

Background: Rac-1 is an integral signaling component of insulin-stimulated GLUT4 traffic in muscle cells.
Results: Insulin-independent Rac-1 superactivation results in Akt and AS160 phosphorylation that drives GLUT4 translocation and rescues insulin resistance.
Conclusion: Rac-1 superactivation fulfills signaling requirements to elicit an insulin-like response on GLUT4 traffic in muscle cells.
Significance: Signaling capacity from Rac-1 superactivation can bypass the defects developed during insulin resistance.
Keywords: Akt, Ceramide, Glut4, Insulin Resistance, Jun N-terminal Kinase (JNK), Rac1, AS160
Abstract
Insulin activates a cascade of signaling molecules, including Rac-1, Akt, and AS160, to promote the net gain of glucose transporter 4 (GLUT4) at the plasma membrane of muscle cells. Interestingly, constitutively active Rac-1 expression results in a hormone-independent increase in surface GLUT4; however, the molecular mechanism and significance behind this effect remain unresolved. Using L6 myoblasts stably expressing myc-tagged GLUT4, we found that overexpression of constitutively active but not wild-type Rac-1 sufficed to drive GLUT4 translocation to the membrane of comparable magnitude with that elicited by insulin. Stimulation of endogenous Rac-1 by Tiam1 overexpression elicited a similar hormone-independent gain in surface GLUT4. This effect on GLUT4 traffic could also be reproduced by acutely activating a Rac-1 construct via rapamycin-mediated heterodimerization. Strategies triggering Rac-1 “superactivation” (i.e. to levels above those attained by insulin alone) produced a modest gain in plasma membrane phosphatidylinositol 3,4,5-trisphosphate, moderate Akt activation, and substantial AS160 phosphorylation, which translated into GLUT4 translocation and negated the requirement for IRS-1. This unique signaling capacity exerted by Rac-1 superactivation bypassed the defects imposed by JNK- and ceramide-induced insulin resistance and allowed full and partial restoration of the GLUT4 translocation response, respectively. We propose that potent elevation of Rac-1 activation alone suffices to drive insulin-independent GLUT4 translocation in muscle cells, and such a strategy might be exploited to bypass signaling defects during insulin resistance.
Introduction
Insulin-stimulated glucose uptake into skeletal muscle is a major determinant of whole body glucose homeostasis as muscle is the primary tissue responsible for the vast majority of dietary glucose disposition (1). Insulin binding to its receptor at the muscle cell surface initiates a complex signaling cascade leading to the net recruitment of glucose transporter 4 (GLUT4)4-containing vesicles from an intracellular compartment to the plasma membrane (2). Identifying the full complement of insulin-triggered signals regulating GLUT4 translocation in muscle could provide a valuable asset in overcoming defects that lead to insulin resistance and its consequent type 2 diabetes.
In L6 muscle cells in culture, the first postreceptor event leading to GLUT4 translocation is the selective tyrosine phosphorylation of isoform 1 of the insulin receptor substrate (IRS-1) (3) with consequent activation of phosphatidylinositol 3-kinase (PI3K) to elevate membrane PI(3,4,5)P3 (4, 5). Downstream of PI3K, insulin signaling in muscle cells bifurcates into two independent pathways characterized, respectively, by activation of the serine/threonine kinase Akt and the Rho family GTPase Rac-1 (6–8). Akt in turn phosphorylates the protein AS160 to inhibit its GTPase-activating protein activity toward Rab GTPases (9–11). This enables the activation (GTP loading) of Rabs 8A and 13 to prevail and provide mobilization cues for GLUT4 vesicle delivery to the muscle cell plasma membrane (12, 13). The other signaling arm involves PI3K-dependent Rac-1 activation (GTP loading) that triggers a cycle of branching and severing of cortical actin filaments (14). The resulting cortical mesh of branched actin filaments may serve as a tether for both signaling molecules and GLUT4 vesicles (4, 7, 14–16). Activation of Rac-1 is thought to depend on either activation of dedicated guanine exchange factors (GEFs) and/or inhibition of dedicated GTPase-activating proteins (17, 18).
In vivo, muscle Akt is readily activated in response to insulin, and Akt-2 gene knock-out mice show reduced insulin-dependent glucose uptake into muscle (19). Similarly, recent studies show that insulin activates Rac-1 in mouse muscle, and mice with muscle-specific Rac-1 gene deletion have diminished insulin-dependent GLUT4 translocation and glucose uptake (20, 21). The latter results parallel observations of abated GLUT4 translocation in L6 muscle cells in culture depleted of Rac-1 via siRNA-dependent gene silencing (7).
Importantly, both Akt and Rac-1 are required for proper insulin-induced GLUT4 translocation as overexpressing dominant-negative mutants of each one or silencing their endogenous gene expression impairs the insulin response of GLUT4 without mutually inhibiting one another (6, 7, 22). Despite this apparent independence of the Akt and Rac-1 signaling arms, overexpression of constitutively active Rac-1 in muscle cells increases surface GLUT4 (23), although the molecular underpinnings are unknown. Here, we build on that observation and unravel the signaling mechanism. Using acute (rapamycin-inducible) and chronic modalities of Rac-1 superactivation (i.e. activation surpassing the activation levels caused by insulin), including activation of a Rac-specific GEF, Tiam1, we achieved an insulin-comparable GLUT4 translocation response without input from insulin. Unexpectedly, this hormone-independent Rac-1 activation generated a modest increase in membrane-associated PI(3,4,5)P3 that in turn caused mild Akt phosphorylation. Downstream signaling was amplified, achieving substantial AS160 phosphorylation and GLUT4 translocation. Rac-1 superactivation bypassed the requirement for IRS-1 phosphorylation. Finally, we show that Rac-1 superactivation relieves the GLUT4 traffic defect imposed by two strategies that cause insulin resistance: activation of the stress kinase JNK and ceramide exposure. Hence, Rac-1 is mechanistically identified as a viable target for the treatment of insulin resistance in a cellular model. By crossover activation of Akt, Rac-1 activation enacts the full complement of insulin-derived distal responses that mobilize GLUT4 in muscle cells independently of IRS-1 participation.
EXPERIMENTAL PROCEDURES
Reagents and Constructs
Polyclonal anti-myc (A-14) antibody, monoclonal anti-FLAG antibody, DMSO, and rapamycin were purchased from Sigma-Aldrich. Monoclonal anti-phosphotyrosine antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-HA antibody was purchased from Covance Inc. (Princeton, NJ). Polyclonal anti-P-Akt(308), P-Akt(473), and phosphorylated Akt substrate were obtained from Cell Signaling Technology (Danvers, MA). Polyclonal anti-GFP antibody was from Abcam Inc. (Cambridge, MA). Monoclonal P-IRS-1(307) antibody was from EMD Millipore (Billerica, MA). Cy3- and Cy5-conjugated donkey anti-rabbit and donkey anti-mouse secondary antibodies and horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Human insulin was purchased from Eli Lilly (Indianapolis, IN). Rhodamine-phalloidin was from Invitrogen. Akt inhibitor 1/2 (Akti1/2), compound C, and latrunculin B (LB) were from Calbiochem. Wortmannin and C2-ceramide were from Enzo Life Science (Farmingdale, NY). The cDNA constructs (Lyn-FRB, YFP-tagged FK506-binding protein (FKBP) (YF), YF-CA-Rac, YF-Tiam1, and YF-DN-Rac) used for rapamycin-induced heterodimerization have been described previously and were generously provided by Dr. Mary Teruel and Dr. Tobias Meyer (24). Wild-type (WT)-Rac- and constitutively active (CA)-Rac-GFP were provided by Dr. Mark R. Philips. HA-IRS1 was provided by Dr. Maria Rozakis. FLAG-CA-JNK1 was provided by Dr. Gokhan Hotamisligil (25). Tiam1-GFP was provided by Dr. Sergio Grinstein. Cyan fluorescent protein-tagged WT-Cdc42 and CA-Cdc42 were provided by Dr. John Brumell.
Cell Culture and Transfection
The rat L6 muscle cell line stably expressing GLUT4 with an exofacial Myc epitope tag (L6-GLUT4myc) was cultured as described previously (26). Transfection of cDNA was performed with Lipofectamine 2000 (Invitrogen) or FuGENE HD (Promega) according to the manufacturers' protocols. Briefly, cDNA-Lipofectamine mixture was applied to the cells for 4–6 h before exchanging to fresh growth medium. Cells were allowed to recover overnight before experiments the next day. Cells were treated with the cDNA-FuGENE mixture for 24 h to achieve higher transfection efficiency. Maximal transfection efficiency was 30%; hence, single cell assays were used to assess the consequence of transfection.
