

Background: In the retina, insulin-like growth factor I (IGF-I) is neuroprotective and essential for vasculature homeostasis.
Results: Transgenic mice overexpressing Igf-I in the retina present chronic gliosis and retinal stress leading to neurodegeneration.
Conclusion: IGF-I induces deleterious processes in the retina that, chronically, may overcome its neurotrophic properties.
Significance: Increased intraocular IGF-I may contribute to the pathogenesis of conditions such as ischemic or diabetic retinopathies.
Keywords: Animal Models, Glia, Insulin-like Growth Factor (IGF), Neurodegeneration, Retina
Abstract
Insulin-like growth factor I (IGF-I) exerts multiple effects on different retinal cell types in both physiological and pathological conditions. Despite the growth factor's extensively described neuroprotective actions, transgenic mice with increased intraocular levels of IGF-I showed progressive impairment of electroretinographic amplitudes up to complete loss of response, with loss of photoreceptors and bipolar, ganglion, and amacrine neurons. Neurodegeneration was preceded by the overexpression of genes related to retinal stress, acute-phase response, and gliosis, suggesting that IGF-I altered normal retinal homeostasis. Indeed, gliosis and microgliosis were present from an early age in transgenic mice, before other alterations occurred, and were accompanied by signs of oxidative stress and impaired glutamate recycling. Older mice also showed overproduction of pro-inflammatory cytokines. Our results suggest that, when chronically increased, intraocular IGF-I is responsible for the induction of deleterious cellular processes that can lead to neurodegeneration, and they highlight the importance that this growth factor may have in the pathogenesis of conditions such as ischemic or diabetic retinopathy.
Introduction
IGF-I4 is a growth factor with mitogenic, differentiating, anti-apoptotic, and metabolic functions (1) that also plays a key role in pathological retinal neovascularization (2). IGF-I is increased in the vitreous of patients with proliferative ocular diseases, such as diabetic retinopathy (3), and pharmacological inhibition or genetic deletion of the Igf-I receptor in the retinal vasculature of rodents prevent neovascularization under hypoxic conditions (4, 5). Similarly, in humans, mutations in genes coding for the growth hormone receptor, Igf-I, or Igf-I receptor are associated with reduced retinal neovascularization (6). IGF-I is a potent inductor of the expression of the pro-angiogenic factor VEGF, through hypoxia-inducible factor 1α stabilization (7). In addition to the effects on VEGF transcription, IGF-I may act cooperatively with VEGF to stabilize newly formed vessels (4).
Besides its actions on retinal vasculature, IGF-I is a pro-survival molecule that can act on neurons (8). Humans carriers of homozygous mutations associated with reduced expression of the Igf-I gene present mental retardation and deafness, associated with severe prenatal growth retardation, postnatal growth failure, and microcephaly, confirming the role of IGF-I as a neurotrophic factor (9, 10). Because of these properties, IGF-I has been used to counteract neurodegeneration in experimental animal models of brain injury (11) and retinal neurodegeneration (12). Moreover, IGF-I-deficient mice develop slow retinal neurodysfunction with loss of ERG responses (13). This evidence demonstrates that IGF-I plays an important role in neuronal development and survival.
Transgenic mice overexpressing Igf-I in photoreceptors (TgIGF-I) have increased intraocular VEGF levels and develop many of the retinal vascular alterations characteristic of human diabetic eye disease, despite not being hyperglycemic (14, 15). Nonproliferative vascular alterations are found in young animals (2–3 months), and the phenotype progresses to overt pre-retinal neovascularization in adult mice (7–8 months) (14). VEGF, which plays a pivotal role in the development of proliferative retinopathies (16), is expressed by glial cells in transgenic retinas, the cell type that also overproduces this factor in diabetic patients (17). Gliosis is observed in Igf-I -overexpressing retinas at nonproliferative stages, before neovascularization occurs, suggesting that Igf-I overexpression is likely causing other alterations on transgenic retinas that contribute to the phenotype.
Given the apparently antagonistic effects of IGF-I on the retina, with deleterious effects on retinal vasculature (6) but positive effects on neuronal survival (12), we evaluated the consequences of the chronic elevation of intraocular IGF-I on retinal functionality in TgIGF-I mice. We observed a progressive decline in ERG amplitudes in transgenic animals, leading to complete loss of response in old animals. This study demonstrates that the chronic overproduction of IGF-I in the retina induces a series of deleterious cellular processes, independent of neovascularization, that eventually surpass IGF-I pro-survival properties leading to neurodegeneration.
EXPERIMENTAL PROCEDURES
Animals
CD1 heterozygous mice overexpressing Igf-I in the retina were used (14). Animal care and experimental procedures were approved by the Ethics Committee in Animal and Human Experimentation of Universitat Autònoma de Barcelona.
Electroretinography
ERG recordings were performed in deeply anesthetized and dark-adapted mice. Recordings were taken between a mouse electrode fixed on a corneal lens over the mouse eye (Burian-Allen electrode, Hansen Ophthalmic Development Lab), a reference electrode located in the mouth, and a ground electrode located in the tail. After pupil dilatation, flash-induced ERG responses were recorded from the right eye in response to light stimuli produced with a Ganzfeld stimulator. Light stimuli intensities ranged from −4 to 2 log cd·s·m−2 and, for each intensity, 4–64 consecutive light presentations were averaged. Each range of flashes allowed recording of particular responses as follows: from −4 to −1.52 for rod-mediated responses; from −1.52 to 0.48 for mixed rod and cone responses; 0.48 and at a recording frequency range of 100–1000 Hz for oscillatory potentials; from −0.52 to 2 log cd·s·m−2 on a rod-saturating background of 30 cd/m2 for cone-mediated responses; and at 1.48 log cd·s·m−2 on a rod-saturating background for flicker responses (20 Hz). The ERG signals were amplified, band filtered between 0.3 and 1000 Hz (Grass CP511 AC amplifier), and digitized at 10 kHz with a Power Lab data acquisition board (ADI Instruments). ERG measurements were taken by an observer blinded to the experimental conditions of the animals.
Histological Analysis
Formalin-fixed, paraffin-embedded eye sections were incubated with tomato lectin Lycopersicon esculentum (Sigma), anti-rhodopsin (Abcam, Cambridge, UK), anti-calretinin (Invitrogen), anti-PKCα (Sigma), anti-Brn3a (Santa Cruz Biotechnology, Santa Cruz, CA), GFAP (Dako Cytomation, Glostrup, Denmark), and anti-cleaved caspase3 (Cell Signaling, Danvers, MA). Nuclei were counterstained with DAPI (Sigma) Images were obtained with a laser-scanning confocal microscope (TCs SP2; Leica Microsystems GmbH, Wetzlar, Germany) or a Nikon Eclipse 90i microscope (Nikon Instruments Inc., Tokyo, Japan).
Morphometric Analysis
Retinal layers (INL and ONL) length was measured with NIS Elements software (Nikon) in six central and six peripheral images (×20) from retinal sections showing the optic nerve. Three measurements were performed in each image for each layer, ensuring perpendicular distances between layer limits. The length of the posterior segment of rod photoreceptors was determined by the same method in central sections. For quantification of specific neuronal populations, total positive cells were counted per 20 images of central retinal sections with optic nerve.
Microarray Samples and Analysis
The protocol for RNA extraction recommended by Affymetrix was followed (Expression Analysis Technical Manual). Microarray analysis was performed by Progenika (Bilbao, Spain). The final gene list contained only those probe sets with a p < 0.05. For the interpretation, cross-checking, and visualization of the data, FatiGO Term Enrichment (release date Nov. 20, 2010), Database for Annotation, Visualization, and Integrated Discovery (DAVID, david.abcc.ncifcrf.gov), and Kyoto Encyclopedia of Genes and Genomes (KEGG database), and GeneCodis were used. Array data have been submitted to GEO database (accession number GSE46246).
