

Background: The mycosin-1 protease (MycP1) is essential for export and cleavage of the type VII-secreted virulence-associated proteins involved in pathogenesis of Mycobacterium tuberculosis and related species.
Results: The x-ray structure of MycP1, with its proposed propeptide, is described.
Conclusion: The proposed propeptide wraps around the perimeter of a subtilisin-like fold, leaving the catalytic center unobstructed.
Significance: MycP1 may operate through a novel mode of regulation.
Keywords: Microbiology, Mycobacteria, Mycobacterium tuberculosis, Protease, Protein Secretion, ESX, Subtilisin, Type VII Secretion System
Abstract
Mycobacteria use specialized type VII (ESX) secretion systems to export proteins across their complex cell walls. Mycobacterium tuberculosis encodes five nonredundant ESX secretion systems, with ESX-1 being particularly important to disease progression. All ESX loci encode extracellular membrane-bound proteases called mycosins (MycP) that are essential to secretion and have been shown to be involved in processing of type VII-exported proteins. Here, we report the first x-ray crystallographic structure of MycP1(24–407) to 1.86 Å, defining a subtilisin-like fold with a unique N-terminal extension previously proposed to function as a propeptide for regulation of enzyme activity. The structure reveals that this N-terminal extension shows no structural similarity to previously characterized protease propeptides and instead wraps intimately around the catalytic domain where, tethered by a disulfide bond, it forms additional interactions with a unique extended loop that protrudes from the catalytic core. We also show MycP1 cleaves the ESX-1 secreted protein EspB from both M. tuberculosis and Mycobacterium smegmatis at a homologous cut site in vitro.
Introduction
Mycobacterium tuberculosis is an intracellular pathogen that specifically requires specialized type VII secretion systems (T7SS)3 to passage proteins across its cell wall, a process required for survival in the human host phagosome (1). Mycobacterial genomes encode up to five nonredundant T7SS (also referred to as ESX-1 to 5). The most well studied system is ESX-1, which M. tuberculosis and related pathogens use to secrete the T cell antigens ESAT-6/CFP-10 in addition to other ESX-secreted-proteins (Esps) that are required for progression of disease (2–6). MycP1 (mycosin-1 protease) is essential for ESX-1 secretion in pathogenic M. tuberculosis and for efficient DNA conjugation in the avirulent saprophyte Mycobacterium smegmatis (7, 8). The other T7SS of known function are involved in iron acquisition (ESX-3) (9) or export of the virulence-related PE/PPE (Pro-Glu/Pro-Pro-Glu) family of proteins (ESX-5) (10, 11). MycP1 is one of six conserved components that are found in all five T7SS and are thus designated MycP1–5 in accordance with their associated systems (12). Although a recent compositional analysis of the ESX-5 T7SS inner membrane complex did not identify MycP5 as part of the assembly (13), knocking out MycP1 abrogates ESX-1 protein secretion, indicating that its presence is essential (7, 14).
Proteins exported by the T7SS have been detected in culture filtrates and in cell wall fractions as lower molecular mass/proteolytically processed species compared with those which are not exported (3, 4, 7, 11, 15). For example, LipY, a substrate of the ESX-5 system, is cleaved between its N-terminal PE domain and its C-terminal lipase domain following secretion (15). Members of this enigmatic PE family, as well as those of the PPE family, have been shown to be required for export to the cell wall (15, 16). It was thus hypothesized that the MycP family may function in removal of these PE/PPE “leader” domains following (or concomitantly with) secretion (15). Ohol and co-workers provided the first evidence of MycP1tb proteolytic activity against a T7SS substrate by complementing a ΔmycP1tb knock-out strain with a mutant in which the predicted serine nucleophile of the enzyme was changed to alanine (MycP1tb-S332A) (7). This catalytically impaired mutant resulted in an altered cleavage pattern of the C-terminal portion of the secreted protein EspBtb which suggested that native MycP1tb mediates cleavage at two sites (7). Intriguingly, the mutant strain also showed a 2-fold increase in secretion of known ESX-1 substrates as well as reduced virulence in mice, leading to the hypothesis that MycP1 activity negatively regulates secretion via EspB (7). Although several groups have documented secretion-coupled cleavage of the EspB C terminus (3, 4, 7), convincing data showing the exact residue at which proteolysis occurs have not been provided.
Proteases are often synthesized with propeptide segments that act as intramolecular chaperones to promote folding as well as regulate activity to prevent untimely and potentially destructive proteolysis in the incorrect cellular compartment (17). These propeptides range from short dipeptides to independent domains and are often proteolytically processed and removed as the enzyme matures (17). Sequence analysis (18, 19) of the MycP family indicates that they have an N-terminal Sec signal sequence followed by a 40-amino acid stretch that, despite showing no sequence homology to propeptides of other subtilisin-like proteases, has previously been proposed to operate as a propeptide (7, 19). Following this is a predicted subtilisin domain which is attached to a predicted C-terminal transmembrane anchor by a ∼30-amino acid proline-rich linker.
Despite more than a decade of investigation, there are minimal data describing maturation events leading to MycP activity. In one study, M. tuberculosis MycP1tb expressed in infected macrophages was assayed by immunoblotting with the enzyme found at a lower apparent molecular mass after 6 weeks compared with samples taken from initial cell cultures; the authors speculated that this electrophoretic shift was related to loss of the putative propeptide (19). Recently, Ohol et al. reported that a recombinant maltose-binding protein fusion construct of M. smegmatis MycP1sm(24–407) is initially inactive (7). However, extended sample aging with Factor Xa resulted in a shortened species as determined by SDS-PAGE relative to the maltose-binding protein fusion as well as an apparent gain in promiscuous proteolytic activity against a range of fluorogenic tetrapeptide substrates (7). The authors attribute the shortened species to loss of the proposed prodomain; however, data showing specific cut sites to validate this hypothesis were not explicitly provided.
