma P. C., Costa C., Taron M., Rosell R., Halmos B., Bivona T. G. (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amann J., Kalyankrishna S., Massion P. P., Ohm J. E., Girard L., Shigematsu H., Peyton M., Juroske D., Huang Y., Stuart Salmon J., Kim Y. H., Pollack J. R., Yanagisawa K., Gazdar A., Minna J. D., Kurie J. M., Carbone D. P. (2005) Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 65, 226–235 [PubMed] [Google Scholar]
- 13. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 14. Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panaretou B., Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., Pearl L. H. (1998) ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 17, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obermann W. M., Sondermann H., Russo A. A., Pavletich N. P., Hartl F. U. (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 143, 901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Welch W. J. (1991) The role of heat-shock proteins as molecular chaperones. Curr. Opin. Cell Biol. 3, 1033–1038 [DOI] [PubMed] [Google Scholar]
- 18. Kundrat L., Regan L. (2010) Balance between folding and degradation for Hsp90-dependent client proteins: a key role for CHIP. Biochemistry 49, 7428–7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Supko J. G., Hickman R. L., Grever M. R., Malspeis L. (1995) Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 36, 305–315 [DOI] [PubMed] [Google Scholar]
- 20. Sausville E. A., Tomaszewski J. E., Ivy P. (2003) Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Curr. Cancer Drug Targets 3, 377–383 [DOI] [PubMed] [Google Scholar]
- 21. Schulte T. W., Neckers L. M. (1998) The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 42, 273–279 [DOI] [PubMed] [Google Scholar]
- 22. Modi S., Stopeck A., Linden H., Solit D., Chandarlapaty S., Rosen N., D'Andrea G., Dickler M., Moynahan M. E., Sugarman S., Ma W., Patil S., Norton L., Hannah A. L., Hudis C. (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 17, 5132–5139 [DOI] [PubMed] [Google Scholar]
- 23. Neckers L., Workman P. (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 18, 64–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santoro M., Carlomagno F., Romano A., Bottaro D. P., Dathan N. A., Grieco M., Fusco A., Vecchio G., Matoskova B., Kraus M. H., Di Fiore P. P. (1995) Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 267, 381–383 [DOI] [PubMed] [Google Scholar]
- 25. García-Cardeña G., Fan R., Shah V., Sorrentino R., Cirino G., Papapetropoulos A., Sessa W. C. (1998) Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392, 821–824 [DOI] [PubMed] [Google Scholar]
- 26. Guida T., Anaganti S., Provitera L., Gedrich R., Sullivan E., Wilhelm S. M., Santoro M., Carlomagno F. (2007) Sorafenib inhibits imatinib-resistant KIT and platelet-derived growth factor receptor β gatekeeper mutants. Clin. Cancer Res. 13, 3363–3369 [DOI] [PubMed] [Google Scholar]
- 27. Gottardi C. J., Dunbar L. A., Caplan M. J. (1995) Biotinylation and assessment of membrane polarity: caveats and methodological concerns. Am. J. Physiol. Renal Physiol. 268, F285–F295 [DOI] [PubMed] [Google Scholar]
- 28. Ghosh A. K., Secreto C., Boysen J., Sassoon T., Shanafelt T. D., Mukhopadhyay D., Kay N. E. (2011) The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood 117, 1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carlomagno F., De Vita G., Berlingieri M. T., de Franciscis V., Melillo R. M., Colantuoni V., Kraus M. H., Di Fiore P. P., Fusco A., Santoro M. (1996) Molecular heterogeneity of RET loss of function in Hirschsprung's disease. EMBO J. 15, 2717–2725 [PMC free article] [PubMed] [Google Scholar]
- 30. Neckers L., Schulte T. W., Mimnaugh E. (1999) Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest. New Drugs 17, 361–373 [DOI] [PubMed] [Google Scholar]
- 31. Xu W., Yuan X., Xiang Z., Mimnaugh E., Marcu M., Neckers L. (2005) Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 12, 120–126 [DOI] [PubMed] [Google Scholar]
- 32. Murata S., Chiba T., Tanaka K. (2003) CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 35, 572–578 [DOI] [PubMed] [Google Scholar]