RNA Extraction and Quantitative Real Time PCR
Retinas were homogenized in 200 μl of Tripure (Roche Applied Science), and retinal RNA was purified using RNeasy mini kit (Qiagen Sciences, Germantown, MD). cDNA was synthesized with transcriptor first strand cDNA synthesis kit (Roche Applied Science). Quantitative real time PCR was performed using LightCycler® 480 SYBR Green I Master (Roche Applied Science) with specific primers.
Protein Analysis
For Western blot, retinas were homogenized in lysis buffer and 50–100 μg of retinal extract were separated by 12% SDS-PAGE. Immunoblot was performed with anti-acetyl-NF-κB (Cell Signaling), anti-ERK1/2 (Cell Signaling), anti-phospho-ERK1/2 (Cell Signaling), anti-GFAP (DAKO Cytomation), anti-MCP-1 (Abcam), or anti-tubulin (Abcam). Detection was performed using Immobilon Western reagent (Millipore). The pixel intensity of the bands obtained was determined with GeneSnap software for Gene Genius Bio Imaging System (Syngene, Synoptics Ltd.), and the ratio protein content/tubulin content was calculated for each sample to allow loading-independent comparison.
Retinal Explants
Wild-type C57 mice were sacrificed, and eyes were enucleated. Retinas were dissected and incubated in DMEM (reference E15-810, PAA) complemented with 10% FBS (PAA, ref. A15–151) and antibiotic/antimycotic (PAA, ref. P11–002) at 37 °C and 5% CO2. Medium was refreshed 24 h later. After 48 h, recombinant IGF-I (Sigma) was added to DMEM without FBS. Between 2 and 5 h prior to IGF-I addition, 5 μl of 200 μm wortmannin (Sigma) or DMSO (Sigma) were added to the culture media. After 48 h of stimulation, retinas were collected and frozen at −80 °C for total RNA isolation.
Glutamine Synthetase Activity and Oxidized Glutathione
Protein extracts were obtained as for Western blot analysis. 40 μl of a 1:10 dilution of the extracts was added to the reaction mixture containing 50 mm sodium glutamate (Sigma), 20 mm MgCl2, 15 mm ATP (Sigma), 100 mm imidazole, pH 7.2, 1 μl of β-mercaptoethanol, 10 mm creatine phosphate (Sigma), 1 μl of creatine phosphokinase (Sigma). After 1–2 min of preincubation of the mixture at 37 °C, the reaction was started by adding 8 μl of 125 mm hydroxylamine. Ten minutes later, the reaction was stopped with 120 μl of ferric chloride solution (0.37 m FeCl3, 0.67 m HCl, 0.20 m trichloroacetic acid) and placed on ice for 5 min. Afterward, the supernatant was collected from the wells, and absorbance was measured at 525 nm. Values were corrected by protein content of each sample and represented as percentage of wild-type values. Oxidized glutathione was measured in retinal extracts using the glutathione (total) detection kit from Enzo Life Sciences (catalog no. ADI-900-160) following the manufacturer's instructions.
Statistical Analysis
Values are expressed as the mean ± S.E. Differences between groups were compared by unpaired Student's t test. Differences were considered statistically significant at p values less than 0.05.
RESULTS
Neuronal Dysfunction in Transgenic Retinas Overexpressing IGF-I
Retinal neurophysiology was assessed in wild-type (WT) and TgIGF-I mice through ERG studies. Both rod and cone-driven responses were evaluated by performing measurements in scotopic and photopic conditions, respectively. At 3 months of age, transgenic mice did not show any difference in comparison with WT littermates in the amplitudes of the responses obtained with different light intensities (Fig. 1, A–F). At 6 months of age, however, responses were heterogeneous among different animals (Fig. 1, A–F). The abnormal responses in TgIGF-I mice became statistically significant at 7.5 months of age (Fig. 1, A–F). By 9 months of age, the average response in transgenic mice was reduced to about 20% of that recorded in WT animals (Fig. 1, A–F). Clearly, retinal dysfunction was progressive, from normal responses at 3 months of age to almost complete loss of response by 9 months.
FIGURE 1.

Progressive alteration of electroretinographic responses in transgenic mice with increased intraocular IGF-I. A–F, evolution over time of ERG responses in TgIGF-I mice (pink lines) compared with healthy littermates (black lines). With age, TgIGF-I showed a progressive decline in the recorded amplitudes in response to all stimuli tested. A, scotopic threshold response, representing highly sensitive responses of rod photoreceptors; B, scotopic b-wave, representing rod responses; C, mixed scotopic, reflecting stimulation of both rod and cones under scotopic conditions; D, oscillatory potentials, which measure INL neuronal activity; E, photopic b-wave, depicting cone responses; F, flicker, repetitive photopic stimulations that analyze cone recovery. There were statistically significant reductions in the responses in animals aged more than 7.5 months. G, ERG responses were recorded 6 months after a single intravitreal administration of AAV2-hPEDF (left eye) and AAV2-null (right eye) vectors to TgIGF-I mice aged 1.5 months. Age-matched WT littermates were used as controls. Scotopic b-wave (Bscot), mixed a and b-waves, oscillatory potentials, and photopic b-wave (Bphot) were analyzed. Despite the counteraction of retinal neovascularization, the overexpression of PEDF was not able to ameliorate the electroretinographic responses of treated eyes, which presented reduced amplitudes in all tests performed, similar to those of null-injected, untreated eyes. Values are expressed as the mean ± S.E. of 5–9 animals/group. *, p <0.05; **, p <0.01; ***, p <0.001.
To evaluate if this progressive retinal neurodegeneration was the consequence of the development of neovascularization, young (1.5-month-old) TgIGF-I mice were treated intravitreally with adenoassociated viral vectors carrying the gene of pigment epithelium-derived factor (PEDF), a well known antiangiogenic factor (18). This treatment leads to long term production of PEDF, normalization of intraocular levels of VEGF, and prevention of neovascularization and retinal detachment in this animal model (19). However, the evaluation of ERG responses at 7.5 month of age, i.e. 6 months after gene transfer, did not reveal any recovery in PEDF-treated animals (Fig. 1G), suggesting that vascular alterations were not the main mechanism underlying neurodegeneration in transgenic retinas.
Decreased Thickness of Retinal Layers in Transgenic Mice
The thickness of retinal layers is a general indicator of the loss of neuronal populations. ONL and INL thicknesses were quantified in both the peripheral and central areas of retinal sections containing the optic nerve (Fig. 2A). In agreement with functional observations, at 3 months, the thickness of retinal layer was preserved in TgIGF-I retinas and only a slight, nonstatistically significant decrease was observed in 6-month-old animals, mainly in the ONL (data not shown). At 7.5 months of age, retinas of TgIGF-I mice were clearly thinner, with a 40% reduction in the width of both ONL and INL (Fig. 2A). These results suggest that a chronic increase in intraocular IGF-I levels resulted in neurodegeneration of photoreceptors and other neuronal cells.
FIGURE 2.