Toward clarifying these issues pertaining to MycP1 maturation in relation to subsequent proteolytic activity, we present the first crystallographic analysis of a mycosin protease, including the proposed propeptide region (which we will herein refer to as the “N-terminal extension”). MycP1sm from M. smegmatis shares 78% sequence identity to its counterpart in M. tuberculosis, making it a suitable model system for studying mycosins of pathogenic mycobacteria. Our structure shows that the N-terminal extension is not a conventional subtilisin foldase, but rather an extended proline-rich, disulfide-tethered appendage that interacts extensively with the subtilisin core. This observed N-terminal extension of MycP1 does not occupy the active site cleft in a manner typical of previously characterized propeptides, suggesting regulation of the catalytic domain through a novel mechanism. Further, we show that MycP1sm with the N-terminal extension intact is slowly active against the C-terminal region of purified EspB from both M. smegmatis and M. tuberculosis species and determine the conserved cut site using mass spectrometry and N-terminal sequencing.
MATERIALS AND METHODS
Expression and Purification of MycP1
The full MycP1sm gene was synthesized with codons optimized for expression in Escherichia coli (Biobasic). Residues 24–407 were cloned into pET28a(+) using the restriction-free method (20) and transformed into E. coli BL21 codon-plus RIPL-competent cells (Stratagene) for use in protein expression. BL21 E. coli cells expressing native His-tagged (MGSSHHHHHHSSGLVPRGSH) MycP1(24–407) were grown as described (21), and SeMet derivative MycP1(24–407) were grown in minimal media as described (22). Induction was initiated using 1 mm isopropyl 1-thio-β-d-galactopyranoside at A600 = 0.6 followed by growth for 20 h. After harvesting by centrifugation, cells were frozen after weighing resuspensions in a 1:1 volume of 2× lysis buffer (100 mm HEPES, pH 7.5, 1 m NaCl, 20 mm imidazole, and 1 mm TCEP). When needed, the pellet was thawed and diluted three times with IMAC buffer (50 mm HEPES, pH 7.5, 500 mm NaCl, 10 mm imidazole, 0.5 mm TCEP) (16) and lysed using an Avisten C5 homogenizer. Lysate was spun at 40,000 rpm for 1 h, and the supernatant was loaded onto a 1-ml HisPur IMAC column (Thermo, 88225) using an AKTA purifier FPLC system, washed with 10 column volumes of 25 mm imidazole, and eluted with 8 column volumes 500 mm imidazole without subsequent cleavage of the His tag. The eluate was immediately concentrated and injected onto a Superdex 75 XK 26/60 column equilibrated with 20 mm HEPES, pH 7.5, 150 mm NaCl, and 1 mm TCEP, and the fractions containing the center of the main UV peak immediately concentrated to 20 mg/ml and set up for crystallization. The N-terminal deletion constructs were created using the QuikChange protocol (Stratagene). Small scale protein purification of the MycP1 truncation mutants was carried out as described previously (21).
Crystallization, Structure Determination, and Analysis
MycP1(24–407) crystals grew from 0.2 m NaF, 25% PEG 3350 and reached maximum size after 1 week. Crystals were harvested and briefly washed in mother liquor containing 23% glycerol and rapidly cooled in liquid nitrogen before screening using a Rigaku RU200 rotating anode and Mar345db detector. SAD data were collected at the Canadian Light Source CMCF2 at a peak wavelength of 0.97895 Å and processed with imosflm and SCALA (23, 24). Phenix AutoSol (25) was used for phasing, with six selenium atoms found in the substructure solution (figure of merit = 0.37). The model was built and refined using Phenix and Coot (26). Data collection and refinement statistics are given in Table 1. The resultant structure served as a molecular replacement search model to solve a second, higher resolution dataset using crystals grown in 0.1 m Bistris propane, pH 6.77, 0.18 m sodium thiocyanate, and 26% PEG 3350, which were cryoprotected as described above. Both structures had one molecule in the asymmetric unit, and no significant model changes were observed despite the differing space groups in each (I222 versus P212121). The structure was analyzed and figures generated using UCSF Chimera (27). Active site cleft sizes for MycP1 and thermitase (PDB ID code 1THM) were determined with DogSiteScorer (28).
TABLE 1.
Data collection and refinement statistics
| 4KPG | 4J94 | |
|---|---|---|
| Crystal parameters | ||
| Phasing method | SeMet SAD (peak) | MR |
| Space group | I 2 2 2 | P 21 21 21 |
| Cell dimensions | ||
| a, b, c (Å) | 81.00, 94.08, 119.60 | 56.49, 77.29, 84.93 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Data collection | ||
| Resolution (Å) | 47.04-2.15 | 37.22-1.86 |
| Completeness (%) | 99.7 (99.0)a | 86.3 (73.03) |
| Redundancy | 4.8 (4.5) | 2.0 (2.0) |
| Wavelength | 0.97895 | 0.97895 |
| f′/f″ | 8.25/5.13 | |
| Rmerge | 0.036 (0.553) | 0.067 (0.179) |
| I/σI | 10.4 (2.7) | 7.50 (3.95) |
| Refinement | ||
| Resolution (Å) | 47.04-2.15 | 37.22-1.86 |
| Rwork/Rfreeb | 0.1690/0.2043 | 0.1553/0.1913 |
| No. reflections | 25,225 | 27,671 (2,296) |
| No. atoms | ||
| Protein | 2,945 | 2,738 |
| Ligand/ion | 0 | 0 |
| Water | 184 | 421 |
| B-factors (Å2) | ||
| Protein | 36.9 | 15.2 |
| Water | 41.9 | 31.8 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.007 | 0.006 |
| Bond angles (°) | 1.103 | 1.11 |
| Ramachandran values | ||
| Favored (%) | 97.4 | 98 |
| Allowed (%) | 100 | 100 |
a Values in parentheses are for the highest resolution shell.
b Fraction of reflections in test set = 0.1.