Cell Surface and Total GLUT4myc Detection by Immunofluorescence Microscopy
Surface GLUT4myc detection by immunofluorescence in adherent L6 myoblasts was measured as described previously (27). Following 3-h serum starvation, cells were stimulated with/without 100 nm insulin for 10 min. Cells were quickly washed twice with cold phosphate-buffered saline supplemented with calcium and magnesium (PBS+), fixed with 3% (v/v) paraformaldehyde, quenched with 0.1 m glycine, and blocked with 5% (v/v) milk. Surface GLUT4myc was stained by incubation with anti-myc primary antibody followed by Cy3-coupled secondary antibody. For co-staining of tagged cDNA constructs, permeabilization with 0.1% Triton X-100 and detection with epitope antibody were applied after labeling of primary myc antibody. For measurement of rapamycin-induced gain in surface GLUT4myc, 10 min of 1 μm rapamycin treatment was applied to cells transfected with the respective components of the rapamycin heterodimerization system. For total GLUT4myc detection, cells were permeabilized with 0.1% Triton X-100 for 15 min following the quenching step prior to blocking and labeling with anti-myc primary antibody. Fluorescence images were taken with a Zeiss LSM 510 laser-scanning confocal microscope (Thornwood, NY). Cells were scanned along the z axis, a single composite image (collapsed xy projection) of the optical cuts per cell was assembled, and the pixel intensity of each cell (≥25 cells per condition) was quantified by ImageJ software.
Rounded-up Myoblast Assay
Rounded-up myoblasts were generated as described previously (15). Adherent cells were incubated in PBS without calcium and magnesium at 37 °C for 10 min to allow detachment. Cells were then resuspended and attached on the coverslip for 10 min with/without insulin or rapamycin treatment at 37 °C followed by fixation.
Detection of Actin Remodeling
Following treatment with/without insulin or rapamycin for 10 min at 37 °C, myoblasts were fixed, permeabilized with 0.1% Triton X-100 in PBS+ for 3 min, blocked with 5% milk, and stained with rhodamine-phalloidin for filamentous (F)-actin. Cells were imaged using a Zeiss LSM 510 laser-scanning confocal microscope.
Cell Lysates, Immunoprecipitation, and Immunoblotting
After transfection and experimental treatments, cells were washed quickly with cold PBS and lysed with 1% Triton X-100. Lysates were passed through a 29-gauge syringe needle 10 times, and supernatants were collected by a 10-min spin at 12,000 × g. The supernatant was subsequently incubated with protein G beads conjugated to 1–2 μg of epitope antibody at 4 °C for at least 4 h before subjecting to three washes with 0.1% Triton X-100. Immunoprecipitated protein samples were eluted with 2× Laemmli sample buffer, resolved by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes (Bio-Rad), and immunoblotted with the respective antibodies. Primary antibodies were detected with the appropriate HRP-conjugated species-specific IgG secondary antibodies. Immunoblotting was completed with Western Lightning Chemiluminescence Reagent Plus and HyBlot CL autoradiography film from Denville Scientific (Denville, NJ).
Rac-GTP Pulldown Assay
The Rac-1 activation assay was performed as described previously (8). Briefly, myoblasts were serum-starved for 3 h before stimulation with/without 100 nm insulin for 10 min. Cell lysates were collected, spun down at 12,000 × g for 1 min, and incubated with GST-Cdc42/Rac interactive binding domain beads under rotation at 4 °C for 30 min. After four washes, the pulled down proteins were eluted with 2× Laemmli sample buffer and resolved by 10% SDS-PAGE for immunoblotting. For Akti1/2 experiments, inhibitor or DMSO was applied in the last 1 h of serum starvation. For Rac activation with WT-Rac-GFP or CA-Rac-GFP, myoblasts were transfected with the respective constructs for 24 h before the experiments.
Statistics
Statistical analyses were carried out using Prism 4.0 software (GraphPad Software, San Diego, CA). Groups were compared using one-way analysis of variance with Newman-Keuls post hoc analysis. p < 0.05 was considered statistically significant.
RESULTS
Rac-1 Activation via Overexpression of CA-Rac-GFP or Tiam1-GFP Induces a Gain in Surface GLUT4 Independently of Insulin
We first verified that elevation of Rac-1 activity would raise GLUT4 surface levels. L6 myoblasts stably expressing myc-tagged GLUT4 (L6-GLUT4myc) were transiently transfected with WT- or CA-Rac-GFP. In control L6-GLUT4myc cells, GFP expression alone did not change the basal surface GLUT4 level, whereas insulin stimulation enhanced it by 2.2-fold (Fig. 1, A and B). WT-Rac-GFP overexpression did not alter surface GLUT4 levels compared with GFP-expressing control cells (Fig. 1, A and B). However, when CA-Rac-GFP was introduced, surface GLUT4 was significantly higher compared with that in neighboring untransfected cells (Fig. 1A). The 2.4-fold increase in basal surface GLUT4 elicited by CA-Rac-GFP overexpression was similar to that caused by insulin stimulation in the GFP-expressing controls (Fig. 1B). Addition of insulin to CA-Rac-GFP-expressing cells did not cause a further enhancement in surface GLUT4 compared with the already augmented amount detected in the absence of insulin (Fig. 1, A and B). This gain in surface GLUT4 was specific to active Rac-1 because neither the overexpression of WT-Cdc42 nor that of CA-Cdc42, a closely related Rho-GTPase, raised surface GLUT4 compared with the level observed with CA-Rac overexpression (supplemental Fig. 1, A and B). More importantly, transient overexpression of CA-Rac-GFP did not alter the total amount of cellular GLUT4myc compared with the GFP controls (supplemental Fig. 1C), eliminating the possibility of elevated GLUT4myc expression contributing to the heightened surface GLUT4 level observed in CA-Rac-GFP-expressing cells.
FIGURE 1.

Overexpression of CA-Rac-GFP or Tiam1-GFP stimulates GLUT4 translocation to the membrane of muscle cells. A and B, GLUT4myc myoblasts were transfected with GFP, WT-Rac-GFP, or CA-Rac-GFP followed by stimulation with/without insulin for 10 min to measure changes in surface GLUT4myc and quantified relative to GFP basal signal (mean ± S.E. (error bars); *, p < 0.05). C, myoblasts transfected with GFP, CA-Rac-GFP, or Tiam1-GFP were permeabilized and stained with rhodamine-phalloidin to visualize changes in F-actin. D and E, GLUT4myc myoblasts expressing GFP or Tiam1-GFP were stimulated with/without insulin for 10 min followed by staining for surface GLUT4myc and quantification relative to GFP basal signal (mean ± S.E. (error bars); *, p < 0.05). Representative images of three independent experiments are shown. Bar, 10 μm.
To validate the effect of transiently transfected CA-Rac-GFP, we activated endogenous Rac-1 through overexpression of a Rac-specific GEF, Tiam1 (28). As shown in the literature (29), in the absence of growth factor stimulation, Tiam1-GFP overexpression generated F-actin-rich ruffles that resembled those evoked by CA-Rac-GFP, suggesting that the endogenous Rac-1 was activated in Tiam1-overexpressing cells (Fig. 1C). Surface GLUT4 levels were significantly higher in Tiam1-overexpressing cells compared with GFP-expressing or untransfected cells (Fig. 1D). The net gain in GLUT4 at the plasma membrane was again similar to the increase induced by insulin in GFP-expressing controls (Fig. 1E). These results mirrored the data obtained in CA-Rac-expressing cells, reinforcing the observation that potent activation of Rac-1 alone exerts a full, insulin-like response on GLUT4 traffic to the plasma membrane.
To compare the level of Rac-1 activation achieved by insulin stimulation and CA-Rac-GFP overexpression, we analyzed the amount of GTP-loaded Rac in WT- or CA-Rac-GFP-expressing myoblasts stimulated with or without insulin. WT-Rac-GFP-overexpressing cells exhibited an increase in Rac-1 activation following 10 min of insulin stimulation (supplemental Fig. 2). However, the level of GTP-loaded Rac-1 evoked by insulin was much lower compared with that in myoblasts overexpressing CA-Rac-GFP. Therefore, the effect achieved by CA-Rac-GFP expression in myoblasts can comparatively be considered to be a Rac-1 superactivation, and this term is used throughout this report.