Reduced thickness of the different retinal layers and decreased neuronal populations in IGF-I-overexpressing retinas. A, thickness of retinal layers was measured at 7.5 months of age. Retinal layers were clearly thinner in TgIGF-I mice, with statistically significant reductions in both ONL and INL thickness in central and peripheral retina. B–E, representative images of the immunolabeling of retinal sections with specific neuronal markers (left) and quantification of labeled populations (right) in 7.5-month-old WT and TgIGF-I mice. B, rhodopsin immunolabeling (red) showing positive staining in the outer segment (OS) of photoreceptors. Outer segment length (arrows) was measured in medial retinal sections. The length of the outer segment was significantly reduced in transgenic retinas. C, representative images of PKCα immunostaining (green). The terminals of bipolar cells located in the outer and inner plexiform layers (upper and lower insets) were strongly positive in WT retinas. Cell bodies spanning from the INL to the GL were also labeled. Transgenic retinas showed reduced PKCα reactivity. Quantification of the number INL PKC-positive cells revealed a significant loss of bipolar PKC+ cells in transgenic mice. D, representative images of calretinin immunolabeling (green) showing reduced staining in both the GL and the INL in TgIGF-I. Quantification of the number of calretinin+ cells demonstrated a reduction in the number of amacrine neurons in transgenic retinas. E, immunofluorescent detection of Brn3a (green), expressed specifically in the nuclei of ganglion neurons (arrowheads). Transgenic retinas showed fewer Brn3a positive nuclei in the GL. Nuclei were counterstained with DAPI (blue). Values are expressed as the mean ± S.E. of 5–9 animals/group. *, p <0.05; **, p <0.01. Scale bar, (A) 35.51 μm; B–E, 18.37 μm.
Loss of Specific Neuronal Populations in TgIGF-I Retinas
To identify which type of neurons was degenerating in transgenic retinas, specific markers were used to detect and quantify different neuronal populations in 7.5-month-old animals. The length of rod photoreceptor outer segment was markedly reduced in transgenic retinas, as determined by morphometric analysis on rhodopsin-immunostained sections (Fig. 2B). Nevertheless, TgIGF-I retinas had normal cone arrestin content (data not shown), indicating that cone photoreceptors were not altered. Collectively, these observations suggested that the reduced ONL thickness observed (Fig. 2A) was likely due to the loss of rods, as rods and cones are the only cell types found in this nuclear layer.
Bipolar and amacrine neurons specifically express the α- isoform of protein kinase C (PKCα) and calretinin, respectively. Retinal sections from 7.5-month-old TgIGF-I animals had a reduced number of PKCα+ cells and calretinin+ neurons (Fig. 2, C and D), in agreement with a thinner INL (Fig. 2A). Bipolar cells were the only cell type for which we detected a statistically significant reduction at earlier time points (6 months, data not shown). No differences were observed in the number of horizontal neurons (data not shown). Finally, the number of ganglion cells with positive immunostaining for the transcription factor Brn3a was also lower (Fig. 2E). Immunofluorescent detection of cleaved caspase-3, a marker of apoptosis, revealed few caspase-3+ cells in the GL of transgenic retinas (Fig. 3). These findings, together with the decline in ERG responses, suggested there was a progressive loss of neurons in transgenic retinas.
FIGURE 3.

Detection of apoptotic cells in the retinas of transgenic mice with increased intraocular IGF-I. Cells positive for the apoptosis marker cleaved caspase 3 (green, arrows) were found in the ganglion cell layer of transgenic retinas at 6 months of age. Cleaved caspase 3-positive cells could not be detected in retinal sections from age-matched wild-type animals. Nuclei were stained with DAPI (blue). Scale bar, 7.6 μm (left panel), and 11.43 μm (right panel).
Analysis of Gene Expression in Young Transgenic Retinas
To identify the changes in gene expression that could account for the neuronal dysfunction induced by increased intraocular IGF-I, we performed microarray analysis on total retinal RNA from 4-month-old animals. At this age, transgenic retinas do not yet show neurodegeneration but only evidence of nonproliferative vascular alterations and gliosis (14). After data normalization and filtering, we detected 37 genes differentially expressed in transgenic retinas, of which 25 were up-regulated and 12 were down-regulated more than 1.5-fold (Table 1). Most of the up-regulated genes were classified in three main categories as follows: retinal stress, gliosis, and angiogenesis, whereas down-regulated genes were mostly associated with angiogenesis and CNS development. Several of the up-regulated genes in the category of retinal stress corresponded to acute-phase proteins, such as α2-macroglobulin, lipocalin2, and components of the complement system. Microarray results were confirmed by quantitative PCR, and the expression of up-regulated genes was studied at different ages to identify the onset of each response (Fig. 4A). For most genes, expression was increased already by 1.5 months of age, and this rise was maintained or even exacerbated through the animal's life (Fig. 4A). Gliosis-related genes, including Gfap, S100b, and Gja1, showed an up-regulation pattern parallel to that of stress and acute-phase protein genes (Fig. 4A). Some of these genes were already up-regulated in retinas from transgenic animals as young as 3 weeks old (Fig. 4B). The expression of the transcription factor CEBP-δ, one of the main transcriptional activators of acute-phase genes (20), was also increased in transgenic retinas (1.35-fold increase, GEO GSE46246). CEBP-δ up-regulation was confirmed by quantitative RT-PCR in both young and old transgenic mice (Fig. 4C).
TABLE 1.
Microarray expression analysis in young transgenic mice
Classification of up- and down-regulated genes in retinas of 4-month-old IGF-I transgenic mice according to their biological function reported in the literature. Up-regulated genes (≥1.5-fold change) were classified in three main categories as follows: retinal stress, gliosis, and angiogenesis. Down-regulated genes (<−1.5-fold change) were classified in two main categories, angiogenesis and CNS formation.
| Gene description | Gene symbol | Fold change | p value | RefSeq |
|---|---|---|---|---|
| Stress (retinal stress) | ||||
| Nuclear protein 1 | Nupr1 | 2.03 | 0.0003 | NM_019738 |
| Lipocalin 2 | Lcn2 | 2.00 | 0.0013 | NM_008491 |
| Oncostatin M receptor | Osmr | 1.59 | 0.0039 | NM_011019 |
| Interferon-induced transmembrane protein 3 | Ifimt3 | 1.58 | 0.0050 | NM_025378 |
| Complement component 4b | C4b | 1.76 | 0.0084 | NM_009780 |
| α2-Macroglobulin | A2m | 1.73 | 0.0090 | NM_175628 |
| Complement component 1, q subcomponent, α-polypeptide | C1qa | 1.55 | 0.0095 | NM_007572 |
| Inhibitor of DNA binding 2 | Id2 | 1.70 | 0.0214 | NM_010496 |
| Histidine decarboxylase | Hdc | 1.50 | 0.0260 | NM_008230 |
| Endothelin 2 | Edn2 | 2.00 | 0.0497 | NM_007902 |
| Gliosis | ||||
| Glial fibrillary acidic protein | Gfap | 3.10 | 0.0014 | NM_010277 |
| S100 protein, β polypeptide | S100b | 2.02 | 0.0060 | NM_009115 |
| Doublecortin-like kinase 1 | Dclk1 | 1.54 | 0.0111 | NM_019978 |
| Gap junction protein, α1 | Gja1 | 2.06 | 0.0173 | NM_010288 |
| Aquaporin 4 | Aqp4 | 1.52 | 0.0199 | NM_009700 |
| Angiogenesis | ||||
| Carbonic anhydrase 3 | Car3 | −1.59 | 0.0005 | NM_007606 |
| EGF-containing fibulin-like extracellular matrix protein 1 | Efemp1 | 1.52 | 0.0056 | NM_146015 |
| Rac GTPase-activating protein 1 | Racgap1 | 1.53 | 0.0068 | NM_012025 |
| α2-Macroglobulin | A2m | 1.73 | 0.0090 | NM_175628 |
| Procollagen C-endopeptidase enhancer protein | Pcolce | 1.60 | 0.0215 | NM_008788 |
| Polycystic kidney disease 1 homolog | Pkd1 | −1.58 | 0.0272 | NM_013630 |
| Progesterone receptor membrane component 2 | Pgrmc2 | −1.58 | 0.0348 | NM_027558 |
| Rho-guanine nucleotide exchange factor | Rgnef | −3.45 | 0.0426 | NM_012026 |
| CNS formation | ||||
| Nardilysin, N-arginine dibasic convertase, NRD convertase 1 | Nrd1 | −1.54 | 0.0002 | NM_146150 |
| Sema domain, immunoglobulin domain (Ig), short basic domain | Sema3f | −1.52 | 0.0354 | NM_011349 |
| Cut-like homeobox 2 | Cux2 | −1.54 | 0.0426 | NM_007804 |
| Miscellaneous | ||||
| Keratin 18 | Krt18 | −1.65 | 0.0027 | NM_010664 |
| CDK2 (cyclin-dependent kinase 2)-associated protein 1 | Cdk2ap1 | −1.89 | 0.0027 | NM_013812 |
| β2-Microglobulin | B2m | 1.52 | 0.0086 | NM_009735 |
| Actin, α2, smooth muscle, aorta | Acta2 | 1.51 | 0.0107 | NM_007392 |
| Hemoglobin α, adult chain 1 | Hba-a1 | 1.66 | 0.0206 | NM_008218 |
| Hemoglobin α, adult chain 2 | Hba-a2 | 1.66 | 0.0206 | NM_001083955 |
| RIKEN cDNA 1200015N20 gene | 1200015N20Rik | 1.55 | 0.0325 | NM_024244 |
| Chromodomain helicase DNA-binding protein 7 | Chd7 | −1.55 | 0.0343 | NM_001081417 |
| RIKEN cDNA 5830417I10 gene | 5830417I10Rik | −2.23 | 0.0345 | NM_027389 |
| Mitochondrial ubiquitin ligase activator of NFKB 1 | Mul1 | −2.32 | 0.0491 | NM_026689 |
FIGURE 4.