MycP1 and EspB Cleavage Assay
Full-length EspBtb(1–460) (Rv3881c) and EspBsm(1–520) (MSMEG_0076) were cloned into pET28a(+), and N-terminally His-tagged (MGSSHHHHHHSSGLVPRGSH) EspB was expressed in E. coli BL21 codon-plus RIPL cells grown and processed as described above for MycP1. Cells were harvested and lysed in 100 mm HEPES, pH 7.5, 500 mm NaCl and purified over HisPur nickel-nitrilotriacetic acid resin (Thermo, 88223) and further purified using a Superdex 75 column with 20 mm HEPES, pH 7.5, 150 mm NaCl as the elution buffer. MycP1 used for activity assays was purified as above except the buffer used in all stages was 20 mm MES, pH 6.0, 50 mm NaCl. MycP1 and EspB were incubated separately (for controls) and mixed together in 20 mm MES, pH 6.0, 50 mm NaCl at a concentration of 0.2 μg/μl in 50-μl reaction volumes and incubated at 37 ºC. 10-μl aliquots were withdrawn at the indicated time points. The samples were analyzed using SDS-PAGE.
In-gel Digestion and Mass Spectrometry Determination of EspB Cleavage Products
Protein bands were excised and in-gel digested with Trypsin Gold (Promega) as described (29). Eluted peptides were acidified with 0.5% formic acid and purified using C18 Stage tips (30) before analysis on a capillary liquid chromatography system (LC Packings; Dionex) coupled to a quadrupole time-of-flight mass spectrometer (QSTAR XL, Applied Biosystems, operated by the UBC Centre for Blood Research Mass Spectrometry Suite). Peptides were separated on a column packed with Magic C18 resin (Michrom Bioresources) using a 0–80% gradient of organic phase over 90 min. MS data were acquired automatically using the Analyst QS software, v1.1 (Applied Biosystems). Peptides were identified from a protein database containing the M. tuberculosis proteome database (Uniprot) with appended M. smegmatis MTB48, common laboratory contaminant sequences and a reverse sequence decoy database, using Mascot v.2.3 (Matrix Science). Search parameters included mass tolerances of 200 ppm for the parent ion and 0.4 Da for the fragment ions, trypsin, or semitrypsin as cleavage specificity with up to two missed cleavages, and carboxyamidomethylation of cysteine residues (+57.02 Da). To be accepted, spectrum-to-sequences matches required a Mascot ion score >25 for tryptic peptides and an ion score >38 for semi-tryptic peptides.
Edman Sequencing
Separated EspB cleavage fragments were transferred from SDS-PAGE gel to a PVDF membrane and stained with 0.1% (w/v) Coomassie Brilliant Blue R-250, 40% methanol, 10% acetic acid for 5 min. The membrane was destained in 40% methanol, 10% acetic acid and then rinsed for 30 s in 90% methanol, 5% acetic acid. The membrane was dried, and EspB cleavage products were cut out. The first six amino acids were determined by Edman sequencing at the Advanced Protein Technology Centre within the Hospital for Sick Children, Toronto.
Peptide Docking
MycP1 was aligned with subtilisin BPN′ (PDB ID code 2SNI) and the CI-2 chain merged into the MycP1 structure. Residues PVGTIVTMEYRIDR were changed to the identified EspBsm cut sequence DPSLGKPASAGGGGG and the remainder of CI-2 deleted. Hydrogens and missing side chains were added to the model which served as the input for FlexPepDock (31). 100 low resolution and 100 high resolution structures were generated, and the peptide with the lowest energy/RMSD was analyzed.
Differential Scanning Fluorometry and Circular Dichroism (CD)
Differential scanning fluorometry experiments were carried out using 0.2 mg/ml protein in 100 mm MES, pH 6.0, 50 mm NaCl with SYPRO Orange (5× final concentration; Invitrogen S6650). The mixture was monitored in MicroAmp Fast optical reaction plates (Applied Biosystems 4346906) using a 25-μl assay volume and an Applied Biosystems StepOnePlus RT-PCR system set to ROX (excitation, 488 nm; emission, 620 nm). CD spectra were acquired using 5 μm protein in 10 mm Tris, pH 7.5, 30 mm NaCl, 1 mm EDTA over wavelengths spanning 195–275 nm.
RESULTS
MycP1 with Its N-terminal Extension Intact Cleaves Two Purified EspB Homologues in the C-terminal Region in Vitro
We first analyzed MycP1sm (residues 24–407, with predicted N-terminal signal sequence and C-terminal transmembrane helix deleted) by mass spectrometry (±3-Da resolution) and measured the molecular mass to be that of the expected construct, indicating the proposed propeptide is stably attached (data not shown). To determine whether MycP1sm(24–407) is active in vitro against M. tuberculosis EspB (EspBtb) as previously described (7), we purified EspBtb and mixed it with MycP1sm at a molar ratio of 1:1 and conducted cleavage assays by observing electrophoretic shift on SDS-PAGE. EspBtb (consistently running as an ∼55-kDa band) migrated at lower molecular mass (∼48 kDa) following overnight incubation with MycP1 at 37 °C; this truncated band was not observed when EspB was incubated with the MycP1sm-S334A catalytic mutant (Fig. 1A). A doublet of two additional closely spaced lower molecular mass EspBtb bands was also produced (∼10 kDa). We further tested and confirmed MycP1sm activity against purified EspB from M. smegmatis (EspBsm) (Fig. 1B). In contrast to the ∼10-kDa doublet band observed for EspBtb, a single ∼12-kDa band was produced when EspBsm was cleaved. At an equimolar ratio under the assay conditions tested, MycP1 required >6 h incubation to completely cleave either EspB homologue (Fig. 1C). To determine where MycP1 cleaves EspBtb, the cleavage products, along with full-length EspB as a control, were excised from the gel and subjected to in-gel digestion by trypsin followed by LC-ESI-MS/MS analysis. Peptides from the large molecular mass band mapped to a discrete N-terminal region (residues 1–356), whereas peptides from the smaller two bands mapped primarily to the C-terminal region (358–460) (Fig. 1D). A similar peptide distribution was also observed in the LC-MS/MS analysis of the EspBsm cleavage products (data not shown). Thus, MycP1sm, with its N-terminal extension intact, cleaves two distinct EspB homologues in the C-terminal region.