Acute Activation of Rac-1 Is Also Capable of Eliciting an Insulin-like Increase in Basal Surface GLUT4
Although overexpression of CA-Rac and Tiam1 served as good models of activating Rac-1 alone, their effects more closely resembled the consequence of a “chronic” Rac-1 activity in the span of 24 h post-transfection. In contrast, insulin induces GTP loading of Rac-1 within 1 min of addition and promotes maximal activation by 10 min (8, 30). Therefore, to explore whether the gain in basal surface GLUT4 observed with chronic Rac-1 activation would be recapitulated in an acute manner, we took advantage of a rapamycin-inducible heterodimerization system (31). The cell-permeating rapamycin has high affinity toward the FKBP, which within cells interacts tightly with the FRB domain of the protein kinase FKBP12-rapamycin-associated protein. Based on this principle, upon addition of rapamycin, a synthetic construct consisting of the protein of interest fused to FKBP can be rapidly recruited to FRB. FRB can further be localized to the plasma membrane via a genetically engineered targeting sequence of Lyn kinase. Because active Rac-1 is normally localized to the plasma membrane due to prenylation of its C-terminal CAAX motif (32, 33), the rapamycin-inducible Rac-1 activation strategy can be used to investigate acute Rac-1 activation by either (a) replacing the C-terminal CAAX motif on CA-Rac with FKBP so that it remains cytosolic until recruited to the membrane by rapamycin or (b) generating an FKBP chimeric protein containing the DH-PH domain of Tiam1 (domain required for the GEF activity toward Rac) (24). Using these two approaches to also eliminate any possible confounding effect arising from the 24-h CA-Rac-GFP expression, we investigated the effect of acute Rac-1 activation on GLUT4 traffic.
As proof of principle, we first examined whether co-expression of Lyn-FRB and YF constructs would allow recruitment of YF to the plasma membrane upon the addition of rapamycin. In rounded-up myoblasts, which retain signaling capability but also provide improved spatial resolution compared with adhered cells (15), YF remained cytosolic in DMSO-treated control (Fig. 2A). In contrast, after 10 min of rapamycin treatment, the YF signal began to redistribute to the plasma membrane, whereas cells without Lyn-FRB co-expression did not display this membrane recruitment (Fig. 2A). Similar rapamycin-triggered movement to the periphery was observed with either YF-CA-Rac lacking the CAAX motif (YF-CA-Rac) or a YF-linked DH-PH domain of Tiam1 (YF-Tiam1) co-expressed with Lyn-FRB (Fig. 2A).
FIGURE 2.

Acute Rac-1 activation via rapamycin-triggered heterodimerization induces GLUT4 translocation in muscle cells. YF, YF-CA-Rac, or YF-Tiam1 was co-transfected with/without Lyn-FRB in myoblasts. Cells were treated with/without rapamycin for 10 min followed by rounded-up myoblast assay and detecting the redistribution of the YFP signal (A), permeabilization and staining with rhodamine-phalloidin to measure changes in F-actin (B), and measurement of surface GLUT4myc (C and D). Changes were compared with those in YFP-expressing myoblasts stimulated with/without insulin and quantified relative to the YFP basal signal (mean ± S.E. (error bars); *, p < 0.05). Representative images of three independent experiments are shown. Bar, 10 μm.
To ascertain that Rac-1 activation only occurred after rapamycin-triggered recruitment of YF-CA-Rac and YF-Tiam1 to the plasma membrane, we probed for the characteristic Rac-mediated membrane ruffles (28). In DMSO-containing controls, neither YF-CA-Rac nor YF-Tiam1 expression generated membrane ruffling (Fig. 2B). However, upon rapamycin addition (10 min), cells displayed extensive actin remodeling evinced by the accumulation of phalloidin-decorated F-actin at sites of membrane ruffles (Fig. 2B). On the contrary, rapamycin did not cause actin rearrangement in myoblasts expressing YF or YF-DN-Rac with Lyn-FRB (supplemental Fig. 3). These results demonstrate the ability of rapamycin to selectively trigger acute Rac-1 activation in myoblasts by recruiting YF-CA-Rac and YF-Tiam1 to the plasma membrane when co-expressed with Lyn-FRB.
Utilizing this rapamycin-inducible Rac-1 activation strategy, we examined the changes in surface GLUT4 following 10 min of acute Rac-1 activation. As shown in Fig. 2C, expression of YF-CA-Rac did not alter the amount of surface GLUT4 detected in the DMSO-containing control. However, upon 10 min of rapamycin treatment, which provided a surge in Rac-1 activity, the corresponding surface GLUT4 was significantly elevated compared with neighboring untransfected cells. This gain in surface GLUT4 was also reproduced by expressing YF-Tiam1 in myoblasts to turn on endogenous Rac-1 following rapamycin treatment, whereas YF-expressing controls failed to exert any change in GLUT4 at the plasma membrane (Fig. 2C). Interestingly, the rapamycin-triggered elevation in surface GLUT4 achieved in cells with YF-CA-Rac or YF-Tiam1 was comparable with that observed in YFP controls after insulin stimulation (Fig. 2, C and D). These data suggested that, similar to the overexpression of CA-Rac-GFP (Fig. 1B), acute Rac-1 activation was sufficient to drive an insulin-like response on GLUT4 translocation.
Rac-1 Superactivation Signals through PI3K, Akt, and AS160 to Increase Surface GLUT4
Because Akt activation is a prerequisite for insulin-dependent GLUT4 translocation, we were surprised that Rac-1 superactivation would cause GLUT4 translocation because in response to insulin both signals appear to dissociate downstream of PI3K (6, 7). Hence, we hypothesized that Rac-1 superactivation might potentially exert signaling cues to Akt and/or to the upstream canonical insulin signaling molecules IRS-1 and PI3K. To start, we analyzed whether IRS-1 is tyrosine phosphorylated upon Rac-1 superactivation. For these experiments, we used the chronic, sustained active Rac-1 approach. Myoblasts were co-transfected with HA-tagged IRS-1 and CA-Rac-GFP (or GFP as control), then IRS-1 was immunoprecipitated via its HA epitope, and the corresponding level of Tyr(P) was examined with anti-Tyr(P) antibody. When GFP was co-expressed with HA-IRS-1, the basal level of Tyr(P) was negligible, whereas insulin stimulation visibly enhanced Tyr(P) in IRS-1 (Fig. 3A). IRS-1 immunoprecipitated from myoblasts expressing CA-Rac-GFP did not show any increase in Tyr(P) compared with the basal amount in GFP-expressing controls (Fig. 3A), implying that IRS-1 is not engaged by Rac-1 activation.
FIGURE 3.

Overexpression of CA-Rac-GFP results in a modest increase in plasma membrane PI(3,4,5)P3 and Akt phosphorylation without engaging IRS-1. A, HA-IRS-1 was co-transfected with GFP or CA-Rac-GFP before stimulating myoblasts with/without insulin for 10 min followed by immunoprecipitation (IP) using anti-HA antibody. Immunoprecipitates were stained with anti-Tyr(P) antibody to detect phosphorylation of IRS-1. B and C, PH-Akt-RFP was co-expressed along with GFP, WT-Rac-GFP, or CA-Rac-GFP. Rounded-up myoblasts were generated and stimulated for 10 min with/without insulin after which the redistribution of the PH-Akt-RFP to the membrane was determined. The plasma membrane to cytosolic (PM/Cyto) ratio of RFP in each condition was quantified and is expressed relative to the percentage of the parallel, maximal insulin response in units normalized to GFP-expressing control cells. D–F, myoblasts expressing HA-Akt + GFP, WT-Rac-GFP, or CA-Rac-GFP were stimulated with/without insulin for 10 min before immunoprecipitation with HA antibody. Immunoprecipitates were analyzed for P-Akt using anti-Thr(P)-308 or -Ser(P)-473 antibodies. Changes in P-Akt/total HA-Akt in each condition were quantified and are presented as the percentage of the parallel, maximal insulin response in units normalized to the GFP-expressing control. G, GLUT4myc myoblasts transfected with Lyn-FRB + YF-CA-Rac were pretreated with DMSO, 100 nm WM, 10 μm Akti1/2, 200 nm LB, or 10 μm compound C (CC) for 20 or 30 min (Akti1/2 and compound C) followed by 10-min rapamycin treatment in the presence of inhibitors. The changes in surface GLUT4myc were quantified and are illustrated as the percentage of the maximal rapamycin-induced response relative to DMSO-treated control cells (mean ± S.E. (error bars); *, p < 0.05 versus DMSO). Representative images of three to four independent experiments are shown. Bar, 10 μm. IB, immunoblot.