Retinal gene expression profile in young TgIGF-I mice. A, analysis of the expression of gliosis, retinal stress, and acute-phase-related genes in WT and transgenic retinas at different ages. Values are expressed as mean ± S.E. of three animals/group. *, p <0.05; **, p <0.01; ***, p <0.001, $, p <0.0001. B, Gfap, S100B, Edn2, and C4b were already up-regulated in 3-week-old transgenic retinas when analyzed by quantitative PCR. Values are expressed as mean ± S.E. of four animals/group. *, p <0.05; **, p <0.01; ***, p <0.001. C, quantitative RT-PCR analysis of the expression of the transcription factor CEBP-δ in WT and transgenic retinas at the indicated ages. Statistically significant increases in the expression of CEBP-δ were observed in transgenic retinas at 3 and 7.5 months of age. Values are expressed as the mean ± S.E. of 4–5 animals/group.*, p <0.05. D, functional analysis of significant enrichment in the target sequences for a specific set of transcription factors in the genes with altered expression in the microarray using the GeneCodis software. The table shows the percentage of altered genes whose promoters contained target sequences for the indicated transcription factors. E, retinal content of the acetylated, nuclear form of NF-κB (a-NF-κB) at 7.5 months of age. tub, tubulin. Transgenic mice presented higher levels of acetyl-NF-κB when compared with age-matched WT retinas, indicating activation of this pathway. Values are expressed as mean ± S.E. of 2–6 animals/group. *, p <0.05.
By performing functional analysis using the GeneCodis web tool (21), we found in the promoter of genes whose expression was altered in the chip analysis a significant enrichment in target sequences for a specific set of transcription factors (listed in Fig. 4D). This analysis revealed that 10.31% of the genes modified in TgIGF-I retinas presented binding sites for CEBP transcription factors, in agreement with CEBP-δ up-regulation (Fig. 4C). The GeneCodis analysis also revealed that 6.25% of the altered genes had in their promoters target sequences for NF-κB, another transcription factor implicated in the activation of acute-phase proteins (22). Although we were unable to detect any differences at 3 months of age (data not shown), the content of the acetylated form of NF-κB, which is the active form in the nucleus, was increased by 20% in transgenic retinas at 7.5 months of age (Fig. 4E).
Progression of Gliosis and Microgliosis in Transgenic Retinas
The results of the microarray analysis agreed with our previous reports describing the presence of gliosis in the retinas of TgIGF-I mice (14, 15). To determine whether there is a temporal progression of glial alterations, GFAP immunohistochemistry was performed at different ages. We observed increased GFAP staining in both astrocytes and Müller cells in TgIGF-I animals as young as 1.5 months of age, and this GFAP up-regulation was maintained and even intensified throughout the animal's life (Fig. 5A). Quantification by Western blot confirmed a marked increase in GFAP retinal content from an early age (Fig. 5B). Another cellular response characteristic of activated Müller cells is activation of the ERK1/2 pathway (23). In agreement with the presence of gliosis in TgIGF-I mice, a modest but statistically significant increase in the phosphorylated form of ERK was observed in 3-month-old animals, with very evident activation of ERK signaling in old animals (Fig. 5C).
FIGURE 5.

Progression of gliosis and microgliosis in TgIGF-I retinas. A, GFAP immunofluorescent detection (green) in retinal sections from WT and TgIGF-I mice at different ages. At all ages analyzed, transgenic retinas showed overexpression of GFAP, especially in radial Müller cells processes spanning the entire retina (arrows), where GFAP is hardly expressed. This pattern of GFAP staining indicated the presence of reactive gliosis in transgenic retinas. Nuclei were stained with DAPI (blue). Scale bar, 31.27 μm. B, GFAP content was analyzed by Western blot in retinal extracts from WT and TgIGF-I mice at the indicated ages. Quantification of blots demonstrated that GFAP levels were increased in transgenic retinas at all ages. AU, arbitrary units. Values are expressed as mean ± S.E. of two animals/group. *, p <0.05. C, ERK phosphorylation was analyzed by Western blot in retinal extracts from WT and TgIGF-I mice at 3 and 7.5 months of age. After normalization by total ERK levels, quantification of blots showed that P-ERK levels were increased in transgenic retinas at both ages. Values are expressed as mean ± S.E. of four animals/group. *, p <0.05. D, immunofluorescent detection with tomato lectin (Lycopersicum esculentum) allowed the identification of microglial cells in retinal sections. The number of lectin-positive cells per section was determined in retinal sections from WT and transgenic retinas at different ages, showing statistically significant differences in retinas of mice aged 3 months and older. Values are expressed as mean ± S.E. of four animals/group. *, p <0.05; **, p <0.01. E, representative images obtained from transgenic retinas at 7.5 months of age. Arrow indicates a round-shaped lectin+ cell, likely an activated microglial cell, and arrowheads indicate a microglial cell with characteristic stellar shape and dendriform ramifications. Scale bar, 12.06 μm (left panel), 8.54 (right panel).
Microglial cells are resident macrophages of the retina and become activated upon retinal injury (24). IGF-I induces proliferation and activation of microglial cells in retina and brain (25, 26). We previously reported that old TgIGF-I mice had increased numbers of bone marrow-derived microglia (15). When analyzed by staining with tomato lectin, we found that the number of microglial cells was higher in TgIGF-I retinas at all ages, confirming increased microglial infiltration starting early in the animal's life (Fig. 5D). Moreover, few of the tomato lectin-positive cells showed a rounded shape, a characteristic morphology of activated microglia (Fig. 5E, insets).
Gliosis Is a Direct Consequence of IGF-I Signaling
Further confirmation of the direct effects of IGF-I on retinal glial cells was obtained by culturing retinas isolated from WT mice with different concentrations of recombinant IGF-I protein. After 48 h of exposure to 100 ng/ml IGF-I, there was a nonstatistically significant increase in retinal expression of Gfap over levels of untreated retinas, which became clearly significant (about 4-fold) when incubated with 200 ng/ml, suggesting a direct activation of glial cells by IGF-I (Fig. 6A). This effect was abrogated in the presence of wortmannin, a specific inhibitor of PI3K, a downstream effector of IGF-I signaling pathway (Fig. 6A). A similar pattern of IGF-I-mediated up-regulation was observed when the expression of S100b was analyzed (Fig. 6B). These data clearly demonstrate the link between IGF-I signaling and gliosis.