FIGURE 1.

MycP1 cleavage of EspB. A, SDS-PAGE gel showing MycP1 cleavage of EspBtb, with the three EspB cleavage products indicated by asterisks. B, SDS-PAGE gel showing MycP1 cleavage of EspBsm, with two EspB cleavage products indicated by asterisks. C, SDS-PAGE gel showing MycP1-dependent EspB degradation over 6 h. D, LC-MS/MS analysis of uncleaved EspBtb compared with the three EspBtb cleavage products. Tryptic peptides (black) and semitryptic peptides in (gray) are mapped against the full EspB sequence (residues 1–460), indicated by the numbered upper black bar. E, EspBtb sequence surrounding the proposed cleavage sites with LC-ESI-MS/MS, Edman sequencing results, and proposed P residues mapped. F, multiple sequence alignment of EspB homologues with the residues involved in conferring specificity in subtilisin-like enzymes indicated by a box.
To locate the MycP1 cleavage site precisely, we repeated the spectrum-to-sequence assignment searches to include semitryptic peptides, i.e. peptides that had been cut by trypsin at one terminus only, which would suggest the remaining terminus resulted from MycP1 protease activity. Analysis of the middle (∼10-kDa) EspBtb cleavage product revealed the most N-terminal semitryptic peptide matching to EspBtb at residues 359–394, indicating a nontryptic cleavage between Ala358 and Ser359 (354AVKAA358↓359SLGGG363, Fig. 1E). Edman sequencing independently confirmed this site as the N terminus of the C-terminal EspBtb fragment, strongly suggesting MycP1 cleavage at this site. No semitryptic peptide could be identified for M. smegmatis EspBsm but Edman sequencing identified the N terminus of the C-terminal cleavage product at (402SLKPA406↓407SAGGG411). For both EspB homologues, the predicted molecular mass of fragments resulting from cleavage at these sites correlated well to their observed apparent molecular mass on the gel. Pairwise sequence alignment of EspBtb and EspBsm indicates this cleavage site is at a homologous location, immediately preceding a polyglycine stretch (Fig. 1F). Multiple sequence alignment indicates that these residues are similar across EspB species (Fig. 1F). According to protease nomenclature, substrate residues upstream of the cleaved scissile bond are denoted as P1–P6 in the sequence, and residues downstream of the scissile bond are termed P1′–P6′ (32). The binding of the P1 side chain to the corresponding S1 pocket of the enzyme is often deemed most critical in aiding in the optimal positioning of the adjacent substrate peptide carbonyl for nucleophillic attack, and thus greatest conservation is typically observed at this position. The P2′, P2, and P4 positions are also typically key specificity determinants in subtilisin-like protease substrate recognition. Thus, it is notable that residues comprising these positions in our identified EspBtb and EspBsm cleavage sites are identical or similar in chemical nature to each other (P4:[L/V], P3:[K], P2:[P/A], P1:[A], and P2′: [L/A]). Interestingly, a sequence resembling these cut sites (384VRPA387) is found 29 residues downstream in the identified EspBtb sequence and may indicate a second cleavage site, which would explain the doublet band we observe when EspBtb is cleaved. Duplex EspBtb cleavage has also been observed previously in the cellular context (7). Indeed, LC-MS/MS analysis of the lower EspBtb cleavage product identified an N terminus at Gly387, and N-terminal sequencing supports cleavage at this second VRPA cut site (Fig. 1E). Both EspBtb sites we identified at Ala358 and Ala386 are within the vicinity but differ from previously proposed cut sites suggested from experiments using cultures of M. tuberculosis and Mycobacterium marinum cells secreting EspB (3, 4, 7).
Overall Architecture of MycP1
To further understand MycP1 substrate specificity as well as the possible role of the N-terminal extension as a regulatory propeptide, we sought to determine its crystal structure. MycP1(24–407) crystallized in the orthorhombic space group P212121, and the structure was determined to 1.86 Å resolution (Table 1, PDB ID code 4J94). As predicted from sequence analysis, residues 62–390 of MycP1 adopts a subtilisin-like α/β-fold centered on a central 7-stranded parallel β-sheet (Fig. 2A), with a DALI (33) search yielding subtilisin BPN′ as its closest structural homologue (root mean square deviation of 0.965 Å over 186 common Cα atoms) (Fig. 2C). Thus, the canonical subtilisin nomenclature will be used to describe the structure (32). The N-terminal extension of MycP1(24–61) (proposed earlier to form the putative propeptide (7, 19)) is observed to be structurally and functionally distinct from typical subtilisin propeptides (32) in that it does not occupy the active site or have a characteristic foldase domain, which acts as a intramolecular chaperone for subtilisn-like enzymes (35). MycP1 also contains several other unique insertions localized around the active site (Fig. 2B). Part of the C-terminal proline-rich membrane linker (390–403) was resolved and is found hugging the subtilisin core in an extended conformation, passing adjacent to the protein N terminus. Coulombic electrostatic surface maps indicate that MycP1 is negatively charged over the majority of its surface, including the active site cleft. In contrast to other subtilisin-like proteases (32), MycP1 does not appear to bind calcium ions to promote structural integrity.