Next we evaluated whether CA-Rac-GFP would increase the levels of PI(3,4,5)P3 at the plasma membrane as an index of PI3K activation. Changes in PI(3,4,5)P3 at the cell periphery were assessed by transfecting a PI(3,4,5)P3 reporter constituted by the PH domain of Akt linked to RFP (PH-Akt-RFP) (34) and imaging rounded-up myoblasts. In unstimulated cells expressing GFP, the PI(3,4,5)P3 reporter remained mostly cytosolic, and only a faint peripheral signal was observed (Fig. 3B). Upon insulin stimulation, the signal at the plasma membrane was clearly evident, corroborating that the hormone triggers PI(3,4,5)P3 production (Fig. 3B). When PH-Akt-RFP was co-expressed with WT-Rac-GFP, its distribution pattern was similar to that displayed in unstimulated cells expressing GFP (Fig. 3B). Notably, however, expression of CA-Rac-GFP caused a clear redistribution of PH-Akt-RFP toward the plasma membrane, albeit lower than that caused by insulin (Fig. 3B). This enrichment of PH-Akt-RFP at the membrane was not due to the increase in membrane folding caused by Rac-dependent actin reorganization because co-expression of RFP alone with CA-Rac-GFP failed to exhibit a similar reporter accumulation (supplemental Fig. 4). Assessment of the ratio of the PH-Akt-RFP signal at the plasma membrane over the cytosolic signal in each condition revealed that Rac-1 superactivation via CA-Rac-GFP expression elevated PI(3,4,5)P3 production to ∼20% of the maximal insulin response, whereas WT-Rac-GFP had no effect (Fig. 3C).
The small gain in PI(3,4,5)P3 in cells with increased Rac-1 activity served as a potential gateway to study Akt activation. Thus, using myoblasts co-expressing HA-Akt and either the GFP, WT-Rac-GFP, or CA-Rac-GFP, we investigated the activation status of Akt by probing for phosphorylation at the Thr-308 and Ser-473 sites necessary for functional Akt (35–37). As expected, insulin stimulation significantly increased the phosphorylation of Akt at Thr-308 and Ser-473 compared with unstimulated cells co-expressing GFP (control) (Fig. 3D). When CA-Rac-GFP was co-expressed in the absence of insulin, the phosphorylation of Thr-308 and Ser-473 was elevated to 26 and 62%, respectively, of the maximal insulin response measured in parallel (Fig. 3, D, E, and F). In contrast, expression of WT-Rac-GFP also induced a 35% enhancement in Ser-473 phosphorylation relative to the maximal action of insulin but with little if any gain in Thr-308 phosphorylation (Fig. 3, D, E, and F).
To ascertain whether the Rac-driven PI3K and Akt participate in the insulin-independent GLUT4 translocation caused by Rac-1 superactivation, we examined the ability of wortmannin (WM) and Akti1/2 to interfere with this response. For these experiments, we used the acute Rac-1 superactivation assay (rapamycin-triggered) to avoid confounding effects of prolonged action of inhibitors. As shown in Fig. 3G, pretreatment of myoblasts with WM or Akti1/2 lowered the effectiveness of rapamycin-induced acute Rac-1 superactivation to elicit an increase in surface GLUT4 by 58 and 40%, respectively. On the contrary, pretreatment with compound C at a concentration shown previously to prevent AMP-activated protein kinase activity (38) did not alter the rapamycin-triggered response, suggesting that AMP-activated protein kinase does not participate in the Rac-1 superactivation-mediated GLUT4 translocation (Fig. 3G). In parallel, we assessed the effect of LB to explore whether actin dynamics is required for acute Rac-1 superactivation to increase surface GLUT4. Fig. 3G illustrates that indeed LB significantly reduced the GLUT4 response, showing that, as in the case of insulin stimulation, actin dynamics is an obligated component of Rac-1 superactivation-driven elevation in surface GLUT4 (27, 39).
It is well documented that submaximal Akt phosphorylation is sufficient to transmit downstream responses and that there is amplification in the Akt pathway leading to GLUT4 translocation (6, 40–42). We therefore explored whether the modest increase in Akt phosphorylation caused by CA-Rac-GFP sufficed to propagate the signal down to AS160, an essential component for the net gain of surface GLUT4 in response to insulin. When FLAG-AS160 was co-expressed with GFP (control), insulin promoted AS160 phosphorylation as detected by the increase in reactivity with anti-phospho-Akt substrate antibody (Fig. 4A). This gain did not occur in unstimulated cells co-expressing WT-Rac-GFP and FLAG-AS160, whereas a substantial FLAG-AS160 phosphorylation, equivalent to 70% of the maximal insulin response, was observed in cells expressing CA-Rac-GFP (Fig. 4, A and B).
FIGURE 4.
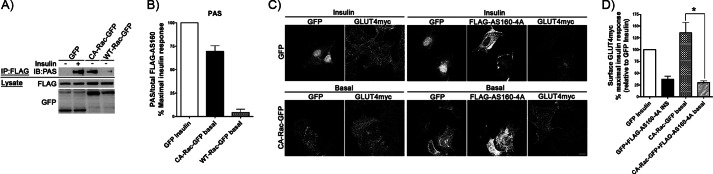
AS160 is phosphorylated upon CA-Rac-GFP overexpression and is required for the Rac-1-induced gain in surface GLUT4. A and B, FLAG-AS160 was co-expressed with GFP, WT-Rac-GFP, or CA-Rac-GFP before immunoprecipitation (IP) with anti-FLAG antibody. Immunoprecipitates were stained with phosphorylated Akt substrate (PAS) antibody to detect changes in AS160 phosphorylation. The intensity of phosphorylated Akt substrate/total FLAG-AS160 in each condition was quantified and is depicted as the percentage of the parallel, maximal insulin response in units normalized to the GFP-expressing control. C and D, FLAG-AS160–4A was co-transfected with GFP or CA-Rac-GFP followed by stimulation with/without insulin (INS) to measure changes in surface GLUT4. Quantification of each condition is presented as the percentage of the parallel, maximal insulin response normalized to GFP-expressing insulin-stimulated cells (mean ± S.E. (error bars); *, p < 0.05). Representative images of three independent experiments are shown. Bar, 10 μm. IB, immunoblot.
Finally, we explored the participation of AS160 in the response of GLUT4 to Rac-1 superactivation. As observed previously (9, 11), overexpression of the AS160-4A mutant (with mutations in four key residues normally phosphorylated by Akt in response to insulin) dampened the insulin-stimulated elevation in surface GLUT4 in control cells (co-expressing GFP) (Fig. 4, C and D). However, the AS160-4A mutant co-expressed with CA-Rac-GFP prevented the characteristic gain in surface GLUT4 exerted by Rac-1 superactivation (Fig. 4, C and D). In fact, AS160-4A had an effect on the response of GLUT4 quantitatively similar to either insulin or Rac-1 superactivation (achieving only 37 and 30% increases in surface GLUT4, respectively) (Fig. 4D). Collectively, these results substantiate the involvement of PI3K, Akt, and AS160 in Rac-1 superactivation-driven GLUT4 translocation.
Rac-1 Superactivation Relieves Defective GLUT4 Translocation Imposed by JNK and Ceramide
Insulin resistance of muscle and of muscle cells is typified by a lower GLUT4 translocation in response to the hormone, consequently reducing glucose uptake (43, 44). During high fat feeding, muscle insulin resistance involves the gain in lipids and their derivatives with resulting activation stress kinases such as JNK that in turn interfere with IRS-1 tyrosine phosphorylation. In particular, JNK activation causes IRS-1 phosphorylation at Ser-307, thereby functionally uncoupling IRS-1 from the insulin receptor and promoting IRS-1 degradation (25, 45, 46). Because the GLUT4 translocation elicited by Rac-1 superactivation illustrated above bypasses a requirement for IRS-1, we hypothesized that this feature might be utilized to restore defective insulin downstream of IRS-1. We therefore reconstituted the JNK-mediated insulin resistance in myoblasts by overexpressing a constitutively active JNK construct (FLAG-CA-JNK) recently shown to cause insulin resistance in vivo due to aborted signaling at the level of IRS-1 (25). As a proof of principle, co-expression of FLAG-CA-JNK and HA-IRS-1 significantly reduced the total level of HA-IRS-1 and enhanced the relative amount of IRS-1 phosphorylation of Ser-307 (Fig. 5A). Under these conditions, FLAG-CA-JNK significantly lowered the insulin-induced gain in surface GLUT4, attesting to the establishment of insulin resistance (Fig. 5, B and C). Notably, however, FLAG-CA-JNK did not prevent the gain in surface GLUT4 elicited by CA-Rac-GFP (Fig. 5, B and C). This observation suggests that the signaling components responsible for Rac-1 superactivation-driven GLUT4 translocation are not hindered by active JNK. Hence, Rac-1 superactivation is not susceptible to inhibition by insults causing insulin resistance at the level of IRS-1.
