

Background: Eukaryotic elongation factor-2 kinase (EF2K) inhibits the elongation phase of protein translation.
Results: EF2K degradation by the ubiquitin-proteasome system (UPS) is regulated by cAMP-PKA signaling and SCFβTRCP.
Conclusion: Degradation of EF2K enables coordination of UPS function and translational control.
Significance: This coordination may be important for achieving proper protein expression to effect cellular adaptations, including synaptic plasticity.
Keywords: Cyclic AMP (cAMP), E3 Ubiquitin Ligase, Proteasome, Translation Control, Translation Elongation Factors, βTRCP, eEF-2, eEF-2 Kinase
Abstract
Protein translation and degradation are critical for proper protein homeostasis, yet it remains unclear how these processes are dynamically regulated, or how they may directly balance or synergize with each other. An important translational control mechanism is the Ca2+/calmodulin-dependent phosphorylation of eukaryotic elongation factor-2 (eEF-2) by eukaryotic elongation factor-2 kinase (EF2K), which inhibits elongation of nascent polypeptide chains during translation. We previously described a reduction of EF2K activity in PC12 cells treated with NGF or forskolin. Here, we show that both forskolin- and IGF-1-mediated reductions of EF2K activity in PC12 cells are due to decreased EF2K protein levels, and this is attenuated by application of the proteasome inhibitor, MG132. We further demonstrate that proteasome-mediated degradation of EF2K occurs in response to A2A-type adenosine receptor stimulation, and that activation of protein kinase A (PKA) or phospho-mimetic mutation of the previously characterized PKA site, Ser-499, were sufficient to induce EF2K turnover in PC12 cells. A similar EF2K degradation mechanism was observed in primary neurons and HEK cells. Expression of a dominant-negative form of Cul1 in HEK cells demonstrated that EF2K levels are regulated by an SCF-type ubiquitin E3 ligase. Specifically, EF2K binds to the F-box proteins, βTRCP1 and βTRCP2, and βTRCP regulates EF2K levels and polyubiquitylation. We propose that the proteasomal degradation of EF2K provides a mechanistic link between activity-dependent protein synthesis and degradation.
Introduction
Mounting the proper cellular response to stimulation requires precise temporal and spatial control of protein abundance. Following gene transcription, this precision is achieved largely via translation of mRNAs into proteins and the degradation of existing proteins by the ubiquitin-proteasome system (UPS).2 Application of either ribosomal or proteasomal inhibitors impairs many types of lasting cellular changes, notably long-term synaptic plasticity and memory (1–4). In the case of long-term potentiation (LTP), concomitant application of both classes of inhibitors rescues the impairments caused by application of either drug alone (5, 6). These findings imply that a balance or coordination of protein synthesis and degradation, rather than engagement of either process per se, may be especially important in control of neuronal plasticity; however the mechanisms by which this coordination is achieved remain to be elucidated.
Eukaryotic elongation factor-2 kinase (EF2K), formerly termed CaMKIII, is a Ca2+/calmodulin-dependent protein kinase that inhibits the elongation phase of translation by phosphorylating eukaryotic elongation factor-2 (eEF-2) at Thr-56. Following peptide bond formation, eEF-2 catalyzes the translocation of the ribosome along the mRNA (7, 8), a function in which phospho-eEF-2 is impaired. The Ca2+-dependence of EF2K enables it to transduce the activation of various receptors in diverse cell types into changes in protein synthesis with high sensitivity and temporal and spatial specificity (9–12). In addition to its regulation by Ca2+, EF2K activity is modulated bi-directionally by phosphorylation by several kinases, including protein kinase A (PKA), AMP-regulated kinase (AMPK), p70 S6 kinase, and p90 RSK (13–17). Thus, regulation of EF2K acts to exert translational control downstream of diverse extracellular stimuli and signal transduction pathways.
Previously, we have shown that stimulation of PC12 cells with nerve growth factor (NGF) or the adenylyl cyclase-activating drug, forskolin (Fsk), decreases EF2K activity (18, 19). These studies, combined with the observation that EF2K is regulated by the UPS in other cell types (20, 21), led us to hypothesize that this reduction of EF2K activity results from its phosphorylation-dependent polyubiquitylation and degradation. Here, we show that EF2K is degraded in a proteasome-dependent manner in response to cAMP elevation in PC12 cells, cortical neurons, and HEK cells. This occurs via a mechanism involving activation of PKA, phosphorylation of Ser-499, and recruitment of the ubiquitin E3 ligase, SCFβTRCP. These data suggest that cAMP-dependent EF2K degradation serves as a focal point to effect a coordination between UPS function and translational control.