FIGURE 6.
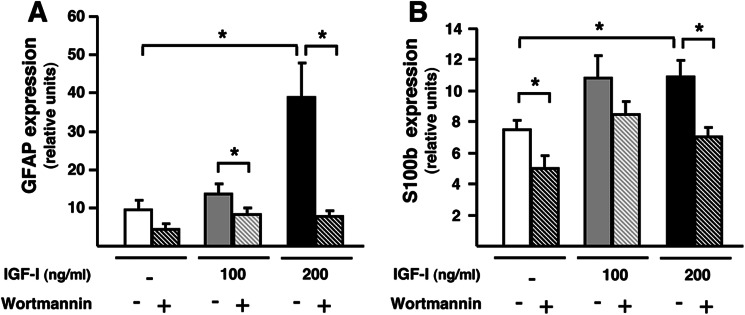
Increased gliosis in wild-type retinas exposed to recombinant IGF-I. A, GFAP; B, S100b expression levels in WT retinas incubated with increasing concentrations of IGF-I in the presence or absence of wortmannin, an inhibitor of IGF-I downstream signaling. The expression of both markers of gliosis was increased in the presence of IGF-I, and this effect was abrogated by the addition of wortmannin. Values are expressed as the mean ± S.E. of 3–4 retinas/group. *, p <0.05.
Increased Oxidative Stress in IGF-I-overexpressing Retinas
Oxidative stress, which can potentially cause neurodegeneration, is especially likely to occur in the retina due to the tissue's constant exposure to radiation and its high oxygen consumption. Glutathione acts as a scavenger of free radicals and reactive oxygen species (ROS) (27). In the retina, glutathione is produced mostly by Müller cells and astrocytes, which rapidly release it to provide neurons with antioxidant equivalents in situations of oxidative stress (27). The levels of oxidized glutathione were increased from an early age in TgIGF-I (Fig. 7A). Moreover, the expression of Nrf2, a transcription factor that activates a battery of antioxidant and cytoprotective genes (28), was also increased in transgenic retinas (Fig. 7B). NADPH oxidase is one of the main enzymes involved in the production of ROS in microglia, astrocytes, and neurons (29). The expression of NADPH oxidase regulatory subunits, p67Phox and p22Phox, was up-regulated in transgenic retinas (Fig. 7, C and D). Depending on the cell type, NADPH oxidase can be formed by different catalytic subunits such as Nox1, Nox2, and Nox4 (29). All three subunits were strikingly up-regulated in transgenic retinas at 7.5 months of age (Fig. 7, E–G). In addition, retinal iNOS expression was also markedly increased at this age (Fig. 7H), suggesting that there might be overproduction of nitric oxide (NO) in transgenic retinas. Altogether, these results suggest a raise in oxidative stress in TgIGF-I retinas.
FIGURE 7.
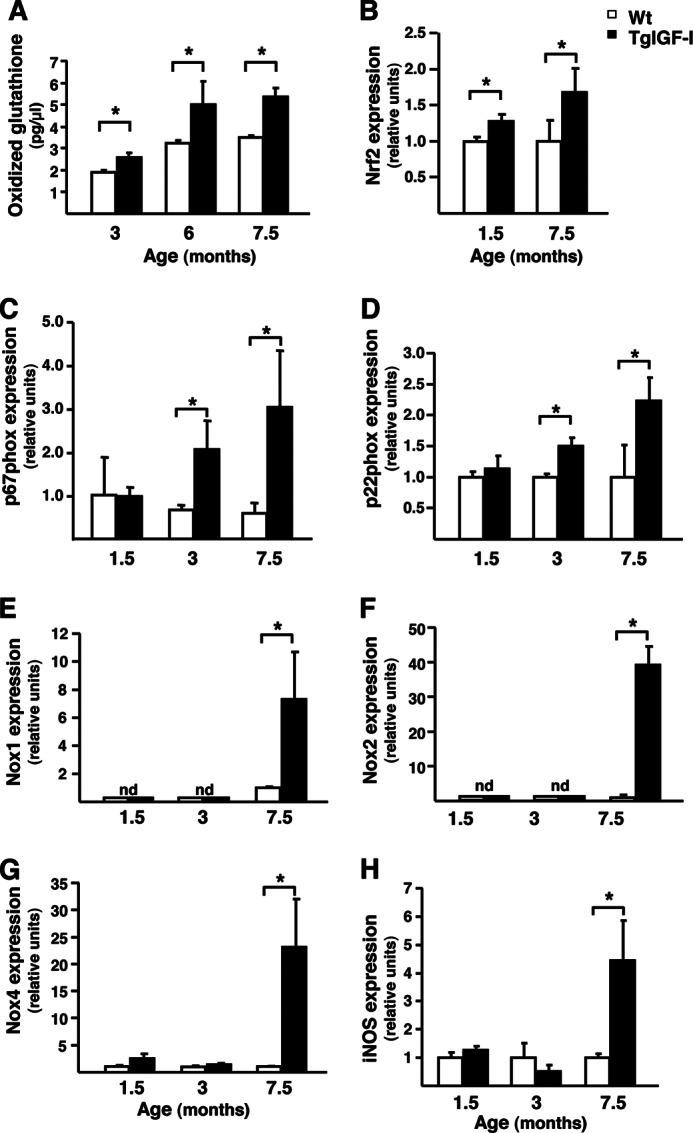
Increased oxidative stress in mice with increased intraocular IGF-I. Analysis of markers of oxidative stress in WT and Tg-IGF-I animals at different ages. A, levels of oxidized glutathione were higher in transgenic retinas at all ages studied. Values are expressed as the mean ± S.E. of five animals/group. *, p <0.05. B, up-regulation of Nrf2 expression in both young and old transgenic retinas, determined by quantitative PCR. Values are expressed as the mean ± S.E. of 4–5 animals/group. *, p <0.05. Retinal expression of the p67Phox (C) and p22Phox (D) regulatory subunits of the NADPH oxidase enzyme, assessed by quantitative RT-PCR. A marked up-regulation of both subunits was observed in transgenic mice at 3 and 7.5 months of age. E–G, retinal expression of the catalytic subunit of NADPH oxidase, which are expressed in different retinal cell types. E, Nox1; F, Nox2, and G, Nox4 were all markedly up-regulated in transgenic retinas at 7.5 months of age. nd, not detected. Values are expressed as mean ± S.E. of 4–5 animals/group. *, p <0.05. H, retinal expression by quantitative RT-PCR of the enzyme iNOS. There was a marked increase in iNOS expression in transgenic retinas at 7.5 months of age. Values are expressed as mean ± S.E. of 4–5 animals/group. *, p <0.05.
Impaired Glutamate Recycling in TgIGF-I
Persistent gliosis can impair the recycling by Müller cells of neurotransmitters, such as glutamate, whose accumulation in the retinal milieu causes neurotoxicity (30). Glutamine synthetase (GS), which converts glutamate into glutamine (30), showed reduced activity in transgenic retinas (Fig. 8A), suggesting impairment in glutamate detoxification.
FIGURE 8.