FIGURE 5.
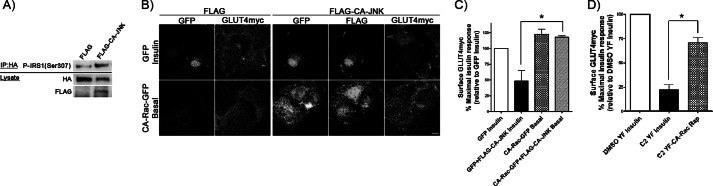
Signaling defects imposed by CA-JNK- and C2-ceramide-induced insulin resistance do not impede the Rac-1-mediated gain in surface GLUT4. A, HA-IRS-1 was co-transfected with FLAG or FLAG-CA-JNK followed by immunoprecipitation (IP) with HA antibody to detect changes in Ser-307 phosphorylation and degradation of IRS-1. B and C, GLUT4myc myoblasts co-expressing FLAG-CA-JNK + GFP or CA-Rac-GFP were stimulated with/without insulin followed by surface GLUT4 staining. Surface GLUT4myc in each condition was quantified and is presented as the percentage of the maximal, parallel insulin response normalized to GFP-expressing, insulin-stimulated cells. D, GLUT4myc myoblasts co-expressing Lyn-FRB + YF or YF-CA-Rac were pretreated with DMSO or 50 μm C2-ceramide for 2 h followed by treatment with/without insulin or rapamycin (Rap) before staining for surface GLUT4. Results in each condition were quantified and are represented as the percentage of the maximal, parallel insulin response relative to DMSO-treated, YF-expressing insulin-stimulated cells (mean ± S.E. (error bars); *, p < 0.05). Representative images of three independent experiments are shown. Bar, 10 μm.
We and others have shown previously that insulin resistance can also arise as a result of intracellular ceramide accumulation (whether exogenously supplied or driven by excess saturated fatty acids), and such insulin resistance involves signaling steps downstream of IRS-1 (7, 47, 48). Delivery of cell-permeating C2-ceramide has been used to recapitulate this model of lipotoxic insulin resistance. C2-ceramide reduces the insulin-induced activation of both Akt and Rac-1 (7, 47, 48). To investigate the susceptibility of the Rac-induced GLUT4 translocation to C2-ceramide, we pretreated myoblasts with C2-ceramide before subjecting them to acute Rac-1 superactivation. Unlike the DMSO-containing YF control whose insulin-mediated GLUT4 translocation significantly dropped to 21% with C2-ceramide treatment, Rac-1-induced GLUT4 gain at the membrane was far less affected as the response was still 71% of the maximal attainable response (Fig. 5D). Presumably, Rac-1 superactivation allowed for sufficient Rac-1 and Akt activity to drive a partial GLUT4 translocation even in the presence of C2-ceramide. We surmise that Rac-1 superactivation may be a principle to be applied in the future to counteract at least in part the defective GLUT4 traffic imposed by excess lipid supply.
DISCUSSION
In response to insulin, Rac-1 acts in concert with the Akt → AS160 signaling cascade to enact the net mobilization of GLUT4 vesicles to the plasma membrane of muscle cells. It is proposed that Rac-mediated actin remodeling results in a dynamic submembrane tether that enriches GLUT4 vesicles and signaling molecules for subsequent fusion (4, 7, 15, 16). Only a fraction of the total cellular Rac-1 complement is activated in response to insulin based on the comparison of Rac-GTP pulled down from cells expressing WT-Rac and CA-Rac (supplemental Fig. 2) or the comparison of the level of Rac-1 activation in insulin-stimulated cells versus GTPγS-treated lysates (8). Although the Akt → AS160 and Rac-1 → actin arms of insulin signaling operate independently of each other (supported by the inability of Rac-1 knockdown and Akti1/2 to inhibit respective Akt phosphorylation (7) and Rac-1 activation (supplemental Fig. 5, A and B)), it has been reported that overexpression of constitutively active Rac-1 can promote GLUT4 translocation (23). However, the underpinning molecular mechanism was not explored, and this question prompted the present study. Surprisingly, we found that Rac-1 superactivation leads to activation of PI3K, Akt, and AS160, and these signals along with the ongoing Rac-mediated actin remodeling mirror the typical response elicited by insulin stimulation resulting in GLUT4 translocation to the cell membrane. AMP-activated protein kinase does not participate in this insulin-independent response because (a) application of compound C did not impair the Rac-1 superactivation-induced GLUT4 translocation (Fig. 3G) and (b) insulin stimulation of CA-Rac-GFP-expressing cells did not cause an additional gain in surface GLUT4, contrasting with the previously reported additive effect of AMP-activated protein kinase and insulin (49, 50). Notably, Rac-1 superactivation restored the GLUT4 response in muscle cells rendered insulin-resistant via JNK activation or ceramide delivery.
Rac-1 Superactivation Leads to PI(3,4,5)P3 Production, Akt, and AS160 Phosphorylation
PI(3,4,5)P3 Accumulation
The finding that PI(3,4,5)P3 production was induced by Rac-1 superactivation (Fig. 3, B and C) was surprising because growth factor-dependent Rac-1 activation is linked to PI(3,4,5)P3-mediated recruitment and activation of Rac GEFs such as Tiam1, P-Rex, and Vav (51–53). Nonetheless, overexpression of CA-Rac and acute Rac-1 superactivation bypassed this prerequisite, and each led to accumulation of a PI(3,4,5)P3-sensitive probe at the plasma membrane of muscle cells (Fig. 3, B and C), fibroblasts, and neutrophils without a need of stimulus (54, 55). On the other hand, expression of CA-Cdc42 did not provoke significant PI(3,4,5)P3 production (54), and this small GTPase was also unable to increase surface GLUT4 (supplemental Fig. 1). Hence, a potential feedback mechanism to elevate PI(3,4,5)P3 appears to be Rac-specific, but the molecular basis for this step remains elusive. A potential explanation is an interaction between Rac-1 and PI3K. A pulldown of cytosolic proteins bound to GST-Rac-1 loaded with GTP was found to contain PI3K activity, and it was subsequently revealed that the p85 subunit of PI3K can interact with Rac-1 in a GTP-dependent manner in vitro (56). Because active Rac-1 is naturally anchored at the plasma membrane through prenylation, such interaction between Rac-1 and PI3K would bring PI3K close to the vicinity of its substrate, phosphatidylinositol 4,5-bisphosphate, at the plasma membrane for Rac-mediated PI(3,4,5)P3 production. However, in vivo evidence of this interaction during growth factor stimulation is lacking.
Another contributing factor for the increased PI(3,4,5)P3 generation by Rac-1 superactivation could be the accumulation of active PI3K units in the Rac-induced branched cortical actin (4). Biochemical evidence supports a PI3K-actin filament interaction as PI3K was enriched in the detergent-insoluble fraction containing polymerized F-actin in chemotaxic Dictyostelium discoideum cells, and its localization to membrane was lost after treatment with latrunculin A (57). More importantly, the catalytic subunit p110 of PI3K accumulated at the site of remodeled cortical actin following insulin stimulation in muscle cells (4). These observations support a role for cortical actin in positioning PI3K close to the membrane. Indeed, when Rac superactivation-mediated actin remodeling was perturbed by actin-disrupting agents, the consequent PI(3,4,5)P3 production at the plasma membrane was blocked (54, 58). This may explain why LB treatment was most effective at blocking the GLUT4 translocation induced by Rac-1 superactivation (Fig. 3G) because disrupting the cortical actin lattice would prevent Akt activation secondarily to impaired PI(3,4,5)P3 production along with the elimination of Rac-mediated actin remodeling.
Akt Phosphorylation
Akt must be phosphorylated at both its activation site (Thr-308) and its regulatory site (Ser-473) to become fully active (35, 36). Thr-308 phosphorylation provides partial activation, whereas Ser-473 phosphorylation on its own allows minimal activity but contributes to potentiation of Akt activity and substrate specificity (36, 59–61). According to these findings, the phosphorylation of Akt at both Thr-308 and Ser-473 must have activated the kinase as reflected by the phosphorylation of its downstream target AS160 (Figs. 3D and 4A). On the other hand, although overexpression of WT-Rac-GFP in myoblasts contributed to slight elevation in Ser-473 phosphorylation (Fig. 3F), no Thr-308 phosphorylation was achieved, and there was no ensuing AS160 phosphorylation (Figs. 3E and 4, A and B).