FIGURE 2.
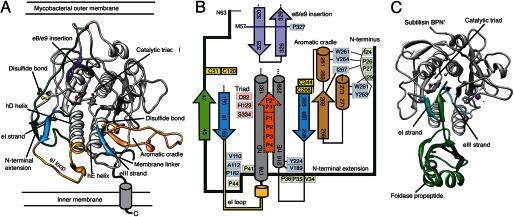
Overall structure of M. smegmatis MycP1. A, crystal structure of M. smegmatis MycP1(24–407) displayed as a ribbon representation, with key features labeled and highlighted by color. B, schematic of active site region illustrating how the unique MycP1 secondary structural features and selected conserved residues interact. Green, N-terminal extension; blue, eI/eIII substrate docking strands; yellow, eI loop; brown, aromatic cradle; purple, e8/e9 insertion; orange, substrate location. C, for comparison, the structure of subtilisin BPN′ with its propeptide foldase domain (PDB ID code 1SBP).
Catalytic Triad
In structures derived from two different space groups, the positioning of the MycP1 catalytic triad is unusual, namely, the side chain conformation of the nucleophile Ser334 is flipped and facing away from the presumed scissile bond location (Fig. 3A). Moreover, the phi angle (ϕ) for His123 in our MycP1 structure is −147°, compared with −60° in subtilisin BPN′. A survey of subtilisin-like enzymes of known structure reveal both ϕ/ψ torsion angles for the catalytic histidine are conserved in all other cases (ϕ = −58° ± 5.8 (S.D.), n = 22), making the MycP1 backbone atypical in this region (Fig. 3B). The increased planarity of this peptide bond alters the back bone scaffold, and in combination with other global structural changes, His123 Cα and its imidazole side chain are positioned ∼1.7 Å further from the active site cleft in comparison to subtilisin BPN′ (Fig. 3, C and D). The other two triad members, Ser334 and Asp92, maintain their hydrogen bonds with His123 (Ser334-Oγ–His123-Nϵ and Asp92-Oδ1–His123-Nδ, respectively), resulting in all three triad residues occupying positions further from the presumed scissile bond position, with perhaps the most important change being the Ser334 side chain adopting a conformation that in our structure appears suboptimal for nucleophilic attack, and a conformational change would be required to properly position the catalytic machinery. The oxyanion hole residue Asn239 appears positioned similarly as in other subtilisin-like enzymes.
FIGURE 3.

MycP1 catalytic triad. A, structural overlay of MycP1 (blue) and subtilisin BPN′ (PDB ID code 2SNI, tan) with catalytic triads displayed. B, Ramachandran plot indicating ϕ/ψ torsion angles for MycP1 and 22 subtilisin-like enzymes. C, differing ϕ angles and His/Ser geometries for MycP1/subtilisin BPN′. D, structural alignment of subtilisin-like enzymes with catalytic triad displayed. The MycP1 triad is highlighted in cyan. An acyl-enzyme structure of subtilisin Carlsberg (PDB ID code 1BE6) indicates the serine location and orientation required for nucleophilic attack. Previously published PDB files used in structural alignment were: 1BE6, 3TI9, 3T41, 3LPC, 3F7M, 3EIF, 2Z30, 2Z2Z, 2QTW, 2IXT, 2B6N, 1Y9Z, 1TO2, 1THM, 1SH7, 1R6V, 1R0R, 1CGI, 1DBI, 1BH6.
Active Site Cleft
The predicted MycP1 catalytic cleft is unusually large (volume = 1083 Å3) compared with typical subtilisin-like enzymes of known structure (e.g. thermitase = 373 Å3) (28) (Fig. 4A). This is in part due to a unique 18-residue insertion after the eI substrate binding strand (termed here the eI loop; Fig. 4B) that creates a wide, elongated groove via its interaction with the N-terminal extension (Fig. 4C). A second mycosin-specific insertion, the e8/e9 β-strands, juts over the cleft to form a “lid” over the catalytic triad. To locate putative subsites (denoted by S) we used Rosetta FlexPepDock (31) to model the newly identified M. smegmatis recognition motif (SLKPASAGG) into the cleft of MycP1sm (Fig. 4D). As mentioned above, P1–P4 and P2′ are typically the most important determinants for specificity in subtilisin-like enzymes. The conserved P1 Ala and P2 Pro side chains are complementary to their respective subsites and contact Ala204 (Cβ) and Thr156 (Cγ), respectively. MycP1 appears to have a strong preference for Lys/Arg at P3, unusual in that this side chain is typically solvated and not a major contributor to specificity in most subtilisin-like enzymes (32). However, MycP1 has evolved a disulfide-stabilized loop that protrudes over top of the cleft to scaffold Asp243, creating a negatively charged S3 site that is optimally positioned to interact with the positively charged P3 side chain. The S2′ site is very similar to that of subtilisin BPN′, an apolar pocket created by Phe292 that could accommodate a hydrophobic residue, and is consistent with the Leu/Ala identified in our cleavage site. Although P4 is [LV] and should reasonably bind to a hydrophobic pocket, an obvious S4 subsite was not evident in the docking experiment. Of final note, the eI/eIII substrate-docking strands that typically orient the peptide substrate for catalysis (32) are separated by ∼15 Å in MycP1 (Fig. 4E) and appear incapable of forming such a triple-stranded ES complex without a conformational change/induced fit mechanism that would bring these active site elements into sufficiently close proximity. Taken together, the MycP1sm S2′, S1, S2, and S3 subsites in the structure appear complementary to the P2′, P1, P2, and P3 residues identified in the EspBsm cleavage site; however, it is possible that additional conformational changes may be needed to optimize the β-strand interactions and formation of downstream subsites involved in binding substrate and subtle repositioning of catalytic groups to promote optimal catalysis.