- 33. Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 34. Murata S., Minami Y., Minami M., Chiba T., Tanaka K. (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2, 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu W., Marcu M., Yuan X., Mimnaugh E., Patterson C., Neckers L. (2002) Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. U.S.A. 99, 12847–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alfano L., Guida T., Provitera L., Vecchio G., Billaud M., Santoro M., Carlomagno F. (2010) RET is a heat shock protein 90 (HSP90) client protein and is knocked down upon HSP90 pharmacological block. J. Clin. Endocrinol. Metab. 95, 3552–3557 [DOI] [PubMed] [Google Scholar]
- 37. Zhou P., Fernandes N., Dodge I. L., Reddi A. L., Rao N., Safran H., DiPetrillo T. A., Wazer D. E., Band V., Band H. (2003) ErbB2 degradation mediated by the co-chaperone protein CHIP. J. Biol. Chem. 278, 13829–13837 [DOI] [PubMed] [Google Scholar]
- 38. Morishima Y., Wang A. M., Yu Z., Pratt W. B., Osawa Y., Lieberman A. P. (2008) CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum. Mol. Genet. 17, 3942–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tikhomirov O., Carpenter G. (2003) Identification of ErbB-2 kinase domain motifs required for geldanamycin-induced degradation. Cancer Res. 63, 39–43 [PubMed] [Google Scholar]
- 40. Xu W., Mimnaugh E., Rosser M. F., Nicchitta C., Marcu M., Yarden Y., Neckers L. (2001) Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem. 276, 3702–3708 [DOI] [PubMed] [Google Scholar]
- 41. Citri A., Alroy I., Lavi S., Rubin C., Xu W., Grammatikakis N., Patterson C., Neckers L., Fry D. W., Yarden Y. (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 21, 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fumo G., Akin C., Metcalfe D. D., Neckers L. (2004) 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood 103, 1078–1084 [DOI] [PubMed] [Google Scholar]
- 43. Matei D., Satpathy M., Cao L., Lai Y. C., Nakshatri H., Donner D. B. (2007) The platelet-derived growth factor receptor α is destabilized by geldanamycins in cancer cells. J. Biol. Chem. 282, 445–453 [DOI] [PubMed] [Google Scholar]
- 44. Farina A. R., Tacconelli A., Cappabianca L., Cea G., Chioda A., Romanelli A., Pensato S., Pedone C., Gulino A., Mackay A. R. (2009) The neuroblastoma tumour-suppressor TrkAI and its oncogenic alternative TrkAIII splice variant exhibit geldanamycin-sensitive interactions with Hsp90 in human neuroblastoma cells. Oncogene 28, 4075–4094 [DOI] [PubMed] [Google Scholar]
- 45. Citri A., Harari D., Shohat G., Ramakrishnan P., Gan J., Lavi S., Eisenstein M., Kimchi A., Wallach D., Pietrokovski S., Yarden Y. (2006) Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 281, 14361–14369 [DOI] [PubMed] [Google Scholar]
- 46. Shimamura T., Lowell A. M., Engelman J. A., Shapiro G. I. (2005) Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 65, 6401–6408 [DOI] [PubMed] [Google Scholar]
- 47. Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K. D., Karras G. I., Lindquist S. (2012) Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y., Ye X., Tan C., Hongo J. A., Zha J., Liu J., Kallop D., Ludlam M. J., Pei L. (2009) Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 28, 3442–3455 [DOI] [PubMed] [Google Scholar]
- 49. Ramalingam S. S., Egorin M. J., Ramanathan R. K., Remick S. C., Sikorski R. P., Lagattuta T. F., Chatta G. S., Friedland D. M., Stoller R. G., Potter D. M., Ivy S. P., Belani C. P. (2008) A phase I study of 17-allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clin. Cancer Res. 14, 3456–3461 [DOI] [PubMed] [Google Scholar]
- 50. Hubbard J., Erlichman C., Toft D. O., Qin R., Stensgard B. A., Felten S., Ten Eyck C., Batzel G., Ivy S. P., Haluska P. (2011) Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Invest. New Drugs 29, 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iyer G., Morris M. J., Rathkopf D., Slovin S. F., Steers M., Larson S. M., Schwartz L. H., Curley T., DeLaCruz A., Ye Q., Heller G., Egorin M. J., Ivy S. P., Rosen N., Scher H. I., Solit D. B. (2012) A phase I trial of docetaxel and pulse-dose 17-allylamino-17-demethoxygeldanamycin in adult patients with solid tumors. Cancer Chemother. Pharmacol. 69, 1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]