Phosphorylation of Akt at Thr-308 is controlled by PDK1 (35). The rise in PI(3,4,5)P3 at the plasma membrane serves as a cue to recruit and enhance PDK1 activity. Because Akt is also enriched at the plasma membrane via its PI(3,4,5)P3-binding PH domain (62), Akt and PDK1 may come in close proximity, favoring Akt phosphorylation. As seen in Fig. 3, B and C, the amount of membrane-bound PI(3,4,5)P3 in CA-Rac-GFP-expressing cells is equivalent to about 20% of the maximal insulin-dependent response. This modest gain correlates well with the 26% increase in phospho-Thr-308 Akt achieved by Rac-1 superactivation relative to the maximal insulin response, suggesting linear transmission of the PI(3,4,5)P3 signal amplitude to PDK1 → Akt (Fig. 3E). Consistent with this calculation, Thr-308 phosphorylation of Akt is considered the limiting determinant for Akt activation (59, 60, 63, 64).
On the other hand, mTORC2 is the kinase responsible for Ser-473 phosphorylation of Akt in response to insulin (65). mTORC2 exists in a complex containing mTOR, mLTST8, and Rictor; however, the mechanism of mTORC2 activation is unknown (66). Recently, it was revealed that Rac interacts with mTOR in the mTORC2 complex in a GTP-independent manner (67). This interaction provides spatial regulation to mTORC2 as enrichment of active Rac at the plasma membrane would bring along mTORC2. Accordingly, Rac-1 activation by CA-Rac-GTP may position mTORC2 near the plasma membrane where Akt is recruited by PI(3,4,5)P3. The close proximity of mTORC2 and Akt might contribute to the increase in phospho-Ser-473 Akt observed with Rac-1 superactivation without the need of insulin stimulation (Fig. 3, D and F).
In addition to PDK1 and mTORC2, the kinase PAK1 can phosphorylate Ser-473 of Akt in vitro (68). Interestingly, PAK1 inhibition reduces IGF-1-stimulated Akt activation and the Akt hyperactivity of cancer cells (68–70). PAK1 can also act as a scaffold for both PDK1 and Akt (71). Because PAK1 is recruited through its Cdc42/Rac interactive binding domain to GTP-loaded Rac-1, an association of PAK1, PDK1, and Akt at the plasma membrane would provide an additional avenue, independent of PI(3,4,5)P3 production, driving Akt phosphorylation following Rac-1 activation.
Because WM and Akti1/2 prevent the production of plasma membrane PI(3,4,5)P3 and sterically bind to the PH domain of Akt, respectively (72), both agents impair Akt activation by hindering the recruitment of Akt to the plasma membrane. However, the above mentioned PAK1-Akt-PDK1 complex and direct Rac-mTORC2 interaction could alleviate the complete reliance on the traditional membrane PI(3,4,5)P3 and PH domain of Akt to trigger Akt activation. This could explain why only a partial inhibitory effect was observed with WM and Akti1/2 on the Rac-1 superactivation-induced GLUT4 translocation (Fig. 3G). Alternatively, the extensive actin remodeling generated during Rac-1 superactivation could be the main driver of this insulin-like translocation response. Because remodeled actin can act as scaffold to position insulin signaling molecules such as Akt close to the plasma membrane (4), the inhibitory effect of WM and Akti1/2 on Akt recruitment would be reduced compared with the normal insulin stimulation. This scenario is consistent with the near complete reduction of rapamycin-triggered GLUT4 translocation with LB (Fig. 3G).
AS160 Phosphorylation
Despite the low level of Rac-1 superactivation-mediated Akt activation relative to the maximal insulin response (26%), downstream phosphorylation of AS160 amounted to ∼70% of the maximal insulin response (Fig. 4B). This signal amplification from Akt to AS160 has also been recently appreciated for the insulin signaling cascade. Insulin dose dependence and Akt inhibitor studies in adipocytes and muscle cells suggest that very little Akt activity is required for near maximal AS160 phosphorylation (40, 41). From those studies, 30% insulin-stimulated phospho-Thr-308 Akt corresponds to ∼80% of maximal phospho-AS160, approximating our measurements of phospho-Thr-308 and phospho-AS160 in response to CA-Rac-GFP (Fig. 4B). Moreover, we have shown that 19 ± 10% Akt activity suffices for maximal GLUT4 translocation in insulin-stimulated muscle cells (6, 42), and similarly, a WM-mediated reduction in phosphor-Thr-308 Akt to 20% of the maximal value allowed for greater than 50% of the maximal GLUT4 translocation (5). These observations reinforce the concept that Akt phosphorylation/activity does not correlate linearly with insulin-mediated GLUT4 translocation, but rather the phosphorylation status of AS160 closely matches GLUT4 translocation. These calculations are in good agreement with the corresponding responses of Akt, AS160, and GLUT4 to Rac-1 superactivation observed in the present study.
Rac-1 Superactivation Overcomes Insulin Resistance
The level of Rac-1-GTP achieved in response to either chronic expression of CA-Rac-GFP or acute superactivation via rapamycin is likely to be much higher than the insulin-mediated activation of endogenous Rac-1 (supplemental Fig. 2). siRNA-mediated silencing of endogenous Rac-1 does not affect insulin-mediated Akt activation, suggesting that Rac-1 activation does not influence Akt during normal insulin stimulation (7). This may be due to the low level of Rac-1 activation and the high level of Akt phosphorylation evoked by the hormone. Much higher levels of Rac-1 activity in the absence of hormonal stimulation, such as those imposed by the chronic and acute strategies described here, are required to visualize the Rac-mediated Akt activation. Nonetheless, Rac-1 superactivation suffices to elicit enough Akt activation to trigger an insulin-like GLUT4 translocation in muscle cells. This feature was explored in this study to counteract the effect of strategies that cause insulin resistance.
Obesity is a leading factor in the development of insulin resistance because of the lipotoxic and low grade inflammatory environment created by excessive accumulation of saturated fatty acids (43, 73). Both saturated fatty acids and inflammatory cytokines impact skeletal muscle to activate stress kinases, including JNK, leading to IRS-1 serine phosphorylation and degradation (45, 74). When this JNK-induced blockage of IRS-1 signaling was reproduced via overexpression of active JNK, insulin-dependent GLUT4 translocation was significantly diminished (Fig. 5, B and C). However, active JNK did not preclude the GLUT4 translocation evoked by Rac-1 superactivation (Fig. 5, B and C) ostensibly because the latter response bypasses the requirement of IRS-1. Therefore, in vivo, when defects at the level of IRS-1 arise, Rac-1 activation may be a gateway to trigger downstream signaling of Akt and AS160 to drive a full GLUT4 translocation response.
Unlike JNK activation, intracellular ceramides produced from saturated fatty acid excess can directly inhibit insulin signaling downstream of IRS-1 (47, 48, 75, 76). Ceramides can inactivate Akt without influencing IRS-1 or PI3K by both preventing recruitment of Akt to the plasma membrane and increasing PP2A activity that dephosphorylates Akt (77, 78). In muscle cells, C2-ceramide not only reduces Akt but also insulin-dependent Rac-1 activation, whereas IRS-1 tyrosine phosphorylation remains intact (7). Interestingly, C2-ceramide only exerted a 29% reduction in superactivated Rac-1-driven GLUT4 translocation compared with a 78% decrease in GLUT4 insulin response (Fig. 5D). This suggests that Rac-1 superactivation provides enough surge in both Rac-1 and Akt activities to partially overcome the barrier imposed by ceramide and hence averts the negative effects of the lipid metabolite.
Although reduced insulin-dependent glucose uptake is a key factor in the development of whole-body insulin resistance leading to type 2 diabetes, none of the currently available treatments of the disease (metformin, sulfonylureas, and dipeptidyl peptidase inhibitors) act directly on muscle, targeting instead hepatic glucose production and insulin secretion. Here we identify that Rac-1 activation alone suffices to trigger an insulin-like GLUT4 translocation without insulin stimulation (Fig. 6). Unlike insulin, Rac-1 superactivation bypasses IRS-1 but triggers signaling through PI3K, Akt, and AS160. This response is resistant to the deleterious effects of JNK activation or ceramide accumulation that cause insulin resistance. IRS-1 is a common target of agents and conditions causing insulin resistance (43, 44); hence, improving insulin action downstream of IRS-1 should be considered strategically advantageous to improve metabolic outcomes compared with approaches that require IRS-1 participation. Compounds that would turn on a Rac-specific GEF or inhibit Rac-specific GTPase-activating protein activity in a time- and tissue-selective manner would be particularly useful tools to design therapies for insulin resistance.