FIGURE 4.

Active site cleft of MycP1. A, MycP1 and thermitase (PDB ID code 1THM) active site cleft regions, as predicted by DogSiteScorer (27), are shown. The catalytic triad is colored green. B, the eI loop and hD helix are unusually long compared with nearly all other subtilisin-like enzymes; the structural alignment of subtilisin-like enzymes highlights how these features are typically conserved in length and position. The outliers are C5a peptidase (red, 3EIF) and MycP1 (green). Note how the eI substrate docking strand is unusually positioned. C, the N-terminal segment (green) and eI loop (gold) interact to form one side of the cleft, the docked EspB peptide (orange) indicates the cleft. D, a docked peptide corresponding to the EspBsm recognition sequence is shown (orange) with P and S sites indicated. E, MycP1 eI/eIII substrate docking strand spacing (left) compared with subtilisin BPN′ (right). Previously published PDB files used in structural alignment were: 3AFG, 3D43, 3EIF, 3BX1, 3F7M, 3QFH, 2ID4, and 3HJR.
N-terminal Extension
The unusual N-terminal extension of MycP1 (residues 24–61) follows a securely anchored extended path, wrapping along the surface of the subtilisin domain (Fig. 5). At the extreme N terminus, a diproline motif (residues 24–29) sits in an aromatic cradle of MycP1 formed by conserved residues Trp272, Trp261, Trp298, and Tyr273. This cradle is the result of an insertion (between the e5/e6 strands) composed of two short 310 helices flanking a β-strand, that effectively creates an outcropping from the catalytic domain (Fig. 5B). The two proline side chains of the N-terminal motif stack favorably onto the tryptophan indole rings (Fig. 5B). This region of the N-terminal extension adopts a polyproline II helix, with characteristic 120° spacing between residues imposed by the conformational rigidity of the proline residues (36). Its interaction with the aromatic cradle is reminiscent of proline recognition motifs of the WW family (37). The N-terminal extension passes underneath the hD/hE active site helices where a second diproline motif forms another polyproline II helix and is anchored by interactions with a patch of hydrophobic amino acids (Fig. 5C). The N-terminal extension is further bound through a series of electrostatic interactions, β-sheet interactions with the eI loop and tethering by the Cys51-Cys120 disulfide bond (Fig. 5A). Generally, the N-terminal extension is rich in prolines, which appear required to facilitate its extended path around the subtilisin domain of MycP1. Sequence alignment indicates that this is likely a feature common to the entire mycosin family with the upstream end exhibiting more conservation than the downstream portion of the N-terminal extension (Fig. 5D). We observed that MycP1 was expressed and folded (albeit with reduced solubility) even with up to 33 of 40 N-terminal extension residues removed, suggesting that its primary function is unrelated to folding but may contribute to stability (Fig. 5, D and E). Differential scanning fluorometry comparing MycP1(24–407) and MycP1(49–407) showed a 7 °C difference in melting temperature (Tm = 64 °C versus 57 °C), indicating that the N-terminal extension contributes to overall stability (Fig. 5F).
FIGURE 5.
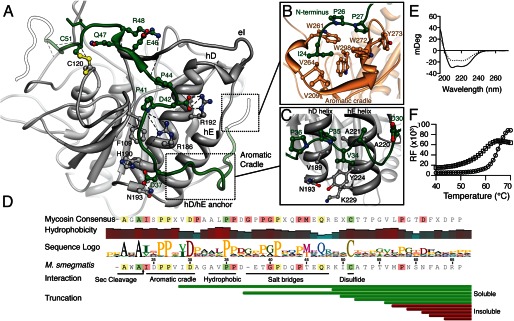
Interactions between the N-terminal extension and subtilisin domain. A, overview of the N-terminal extension path emphasizing electrostatic interaction, β-sheet formation with the eI loop, and the Cys51-Cys120 disulfide bond. B, the N-terminal region of MycP1 adopting a polyproline helix that binds to a proline recognition motif-like aromatic cradle. The IDPP stretch interacts with a triple-tryptophan arrangement, where the proline rings pack parallel to two of the tryptophan residues. Ile24 and Ile29 pack into adjacent hydrophobic pockets. C, N-terminal extension forming a second polyproline II helix that binds to the base of the hD and hE helices. Asp30 is strictly conserved and faces the solvent. D, N-terminal extension sequence conservation with the mycosin consensus. A hydrophobicity plot shows four distinct hydrophobic patches on the N-segment (sliding window average = 3). The N-terminal portion is more highly conserved across all mycosins than C-terminal portion. Results of expression of N-terminal extension truncations are shown as soluble (green bars) or insoluble (red bars). E, circular dichroism of MycP1(24–407) (solid line) and the N-terminal segment truncation mutant MycP1(49–407) (dashed line). F, differential scanning fluorometry experiment of MycP1(24–407) (circles) and MycP1(49–407) (squares).