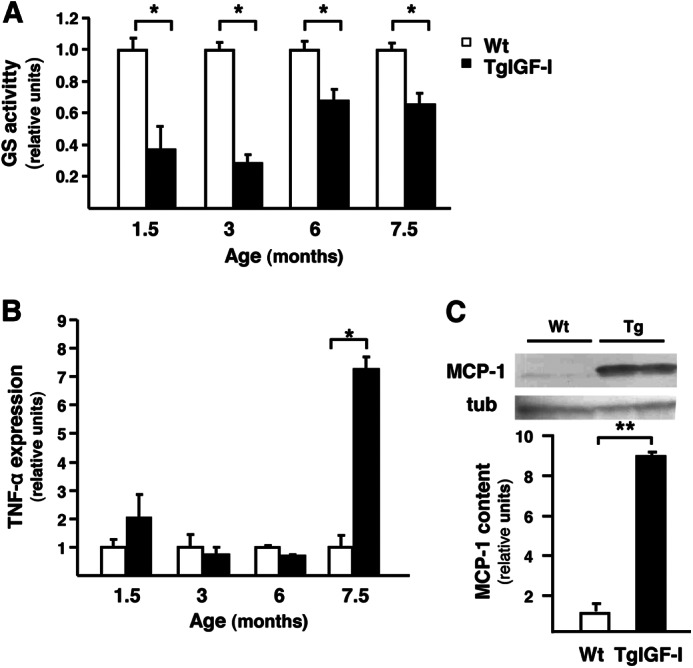
Impaired retinal glial functionality in mice with increased intraocular IGF-I. A, retinal GS activity was assayed in WT and TgIGF-I mice by spectrophotometric monitoring of γ-glutamyl hydroxamate. GS activity was significantly reduced in transgenic animals from an early age. Values are expressed as mean ± S.E. of 8–13 animals/group. *, p <0.05. B, follow up of retinal TNF-α expression by quantitative PCR. TNF-α was noticeably increased in TgIGF-I at 7.5 months of age. Values are expressed as mean ± S.E. of 4–5 animals/group. *, p <0.05. C, retinal MCP-1 content in 7.5 month-old WT and TgIGF-I mice analyzed by Western blot. MCP-1 levels were significantly higher in transgenic retinas. Values are expressed as mean ± S.E. of four animals/group. **, p <0.01. tub, tubulin.
Increased Production of Pro-inflammatory Cytokines in Transgenic Retinas
Activated Müller and microglial cells have direct neurotoxic effects through the release of pro-inflammatory cytokines such as TNF-α and MCP-1, which can cause the death of ganglion neurons (31) and photoreceptors (32). Retinal Tnf-α expression was not altered in younger transgenic mice, but increased substantially (about 7-fold) by 7.5 months of age (Fig. 8B). Similarly, the measurement of MCP-1 content at 7.5 months of age showed much higher levels (9-fold increase) in transgenic retinas (Fig. 8C).
DISCUSSION
IGF-I is a pleiotropic growth factor involved in multiple biological and pathological processes. In addition to the diabetes-like vascular alterations previously reported for transgenic mice with increased intraocular IGF-I (14, 15), this study has unraveled a complex phenotype in transgenic retinas involving neuronal and glial cells. TgIGF-I mice showed a progressive decline in electroretinographic responses and a decrease in specific neuronal populations that resulted in significantly impaired neuronal functionality. Markers of retinal stress, gliosis, and microgliosis were detected at early ages, before major vascular alterations occur in this model. Transgenic mice also showed signs of oxidative stress and alterations in glutamate metabolism, likely secondary to gliosis. Alterations showed a progressive pattern, worsening as animals aged. These changes in normal retinal metabolism may underlie the neuronal dysfunction observed in transgenic retinas, which would be further exacerbated by the increased production of pro-inflammatory cytokines such as TNF-α and MCP-1.
In TgIGF-I mice, ERG recordings showed a pattern compatible with progressive retinal neurodegeneration, from normal responses at 3 months of age to alterations of all recorded parameters in both scotopic and photopic conditions by 6–7 months and flat ERG in old animals. Histological analysis showed loss of retinal neurons (rod photoreceptors, bipolar, ganglion, and amacrine cells) in aged mice, consistent with the ERGs. Overt retinal dysfunction occurred at an age, 7.5 months, at which ≈75% of TgIGF-I mice present retinal detachment (14, 19). However, when neovascularization and retinal detachment are prevented by long term overexpression of the anti-angiogenic factor Pedf in the retina (19), ERG responses do not improve. This observation suggests that additional causes contribute to the dysfunction observed in transgenic retinas.
The analysis of the gene expression profile before onset of neuronal dysfunction in Tg-IGF-I retinas revealed that the genes whose expression was altered fell into three main categories: retinal stress, which included genes involved in the acute-phase response (APR), gliosis, and angiogenesis. Nearly every major disease of the retina, including retinitis pigmentosa and diabetic retinopathy, is associated with reactive gliosis involving Müller cells (33). This physiological response contributes to tissue repair and neuroprotection but, if persistent, may also contribute to tissue damage (33). Likewise, the acute-phase response is part of the body's early defense mechanisms after infection, tissue injury, stress, or neoplasia (34). However, chronic APR can lead to disturbances in normal physiology (35). Increased production of acute-phase proteins has been reported for animal models of ocular pathologies, such as glaucoma, experimental diabetes, and photoreceptor degeneration (36–38). In our animal model, the increase in APR gene expression may have resulted from the up-regulation, at an early age, of the transcription factor CEBP-δ, one of the main transcriptional regulators of acute-phase genes (20). IGF-I is also a well known activator of the transcription factor NF-κB (39), through several signaling pathways, including the Akt pathway, which is activated in transgenic retinas (15). Promoter analysis suggested that 6% of the genes altered in the microarray had target sequences for NF-κB. Activation of NF-κB contributes to the early activation of APR genes, maintained in later stages by the interaction between CEBP-β and CEBP-δ (20).
In our transgenic mice, high intraocular IGF-I led to increased expression of acute-phase proteins and markers of gliosis early in the course of the animal's life (3 weeks), before neuronal or vascular phenotypes appear (14, 15), suggesting that these alterations were a direct consequence of IGF-I and not secondary responses. The observation of up-regulated expression of Gfap and S100b in wild-type retinas exposed to recombinant IGF-I confirmed this hypothesis. Noticeably, increased levels of S100b, produced by reactive astrocytes, have been associated with a wide range of pathological conditions of the CNS, including Alzheimer, epilepsy, and diabetic retinopathy (40–42). S100b, through its binding to the receptor for AGEs (RAGE), can activate NF-κB leading to production of pro-inflammatory cytokines and ROS in both glial and microglial cells (42, 43). Given that the retinal expression of this gene is increased early in the life of TgIGF-I (3 weeks), we hypothesize that S100b plays an important role in the retinal alterations of this animal model. Moreover, the fact that S100b exerts its action through the RAGE the activation of which occurs in diabetic retinopathy by AGEs (42, 44), may explain in part the similarities between the ocular phenotype of normoglycemic, IGF-I-overexpressing transgenic mice, and diabetic retinopathy.
Oxidative stress is a causative factor of neuronal death in neurodegenerative diseases. Oxidized glutathione was significantly increased in transgenic retinas at all ages, suggesting the presence of oxidative stress. Given that TgIGF-I mice are not hyperglycemic, the source of any oxidative stress in transgenic retinas would probably result from IGF-I-mediated cell dysfunction. A strong correlation between increased IGF-I signaling and ROS formation has been established in several cell types, including endothelial cells and neurons, through the activation of NADPH oxidase (45). The regulatory and also the catalytic subunits of this enzyme were up-regulated in IGF-I transgenic retinas. NADPH oxidase is also overexpressed in activated microglia (46) and astrocytes (29), and NF-κB has been postulated to be responsible for this overexpression in reactive astrocytes in the ischemia/reperfusion model (47). Moreover, the up-regulation of iNOS in transgenic eyes suggests increased production of NO, probably by activated Müller and/or microglial cells, contributing to the formation of reactive species. Finally, the up-regulation of Nrf2, a master regulator of endogenous antioxidant protection involved in the transcriptional control of detoxifying enzymes, may be considered a compensatory mechanism against oxidative stress in IGF-I-overexpressing retinas. Indeed, the up-regulation of Nrf2 has been reported after prolonged activation of NF-κB (48).