FIGURE 6.

Model of Rac-1 superactivation-induced GLUT4 translocation in muscle cells. Left, normal insulin response observed in cells expressing endogenous levels of Rac-1 (see Refs. 42 and 79). Right, Rac-1 superactivation promotes membrane accumulation of PI(3,4,5)P3, which results in moderate phosphorylation of Akt and significant phosphorylation of AS160. These signaling components together with actin remodeling satisfied the molecular requirements for GLUT4 translocation without input of insulin.
Acknowledgments
We thank Drs. M. Teruel, T. Meyer, M. R. Philips, G. Hotamisligil, M. Rozakis, J. Brumell, and S. Grinstein for kindly providing various constructs used in this study. We thank Dr. Philip J. Bilan for useful input throughout the course of this study and Zhi Liu for helpful technical assistance.
This work was supported in part by Canadian Institutes of Health Research (CIHR) Grant 7307 (to A. K.).

This article contains supplemental Figs. 1–5.
- GLUT4
- glucose transporter 4
- IRS-1
- isoform 1 of the insulin receptor substrate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- GEF
- guanine exchange factor
- P-
- phospho-
- Akti1/2
- Akt inhibitor 1/2
- LB
- latrunculin B
- YF
- YFP-tagged FKBP
- FKBP
- FK506-binding protein
- FRB
- FKBP12-rapamycin binding
- CA
- constitutively active
- DH
- Dbl homology
- PH
- pleckstrin homology
- RFP
- red fluorescent protein
- WM
- wortmannin
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Baron A. D., Brechtel G., Wallace P., Edelman S. V. (1988) Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. Endocrinol. Metab. 255, E769–E774 [DOI] [PubMed] [Google Scholar]
- 2. Foley K., Boguslavsky S., Klip A. (2011) Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50, 3048–3061 [DOI] [PubMed] [Google Scholar]
- 3. Huang C., Thirone A. C., Huang X., Klip A. (2005) Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J. Biol. Chem. 280, 19426–19435 [DOI] [PubMed] [Google Scholar]
- 4. Patel N., Rudich A., Khayat Z. A., Garg R., Klip A. (2003) Intracellular segregation of phosphatidylinositol-3,4,5-trisphosphate by insulin-dependent actin remodeling in L6 skeletal muscle cells. Mol. Cell. Biol. 23, 4611–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Somwar R., Niu W., Kim D. Y., Sweeney G., Randhawa V. K., Huang C., Ramlal T., Klip A. (2001) Differential effects of phosphatidylinositol 3-kinase inhibition on intracellular signals regulating GLUT4 translocation and glucose transport. J. Biol. Chem. 276, 46079–46087 [DOI] [PubMed] [Google Scholar]
- 6. Wang Q., Somwar R., Bilan P. J., Liu Z., Jin J., Woodgett J. R., Klip A. (1999) Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol. Cell. Biol. 19, 4008–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. JeBailey L., Wanono O., Niu W., Roessler J., Rudich A., Klip A. (2007) Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 56, 394–403 [DOI] [PubMed] [Google Scholar]
- 8. JeBailey L., Rudich A., Huang X., Di Ciano-Oliveira C., Kapus A., Klip A. (2004) Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol. Endocrinol. 18, 359–372 [DOI] [PubMed] [Google Scholar]
- 9. Thong F. S., Bilan P. J., Klip A. (2007) The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 56, 414–423 [DOI] [PubMed] [Google Scholar]
- 10. Bruss M. D., Arias E. B., Lienhard G. E., Cartee G. D. (2005) Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 54, 41–50 [DOI] [PubMed] [Google Scholar]
- 11. Sano H., Kane S., Sano E., Mîinea C. P., Asara J. M., Lane W. S., Garner C. W., Lienhard G. E. (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278, 14599–14602 [DOI] [PubMed] [Google Scholar]
- 12. Sun Y., Bilan P. J., Liu Z., Klip A. (2010) Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 107, 19909–19914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ishikura S., Klip A. (2008) Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am. J. Physiol. Cell Physiol. 295, C1016–C1025 [DOI] [PubMed] [Google Scholar]
- 14. Chiu T. T., Patel N., Shaw A. E., Bamburg J. R., Klip A. (2010) Arp2/3- and cofilin-coordinated actin dynamics is required for insulin-mediated GLUT4 translocation to the surface of muscle cells. Mol. Biol. Cell 21, 3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Randhawa V. K., Ishikura S., Talior-Volodarsky I., Cheng A. W., Patel N., Hartwig J. H., Klip A. (2008) GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J. Biol. Chem. 283, 27208–27219 [DOI] [PubMed] [Google Scholar]
- 16. Talior-Volodarsky I., Randhawa V. K., Zaid H., Klip A. (2008) α-Actinin-4 is selectively required for insulin-induced GLUT4 translocation. J. Biol. Chem. 283, 25115–25123 [DOI] [PubMed] [Google Scholar]
- 17. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 18. Tcherkezian J., Lamarche-Vane N. (2007) Current knowledge of the large RhoGAP family of proteins. Biol. Cell 99, 67–86 [DOI] [PubMed] [Google Scholar]
- 19. Cho H., Mu J., Kim J. K., Thorvaldsen J. L., Chu Q., Crenshaw E. B., 3rd, Kaestner K. H., Bartolomei M. S., Shulman G. I., Birnbaum M. J. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292, 1728–1731 [DOI] [PubMed] [Google Scholar]
- 20. Ueda S., Kitazawa S., Ishida K., Nishikawa Y., Matsui M., Matsumoto H., Aoki T., Nozaki S., Takeda T., Tamori Y., Aiba A., Kahn C. R., Kataoka T., Satoh T. (2010) Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 24, 2254–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sylow L., Jensen T. E., Kleinert M., Højlund K., Kiens B., Wojtaszewski J., Prats C., Schjerling P., Richter E. A. (2013) Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin resistant murine and human skeletal muscle. Diabetes, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khayat Z. A., Tong P., Yaworsky K., Bloch R. J., Klip A. (2000) Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113, 279–290 [DOI] [PubMed] [Google Scholar]
- 23. Ueda S., Kataoka T., Satoh T. (2008) Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 100, 645–657 [DOI] [PubMed] [Google Scholar]
- 24. Inoue T., Heo W. D., Grimley J. S., Wandless T. J., Meyer T. (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henstridge D. C., Bruce C. R., Pang C. P., Lancaster G. I., Allen T. L., Estevez E., Gardner T., Weir J. M., Meikle P. J., Lam K. S., Xu A., Fujii N., Goodyear L. J., Febbraio M. A. (2012) Skeletal muscle-specific overproduction of constitutively activated c-Jun N-terminal kinase (JNK) induces insulin resistance in mice. Diabetologia 55, 2769–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ueyama A., Yaworsky K. L., Wang Q., Ebina Y., Klip A. (1999) GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 277, E572–E578 [DOI] [PubMed] [Google Scholar]
- 27. Török D., Patel N., Jebailey L., Thong F. S., Randhawa V. K., Klip A., Rudich A. (2004) Insulin but not PDGF relies on actin remodeling and on VAMP2 for GLUT4 translocation in myoblasts. J. Cell Sci. 117, 5447–5455 [DOI] [PubMed] [Google Scholar]
- 28. Michiels F., Habets G. G., Stam J. C., van der Kammen R. A., Collard J. G. (1995) A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature 375, 338–340 [DOI] [PubMed] [Google Scholar]
- 29. Michiels F., Stam J. C., Hordijk P. L., van der Kammen R. A., Ruuls-Van Stalle L., Feltkamp C. A., Collard J. G. (1997) Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and c-Jun NH2-terminal kinase activation. J. Cell Biol. 137, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishikura S., Koshkina A., Klip A. (2008) Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic. Acta Physiol. 192, 61–74 [DOI] [PubMed] [Google Scholar]
- 31. Bohdanowicz M., Fairn G. D. (2011) Rapamycin-based inducible translocation systems for studying phagocytosis. Methods Mol. Biol. 748, 183–193 [DOI] [PubMed] [Google Scholar]
- 32. Seabra M. C. (1998) Membrane association and targeting of prenylated Ras-like GTPases. Cell. Signal. 10, 167–172 [DOI] [PubMed] [Google Scholar]
- 33. Kinsella B. T., Erdman R. A., Maltese W. A. (1991) Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J. Biol. Chem. 266, 9786–9794 [PubMed] [Google Scholar]
- 34. Gray A., Van Der Kaay J., Downes C. P. (1999) The pleckstrin homology domains of protein kinase B and GRP1 (general receptor for phosphoinositides-1) are sensitive and selective probes for the cellular detection of phosphatidylinositol 3,4-bisphosphate and/or phosphatidylinositol 3,4,5-trisphosphate in vivo. Biochem. J. 344, 929–936 [PMC free article] [PubMed] [Google Scholar]