DISCUSSION
MycP1(24–407) was crystallized with a native catalytic triad that failed to hydrolyze the proposed prodomain, indicating this full-length enzyme is highly stable over the course of the purification procedure and crystallization experiment. In contrast to subtilisin prodomains, which form a typical foldase fold that occupies the active site within reach of the Ser-His-Asp catalytic triad, the structurally distinct MycP1 N-terminal extension is not bound within the cleft and is >13 Å away from the Ser334 nucleophile. This rules out the possibility of autoproteolytic cleavage of this region in cis, barring conformational rearrangement. Crystallization of wild type subtilisin-like enzymes with their uncleaved prodomains is rare (a catalytically impaired form is typically required), the only other example to our knowledge is the proprotein convertase PCSK9 (38). Further, our mass spectrometry data showing MycP1, with its N-terminal extension intact, is able to cleave EspB (albeit slowly) at a [L/V]K[P/A]A ↓ S[L/A]GG site adds further support that this appendage is not a propeptide. Thus, it is possible that the N-terminal extension may not be cleaved but may modulate proteolysis in response to an appropriate trigger. This mode of operation may be comparable with the intracellular subtilisin proteases, which have a short N-terminal extension that operates through a combined active site blocking/catalytic triad rearrangement mechanism (39). Another possibility is that cleavage of the N-terminal extension may occur in a cellular context, but the appendage could remain anchored to the subtilisin domain through the disulfide bond (e.g. as with chymotrypsin (17)), which could help explain previous reports of activation resulting from addition of Factor Xa protease (7). We have shown that the majority of the N-terminal extension is not required for folding, but does contribute to overall stability. Given that the extension interacts with several features surrounding the active site (the eI strand (residues 159–163), aromatic cradle (residues 261–274), the Cys51-Cys120 disulfide bond, and e8/e9 extension (residues 320–328), we suspect a role in modulating catalysis or conferring specificity. Another possibility is that the N-terminal extension is serving as a placeholder in obstructing binding surfaces, and if removed, these surfaces would have the potential to target substrates or other components of the secretion system, perhaps through the same proline recognition motifs which anchor the uniquely pro-rich N-terminal extension in the structure.
As MycP1 is capable yet inefficient at cleaving EspB in vitro, we hypothesize the MycP1 structure presented here is an isolated T7SS component in its preassembly form and could still represent a zymogen with “leaky” activity. Given that the previous in vitro fluorogenic substrate profiling data indicated that an uncharacterized form of MycP1 is capable of catalysis with broad specificity (7), it is unsurprising that MycP1 is synthesized with the intrinsic capacity to prevent rapid premature proteolysis, which could cause damaging nonspecific cleavage. It is well understood that nucleophilic cleavage of peptide bonds requires a precisely defined geometric arrangement (40). Our analysis of the MycP1 native catalytic triad allows us to propose one possible mechanism by which a subtle conformational change, for example through N-terminal segment cleavage or binding to a cofactor or binding partner, could twist the His123 ϕ torsion angle and Ser334 side chain into positions more similar to that of catalytically competent proteases. MycP1 may have extrinsically low activity in the absence of unknown cofactors including the lipidic membrane environment, other ESX components such as the predicted EccBCDE ATPase-translocon complex, or as-of-yet uncharacterized components of the extracellular portion of the secretion apparatus. Perhaps an undefined temporal trigger mechanism may ensure proteolysis only occurs as ESX substrates are crossing the membrane.
Due to its essential action in virulence and relatively accessible nature, MycP1 is an attractive extracellular anti-tuberculosis drug target. Additionally exciting is the mycosin potential as an engineerable factor in rational design of more effective tuberculosis vaccines due to its influence on processing and presumably the downstream function of highly antigenic/virulence-associated EspB and the PE/PPE protein families (7). Indeed, manipulation of T7SS have driven major advancements in mycobacterial vaccinology (34, 41), and we have provided the first structural template for engineering vaccine strains that perturb the influence of this protease on regulation, secretion, and processing of secreted tuberculosis antigens.
Acknowledgments
We thank the staff at the Canadian Light Source beamline CMCF-1 for assistance with data collection, the Canadian Foundation of Innovation, the British Columbia Knowledge Development Fund for infrastructure funding, Marija Vuckovic for technical aid, and Julien Bergeron and Antonio Ruzzini for helpful discussion.
This work was supported in part by operating grants from the Canadian Institute of Health Research and the Howard Hughes Medical Institute International Senior Scholar Program.
The atomic coordinates and structure factors (codes 4J94 and 4KPG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- T7SS
- type VII secretion system
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- ESI
- electrospray ionization
- Esp
- ESX-secreted protein
- MS/MS
- tandem MS
- MycP1
- mycosin-1 protease
- PDB
- Protein Data Bank
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1. Abdallah A. M., Gey van Pittius N. C., Champion P. A., Cox J., Luirink J., Vandenbroucke-Grauls C. M., Appelmelk B. J., Bitter W. (2007) Type VII secretion: mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 [DOI] [PubMed] [Google Scholar]
- 2. Fortune S. M., Jaeger A., Sarracino D. A., Chase M. R., Sassetti C. M., Sherman D. R., Bloom B. R., Rubin E. J. (2005) Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U.S.A. 102, 10676–10681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu J., Laine O., Masciocchi M., Manoranjan J., Smith J., Du S. J., Edwards N., Zhu X., Fenselau C., Gao L. Y. (2007) A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol. Microbiol. 66, 787–800 [DOI] [PubMed] [Google Scholar]