Müller cells maintain the homeostasis of the retinal milieu, controlling pH, the concentrations of ions, and the recycling of neurotransmitters, such as glutamate (33). However, all these essential functions are impaired when gliosis is established. Glutamate accumulation causes neurotoxicity (30). The retinal activity of the enzyme GS, which detoxifies glutamate to glutamine, is decreased in experimental models of retinal injury (49). Similarly, this activity was significantly reduced in transgenic retinas from an early age, suggesting impaired glutamate detoxification. In addition, in situations of retinal injury and gliosis, activated Müller and microglial cells can produce and secrete pro-inflammatory cytokines, which may contribute to the dysfunction of the blood-retinal barrier and also cause neuronal death (31, 50). Among cytokines, TNF-α and MCP-1 directly contribute to neuronal loss (31, 32). In agreement with the extensive gliosis and microgliosis observed in IGF-I mice, the levels of TNF-α and MCP-1 were considerably increased at 7.5 months of age, when severe loss of vision occurs, according to ERG measurements and the marked reduction of retinal neurons. The increased production of cytokines may act synergistically with the impairment of glial supportive functions to cause neuronal loss in TgIGF-I retinas.
In our model, glial alterations are observed early in the animals' life, whereas neuronal loss and functional impairment do not become evident until mice are 6 months or older. This probably results from the fact that IGF-I is a pro-survival molecule for neurons (8). Despite the profound alterations in retinal homeostasis caused by glial dysfunction and oxidative stress, it is likely that IGF-I overexpression delayed neuronal death in this environment, preventing neuronal functional alterations in young TgIGF-I retinas. As animals aged, the pro-survival effects of IGF-I in the retina were progressively overcome leading to neurodegeneration (Fig. 9).
FIGURE 9.
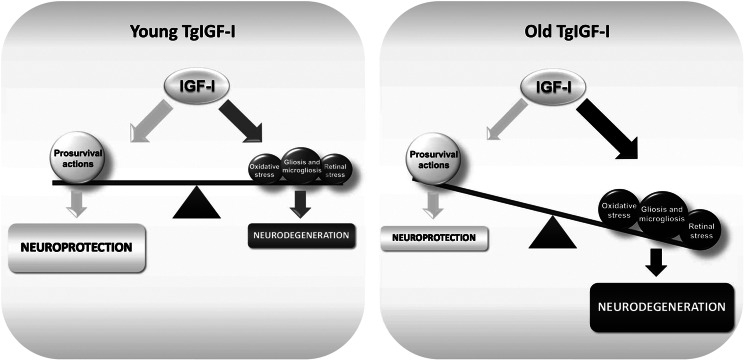
Schematic representation summarizing IGF-I actions on IGF-I transgenic retinas. IGF-I has well described prosurvival properties that are essential for normal neuronal functionality and protection. However, excess IGF-I stimulation induces deleterious processes (gliosis and microgliosis, retinal and oxidative stress) that contribute to the impairment of neuronal functionality and viability, leading to neurodegeneration. IGF-I pro-survival actions are unable to counteract neurodegeneration as animals age.
In conclusion, high intraocular levels of IGF-I trigger a series of cellular processes that bring about retinal stress, impair homeostatic functions of the glia, and lead to neuronal dysfunction and death. This work highlights the importance that increased intraocular levels of IGF-I may have in the progression of diseases such as ischemic or diabetic retinopathy.
Acknowledgments
We thank Prof. Malcom Watford for helpful discussions and M. Moya and D. Ramos for technical assistance.
This work was supported in part by Instituto de Salud Carlos III Grant PI 061417, Plan Nacional I+D+I Grant SAF2011-24698, Generalitat de Catalunya Grant 2009 SGR224, Spain, and European Community Grant CLINIGENE LSHB-CT-2006-018933.
- IGF-I
- insulin-like growth factor I
- AGE
- advanced glycation end product
- GFAP
- glial fibrillary acidic protein
- ROS
- reactive oxygen species
- TgIGF-I
- transgenic IGF-I
- ERG
- electroretinogram
- cd
- candela
- ONL
- outer nuclear layer
- INL
- inner nuclear layer
- PEDF
- pigment epithelium-derived factor
- iNOS
- inducible nitric-oxide synthase
- GS
- glutamine synthetase
- APR
- acute-phase response
- GL
- ganglion cell layer
- CEBP
- CCAAT enhancer-binding protein.
REFERENCES
- 1. Laviola L., Natalicchio A., Giorgino F. (2007) The IGF-I signaling pathway. Curr. Pharm. Des. 13, 663–669 [DOI] [PubMed] [Google Scholar]
- 2. Shaw L. C., Grant M. B. (2004) Insulin-like growth factor-1 and insulin-like growth factor binding proteins: their possible roles in both maintaining normal retinal vascular function and in promoting retinal pathology. Rev. Endocr. Metab. Disord. 5, 199–207 [DOI] [PubMed] [Google Scholar]
- 3. Inokuchi N., Ikeda T., Imamura Y., Sotozono C., Kinoshita S., Uchihori Y., Nakamura K. (2001) Vitreous levels of insulin-like growth factor-I in patients with proliferative diabetic retinopathy. Curr. Eye Res. 23, 368–371 [DOI] [PubMed] [Google Scholar]
- 4. Smith L. E., Shen W., Perruzzi C., Soker S., Kinose F., Xu X., Robinson G., Driver S., Bischoff J., Zhang B., Schaeffer J. M., Senger D. R. (1999) Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 5, 1390–1395 [DOI] [PubMed] [Google Scholar]
- 5. Kondo T., Vicent D., Suzuma K., Yanagisawa M., King G. L., Holzenberger M., Kahn C. R. (2003) Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J. Clin. Invest. 111, 1835–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hellström A., Carlsson B., Niklasson A., Segnestam K., Boguszewski M., de Lacerda L., Savage M., Svensson E., Smith L., Weinberger D., Albertsson Wikland K., Laron Z. (2002) IGF-I is critical for normal vascularization of the human retina. J. Clin. Endocrinol. Metab 87, 3413–3416 [DOI] [PubMed] [Google Scholar]
- 7. Treins C., Giorgetti-Peraldi S., Murdaca J., Monthouël-Kartmann M. N., Van Obberghen E. (2005) Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol. Endocrinol. 19, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 8. D'Ercole A. J., Ye P., Calikoglu A. S., Gutierrez-Ospina G. (1996) The role of the insulin-like growth factors in the central nervous system. Mol. Neurobiol. 13, 227–255 [DOI] [PubMed] [Google Scholar]
- 9. Bonapace G., Concolino D., Formicola S., Strisciuglio P. (2003) A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J. Med. Genet. 40, 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woods K. A., Camacho-Hübner C., Savage M. O., Clark A. J. (1996) Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367 [DOI] [PubMed] [Google Scholar]
- 11. Saatman K. E., Contreras P. C., Smith D. H., Raghupathi R., McDermott K. L., Fernandez S. C., Sanderson K. L., Voddi M., McIntosh T. K. (1997) Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp. Neurol. 147, 418–427 [DOI] [PubMed] [Google Scholar]
- 12. Kermer P., Klöcker N., Labes M., Bähr M. (2000) Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 in vivo. J. Neurosci. 20, 2–8 [PubMed] [Google Scholar]
- 13. Rodriguez-de la Rosa L., Fernandez-Sanchez L., Germain F., Murillo-Cuesta S., Varela-Nieto I., de la Villa P., Cuenca N. (2012) Age-related functional and structural retinal modifications in the Igf1−/− null mouse. Neurobiol. Dis. 46, 476–485 [DOI] [PubMed] [Google Scholar]
- 14. Ruberte J., Ayuso E., Navarro M., Carretero A., Nacher V., Haurigot V., George M., Llombart C., Casellas A., Costa C., Bosch A., Bosch F. (2004) Increased ocular levels of IGF-1 in transgenic mice lead to diabetes-like eye disease. J. Clin. Invest. 113, 1149–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haurigot V., Villacampa P., Ribera A., Llombart C., Bosch A., Nacher V., Ramos D., Ayuso E., Segovia J. C., Bueren J. A., Ruberte J., Bosch F. (2009) Increased intraocular insulin-like growth factor-I triggers blood-retinal barrier breakdown. J. Biol. Chem. 284, 22961–22969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aiello L. P., Avery R. L., Arrigg P. G., Keyt B. A., Jampel H. D., Shah S. T., Pasquale L. R., Thieme H., Iwamoto M. A., Park J. E. (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 331, 1480–1487 [DOI] [PubMed] [Google Scholar]