- 35. Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 36. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 37. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 38. Niu W., Bilan P. J., Ishikura S., Schertzer J. D., Contreras-Ferrat A., Fu Z., Liu J., Boguslavsky S., Foley K. P., Liu Z., Li J., Chu G., Panakkezhum T., Lopaschuk G. D., Lavandero S., Yao Z., Klip A. (2010) Contraction-related stimuli regulate GLUT4 traffic in C2C12-GLUT4myc skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 298, E1058-E1071 [DOI] [PubMed] [Google Scholar]
- 39. Tong P., Khayat Z. A., Huang C., Patel N., Ueyama A., Klip A. (2001) Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J. Clin. Investig. 108, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan S. X., Ng Y., Meoli C. C., Kumar A., Khoo P. S., Fazakerley D. J., Junutula J. R., Vali S., James D. E., Stöckli J. (2012) Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J. Biol. Chem. 287, 6128–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ng Y., Ramm G., Burchfield J. G., Coster A. C., Stöckli J., James D. E. (2010) Cluster analysis of insulin action in adipocytes reveals a key role for Akt at the plasma membrane. J. Biol. Chem. 285, 2245–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bilan P. J., Samokhvalov V., Koshkina A., Schertzer J. D., Samaan M. C., Klip A. (2009) Direct and macrophage-mediated actions of fatty acids causing insulin resistance in muscle cells. Arch. Physiol. Biochem. 115, 176–190 [DOI] [PubMed] [Google Scholar]
- 43. Savage D. B., Petersen K. F., Shulman G. I. (2007) Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 87, 507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garvey W. T., Huecksteadt T. P., Matthaei S., Olefsky J. M. (1988) Role of glucose transporters in the cellular insulin resistance of type II non-insulin-dependent diabetes mellitus. J. Clin. Investig. 81, 1528–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 46. Tanti J. F., Jager J. (2009) Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 9, 753–762 [DOI] [PubMed] [Google Scholar]
- 47. Hajduch E., Balendran A., Batty I. H., Litherland G. J., Blair A. S., Downes C. P., Hundal H. S. (2001) Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia 44, 173–183 [DOI] [PubMed] [Google Scholar]
- 48. Summers S. A., Garza L. A., Zhou H., Birnbaum M. J. (1998) Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 18, 5457–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fazakerley D. J., Holman G. D., Marley A., James D. E., Stöckli J., Coster A. C. (2010) Kinetic evidence for unique regulation of GLUT4 trafficking by insulin and AMP-activated protein kinase activators in L6 myotubes. J. Biol. Chem. 285, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Habegger K. M., Hoffman N. J., Ridenour C. M., Brozinick J. T., Elmendorf J. S. (2012) AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 153, 2130–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Das B., Shu X., Day G. J., Han J., Krishna U. M., Falck J. R., Broek D. (2000) Control of intramolecular interactions between the pleckstrin homology and Dbl homology domains of Vav and Sos1 regulates Rac binding. J. Biol. Chem. 275, 15074–15081 [DOI] [PubMed] [Google Scholar]
- 52. Crompton A. M., Foley L. H., Wood A., Roscoe W., Stokoe D., McCormick F., Symons M., Bollag G. (2000) Regulation of Tiam1 nucleotide exchange activity by pleckstrin domain binding ligands. J. Biol. Chem. 275, 25751–25759 [DOI] [PubMed] [Google Scholar]
- 53. Welch H. C., Coadwell W. J., Ellson C. D., Ferguson G. J., Andrews S. R., Erdjument-Bromage H., Tempst P., Hawkins P. T., Stephens L. R. (2002) P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell 108, 809–821 [DOI] [PubMed] [Google Scholar]
- 54. Srinivasan S., Wang F., Glavas S., Ott A., Hofmann F., Aktories K., Kalman D., Bourne H. R. (2003) Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang H. W., Shin M. G., Lee S., Kim J. R., Park W. S., Cho K. H., Meyer T., Do Heo W. (2012) Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 47, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bokoch G. M., Vlahos C. J., Wang Y., Knaus U. G., Traynor-Kaplan A. E. (1996) Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem. J. 315, 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sasaki A. T., Chun C., Takeda K., Firtel R. A. (2004) Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J. Cell Biol. 167, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Servant G., Weiner O. D., Herzmark P., Balla T., Sedat J. W., Bourne H. R. (2000) Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science 287, 1037–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McManus E. J., Collins B. J., Ashby P. R., Prescott A. R., Murray-Tait V., Armit L. J., Arthur J. S., Alessi D. R. (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23, 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Williams M. R., Arthur J. S., Balendran A., van der Kaay J., Poli V., Cohen P., Alessi D. R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10, 439–448 [DOI] [PubMed] [Google Scholar]
- 61. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 62. Andjelković M., Alessi D. R., Meier R., Fernandez A., Lamb N. J., Frech M., Cron P., Cohen P., Lucocq J. M., Hemmings B. A. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272, 31515–31524 [DOI] [PubMed] [Google Scholar]
- 63. Vincent E. E., Elder D. J., Thomas E. C., Phillips L., Morgan C., Pawade J., Sohail M., May M. T., Hetzel M. R., Tavaré J. M. (2011) Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J. Cancer 104, 1755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gallay N., Dos Santos C., Cuzin L., Bousquet M., Simmonet Gouy V., Chaussade C., Attal M., Payrastre B., Demur C., Récher C. (2009) The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23, 1029–1038 [DOI] [PubMed] [Google Scholar]
- 65. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 66. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 67. Saci A., Cantley L. C., Carpenter C. L. (2011) Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 42, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mao K., Kobayashi S., Jaffer Z. M., Huang Y., Volden P., Chernoff J., Liang Q. (2008) Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J. Mol Cell. Cardiol. 44, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huynh N., Liu K. H., Baldwin G. S., He H. (2010) P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim. Biophys. Acta 1803, 1106–1113 [DOI] [PubMed] [Google Scholar]
- 70. Arias-Romero L. E., Villamar-Cruz O., Pacheco A., Kosoff R., Huang M., Muthuswamy S. K., Chernoff J. (2010) A Rac-Pak signaling pathway is essential for ErbB2-mediated transformation of human breast epithelial cancer cells. Oncogene 29, 5839–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Higuchi M., Onishi K., Kikuchi C., Gotoh Y. (2008) Scaffolding function of PAK in the PDK1-Akt pathway. Nat. Cell Biol. 10, 1356–1364 [DOI] [PubMed] [Google Scholar]
- 72. Calleja V., Laguerre M., Parker P. J., Larijani B. (2009) Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 7, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gregor M. F., Hotamisligil G. S. (2011) Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 [DOI] [PubMed] [Google Scholar]
- 74. Kewalramani G., Fink L. N., Asadi F., Klip A. (2011) Palmitate-activated macrophages confer insulin resistance to muscle cells by a mechanism involving protein kinase C θ and ϵ. PLoS One 6, e26947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chavez J. A., Knotts T. A., Wang L. P., Li G., Dobrowsky R. T., Florant G. L., Summers S. A. (2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 278, 10297–10303 [DOI] [PubMed] [Google Scholar]
- 76. Schmitz-Peiffer C., Craig D. L., Biden T. J. (1999) Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 274, 24202–24210 [DOI] [PubMed] [Google Scholar]
- 77. Stratford S., DeWald D. B., Summers S. A. (2001) Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem. J. 354, 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Teruel T., Hernandez R., Lorenzo M. (2001) Ceramide mediates insulin resistance by tumor necrosis factor-α in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 50, 2563–2571 [DOI] [PubMed] [Google Scholar]
- 79. Zaid H., Antonescu C. N., Randhawa V. K., Klip A. (2008) Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem. J. 413, 201–215 [DOI] [PubMed] [Google Scholar]