- 4. McLaughlin B., Chon J. S., MacGurn J. A., Carlsson F., Cheng T. L., Cox J. S., Brown E. J. (2007) A Mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 3, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Millington K. A., Fortune S. M., Low J., Garces A., Hingley-Wilson S. M., Wickremasinghe M., Kon O. M., Lalvani A. (2011) Rv3615c is a highly immunodominant RD1 (region of difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U.S.A. 108, 5730–5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raghavan S., Manzanillo P., Chan K., Dovey C., Cox J. S. (2008) Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454, 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohol Y. M., Goetz D. H., Chan K., Shiloh M. U., Craik C. S., Cox J. S. (2010) Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 7, 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coros A., Callahan B., Battaglioli E., Derbyshire K. M. (2008) The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol. Microbiol. 69, 794–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siegrist M. S., Unnikrishnan M., McConnell M. J., Borowsky M., Cheng T. Y., Siddiqi N., Fortune S. M., Moody D. B., Rubin E. J. (2009) Mycobacterial ESX-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. U.S.A. 106, 18792–18797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdallah A. M., Verboom T., Hannes F., Safi M., Strong M., Eisenberg D., Musters R. J., Vandenbroucke-Grauls C. M., Appelmelk B. J., Luirink J., Bitter W. (2006) A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol. Microbiol. 62, 667–679 [DOI] [PubMed] [Google Scholar]
- 11. Abdallah A. M., Verboom T., Weerdenburg E. M., Gey van Pittius N. C., Mahasha P. W., Jiménez C., Parra M., Cadieux N., Brennan M. J., Appelmelk B. J., Bitter W. (2009) PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol. Microbiol. 73, 329–340 [DOI] [PubMed] [Google Scholar]
- 12. Bitter W., Houben E. N., Bottai D., Brodin P., Brown E. J., Cox J. S., Derbyshire K., Fortune S. M., Gao L. Y., Liu J., Gey van Pittius N. C., Pym A. S., Rubin E. J., Sherman D. R., Cole S. T., Brosch R. (2009) Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 5, e1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Houben E. N., Bestebroer J., Ummels R., Wilson L., Piersma S. R., Jiménez C. R., Ottenhoff T. H., Luirink J., Bitter W. (2012) Composition of the type VII secretion system membrane complex. Mol. Microbiol. 86, 472–484 [DOI] [PubMed] [Google Scholar]
- 14. Converse S. E., Cox J. S. (2005) A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J. Bacteriol. 187, 1238–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daleke M. H., Cascioferro A., de Punder K., Ummels R., Abdallah A. M., van der Wel N., Peters P. J., Luirink J., Manganelli R., Bitter W. (2011) Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J. Biol. Chem. 286, 19024–19034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cascioferro A., Delogu G., Colone M., Sali M., Stringaro A., Arancia G., Fadda G., Palù G., Manganelli R. (2007) PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol. Microbiol. 66, 1536–1547 [DOI] [PubMed] [Google Scholar]
- 17. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown G. D., Dave J. A., Gey van Pittius N. C., Stevens L., Ehlers M. R., Beyers A. D. (2000) The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene 254, 147–155 [DOI] [PubMed] [Google Scholar]
- 19. Dave J. A., Gey van Pittius N. C., Beyers A. D., Ehlers M. R., Brown G. D. (2002) Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol. 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van den Ent F., Löwe J. (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 [DOI] [PubMed] [Google Scholar]
- 21. Structural Genomics Consortium, China Structural Genomics Consortium, Northeast Structural Genomics Consortium, Gräslund S., Nordlund P., Weigelt J., Hallberg B. M., Bray J., Gileadi O., Knapp S., Oppermann U., Arrowsmith C., Hui R., Ming J., dhe-Paganon S., Park H. W., Savchenko A., Yee A., Edwards A., Vincentelli R., Cambillau C., Kim R., Kim S. H., Rao Z., Shi Y., Terwilliger T. C., Kim C. Y., Hung L. W., Waldo G. S., Peleg Y., Albeck S., Unger T., Dym O., Prilusky J., Sussman J. L., Stevens R. C., Lesley S. A., Wilson I. A., Joachimiak A., Collart F., Dementieva I., Donnelly M. I., Eschenfeldt W. H., Kim Y., Stols L., Wu R., Zhou M., Burley S. K., Emtage J. S., Sauder J. M., Thompson D. (2008) Protein production and purification. Nat. Methods 5, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Larsson A. M. (2009) in Protein Crystallization, (Bergfors T. M., ed) 2nd Ed., pp. 137–154, International University Line, La Jolla, CA [Google Scholar]
- 23. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 28. Volkamer A., Kuhn D., Grombacher T., Rippmann F., Rarey M. (2012) Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 52, 360–372 [DOI] [PubMed] [Google Scholar]
- 29. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 30. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 31. London N., Raveh B., Cohen E., Fathi G., Schueler-Furman O. (2011) Rosetta FlexPepDock web server: high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 39, W249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siezen R. J., Leunissen J. A. (1997) Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6, 501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sweeney K. A., Dao D. N., Goldberg M. F., Hsu T., Venkataswamy M. M., Henao-Tamayo M., Ordway D., Sellers R. S., Jain P., Chen B., Chen M., Kim J., Lukose R., Chan J., Orme I. M., Porcelli S. A., Jacobs W. R., Jr. (2011) A recombinant Mycobacterium smegmatis induces potent bactericidal immunity against Mycobacterium tuberculosis. Nat. Med. 17, 1261–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shinde U., Inouye M. (1995) Folding mediated by an intramolecular chaperone: autoprocessing pathway of the precursor resolved via a substrate assisted catalysis mechanism. J. Mol. Biol. 247, 390–395 [DOI] [PubMed] [Google Scholar]
- 36. Ball L. J., Kühne R., Schneider-Mergener J., Oschkinat H. (2005) Recognition of proline-rich motifs by protein-protein-interaction domains. Angew Chem. Int. Ed. Engl. 44, 2852–2869 [DOI] [PubMed] [Google Scholar]
- 37. Zarrinpar A., Lim W. A. (2000) Converging on proline: the mechanism of WW domain peptide recognition. Nat. Struct. Biol. 7, 611–613 [DOI] [PubMed] [Google Scholar]
- 38. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 39. Vévodová J., Gamble M., Künze G., Ariza A., Dodson E., Jones D. D., Wilson K. S. (2010) Crystal structure of an intracellular subtilisin reveals novel structural features unique to this subtilisin family. Structure 18, 744–755 [DOI] [PubMed] [Google Scholar]
- 40. Dodson G., Wlodawer A. (1998) Catalytic triads and their relatives. Trends Biochem. Sci. 23, 347–352 [DOI] [PubMed] [Google Scholar]
- 41. Pym A. S., Brodin P., Majlessi L., Brosch R., Demangel C., Williams A., Griffiths K. E., Marchal G., Leclerc C., Cole S. T. (2003) Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 9, 533–539 [DOI] [PubMed] [Google Scholar]