- 17. Amin R. H., Frank R. N., Kennedy A., Eliott D., Puklin J. E., Abrams G. W. (1997) Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 38, 36–47 [PubMed] [Google Scholar]
- 18. Dawson D. W., Volpert O. V., Gillis P., Crawford S. E., Xu H., Benedict W., Bouck N. P. (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285, 245–248 [DOI] [PubMed] [Google Scholar]
- 19. Haurigot V., Villacampa P., Ribera A., Bosch A., Ramos D., Ruberte J., Bosch F. (2012) Long-term retinal PEDF overexpression prevents neovascularization in a murine adult model of retinopathy. PLoS One 7, e41511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poli V. (1998) The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 273, 29279–29282 [DOI] [PubMed] [Google Scholar]
- 21. Nogales-Cadenas R., Carmona-Saez P., Vazquez M., Vicente C., Yang X., Tirado F., Carazo J. M., Pascual-Montano A. (2009) GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 37, W317–W322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moshage H. (1997) Cytokines and the hepatic acute phase response. J. Pathol. 181, 257–266 [DOI] [PubMed] [Google Scholar]
- 23. Takeda M., Takamiya A., Yoshida A., Kiyama H. (2002) Extracellular signal-regulated kinase activation predominantly in Müller cells of retina with endotoxin-induced uveitis. Invest. Ophthalmol. Vis. Sci. 43, 907–911 [PubMed] [Google Scholar]
- 24. Karlstetter M., Ebert S., Langmann T. (2010) Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology 215, 685–691 [DOI] [PubMed] [Google Scholar]
- 25. Zelinka C. P., Scott M. A., Volkov L., Fischer A. J. (2012) The reactivity, distribution, and abundance of non-astrocytic inner retinal glial (NIRG) cells are regulated by microglia, acute damage, and IGF1. PLoS One 7, e44477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Donnell S. L., Frederick T. J., Krady J. K., Vannucci S. J., Wood T. L. (2002) IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39, 85–97 [DOI] [PubMed] [Google Scholar]
- 27. Schütte M., Werner P. (1998) Redistribution of glutathione in the ischemic rat retina. Neurosci. Lett. 246, 53–56 [DOI] [PubMed] [Google Scholar]
- 28. Wakabayashi N., Slocum S. L., Skoko J. J., Shin S., Kensler T. W. (2010) When NRF2 talks, who's listening? Antioxid. Redox Signal. 13, 1649–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bedard K., Krause K. H. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 [DOI] [PubMed] [Google Scholar]
- 30. Bringmann A., Pannicke T., Biedermann B., Francke M., Iandiev I., Grosche J., Wiedemann P., Albrecht J., Reichenbach A. (2009) Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 54, 143–160 [DOI] [PubMed] [Google Scholar]
- 31. Lebrun-Julien F., Duplan L., Pernet V., Osswald I., Sapieha P., Bourgeois P., Dickson K., Bowie D., Barker P. A., Di Polo A. (2009) Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 29, 5536–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakazawa T., Hisatomi T., Nakazawa C., Noda K., Maruyama K., She H., Matsubara A., Miyahara S., Nakao S., Yin Y., Benowitz L., Hafezi-Moghadam A., Miller J. W. (2007) Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc. Natl. Acad. Sci. U.S.A. 104, 2425–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bringmann A., Pannicke T., Grosche J., Francke M., Wiedemann P., Skatchkov S. N., Osborne N. N., Reichenbach A. (2006) Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 25, 397–424 [DOI] [PubMed] [Google Scholar]
- 34. Gabay C., Kushner I. (1999) Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454 [DOI] [PubMed] [Google Scholar]
- 35. Venteclef N., Jakobsson T., Steffensen K. R., Treuter E. (2011) Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol. Metab. 22, 333–343 [DOI] [PubMed] [Google Scholar]
- 36. Samardzija M., Wariwoda H., Imsand C., Huber P., Heynen S. R., Gubler A., Grimm C. (2012) Activation of survival pathways in the degenerating retina of rd10 mice. Exp. Eye Res. 99, 17–26 [DOI] [PubMed] [Google Scholar]
- 37. Gerhardinger C., Costa M. B., Coulombe M. C., Toth I., Hoehn T., Grosu P. (2005) Expression of acute-phase response proteins in retinal Müller cells in diabetes. Invest. Ophthalmol. Vis. Sci. 46, 349–357 [DOI] [PubMed] [Google Scholar]
- 38. Steele M. R., Inman D. M., Calkins D. J., Horner P. J., Vetter M. L. (2006) Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest. Ophthalmol. Vis. Sci. 47, 977–985 [DOI] [PubMed] [Google Scholar]
- 39. Salminen A., Kaarniranta K. (2010) Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-κB signaling. Cell. Signal. 22, 573–577 [DOI] [PubMed] [Google Scholar]
- 40. Mrak R. E., Griffinbc W. S. (2001) The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol. Aging 22, 915–922 [DOI] [PubMed] [Google Scholar]
- 41. Van Eldik L. J., Wainwright M. S. (2003) The Janus face of glial-derived S100B: beneficial and detrimental functions in the brain. Restor. Neurol. Neurosci. 21, 97–108 [PubMed] [Google Scholar]
- 42. Zong H., Ward M., Madden A., Yong P. H., Limb G. A., Curtis T. M., Stitt A. W. (2010) Hyperglycaemia-induced pro-inflammatory responses by retinal Müller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia 53, 2656–2666 [DOI] [PubMed] [Google Scholar]
- 43. Bianchi R., Giambanco I., Donato R. (2010) S100B/RAGE-dependent activation of microglia via NF-κB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1β, and TNF-α. Neurobiol. Aging 31, 665–677 [DOI] [PubMed] [Google Scholar]
- 44. Zong H., Ward M., Stitt A. W. (2011) AGEs, RAGE, and diabetic retinopathy. Curr. Diab. Rep. 11, 244–252 [DOI] [PubMed] [Google Scholar]
- 45. Vardatsikos G., Sahu A., Srivastava A. K. (2009) The insulin-like growth factor family: molecular mechanisms, redox regulation, and clinical implications. Antioxid. Redox Signal. 11, 1165–1190 [DOI] [PubMed] [Google Scholar]
- 46. Block M. L., Zecca L., Hong J. S. (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 47. Barakat D. J., Dvoriantchikova G., Ivanov D., Shestopalov V. I. (2012) Astroglial NF-κB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. J. Neurochem. 120, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nair S., Doh S. T., Chan J. Y., Kong A. N., Cai L. (2008) Regulatory potential for concerted modulation of Nrf2- and Nfκb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 99, 2070–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lieth E., LaNoue K. F., Antonetti D. A., Ratz M. (2000) Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. The Penn State Retina Research Group. Exp. Eye Res. 70, 723–730 [DOI] [PubMed] [Google Scholar]
- 50. Krady J. K., Basu A., Allen C. M., Xu Y., LaNoue K. F., Gardner T. W., Levison S. W. (2005) Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 54, 1559–1565 [DOI] [PubMed] [Google Scholar]