EXPERIMENTAL PROCEDURES
Immunoblotting and Antibodies
Cell lysates were diluted with 5× sample buffer, and samples were run on pre-cast 4–12% polyacrylamide Tris-glycine gradient gels (Invitrogen), except for ubiquitylation assays, wherein 6% gels were used. Gels were transferred to nitrocellulose membranes (Bio-Rad) and blocked with 5% milk in PBS. Membranes were incubated with primary antibodies at 4 °C overnight. All antibodies were diluted in a 1:1 mixture of Odyssey blocking buffer (Licor) and PBS with 1% Tween-20. The primary antibodies used were: rabbit anti-EF2K (Angus Nairn, 1:500), rabbit anti-p-eEF-2 Thr-56 (Cell Signaling, 1:1000), mouse anti-GAPDH (Advanced Immuno Chemicals, 1:10000), mouse anti-FLAG (Sigma, 1:1000), rabbit anti-GFP (AbCam, 1:10,000), mouse anti-GST (Cell Signaling, 1:1000), rabbit anti-p-tyrosine hydroxylase (1:1000, Cell Signaling), mouse anti-p-Akt Ser-473 (Cell Signaling, 1:1000), rabbit anti-Akt (Cell Signaling, 1:1000), mouse anti-p-ERK1/2 Thr-202/Tyr-204 (Cell Signaling, 1:1000), or rabbit anti-ERK1/2 (Cell Signaling, 1:1000). Membranes were then washed three times with PBS-T, incubated for 1 h at 4 °C with IRDye-800-conjugated goat anti-mouse (Rockland, 1:5000) and IRDye-680-conjugated goat anti-rabbit (Licor, 1:5000), and washed three more times with PBS-T. Blots were imaged with a Licor Odyssey Infrared Scanner, and quantification was performed using Licor Odyssey software. When necessary, membranes were stripped using a solution containing 25 mm glycine and 2% SDS, pH 2.0, at 55 °C. Upon confirmation that the signal had been removed, stripped membranes were re-blocked, and re-probed with primary and secondary antibodies. In all cases, data shown for EF2K levels were normalized to GAPDH levels measured in the same sample.
Cell Culture
For PC12 cells (ATCC), 6- or 12-well plates were coated with a solution of 0.05 mg/ml of collagen-I from rat tail (Sigma) in 0.02 N acetic acid for at least 1 h and washed with PBS prior to plating. Cells were grown in RPMI 1640, supplemented with 10% horse serum and 5% FBS (all from Invitrogen). HEK 293-T cells (ATCC) were grown on un-coated plates in DMEM (Invitrogen), supplemented with 10% FBS.
Stimulation of PC12 Cells or HEK Cells
PC12 cells were transferred to serum-free media 1 h prior to the addition of any reagents. MG132 (Tocris, 25 μm in DMSO) was added 30 min prior to the start of stimulation. Cells were stimulated with either forskolin (Tocris, 10 μm in DMSO), IGF-1 (Millipore, 100 ng/ml in water), CGS 21680 hydrochloride (Tocris, 100 nm in DMSO), or 6-BNZ-cAMP (Sigma, 100 μm in water) for up to 48 h and lysed in buffer containing 1% Nonidet P-40, 50 mm Tris, pH 7.4, 200 mm NaCl, 1 mm EDTA, and protease and phosphatase inhibitor cocktails (Sigma, 1:100). For protein turnover experiments, cells were treated with cycloheximide (Chx, Sigma, 30 μg/ml in DMSO) in serum-containing media and lysed at the indicated times. Stimulation experiments in HEK cells were performed 48 h post-transfection. Cells were transferred to media containing serum and cycloheximide (30 μg/ml) in the presence or absence of forskolin (10 or 50 μm) and lysed at the indicated times.
Culture and Stimulation of Primary Cortical Neurons
Cortical cultures were prepared from rat embryos according to standard protocols. All procedures were approved by the Yale Animal Care and Use Committee (YACUC) and followed the NIH Guide for the Care and Use of Animals. Pregnant rats (E18) were euthanized with CO2 asphyxiation. Embryos were isolated and brains dissected and bathed in Hank's Balanced Salt Solution (HBSS) containing 1% penicillin-streptomycin (both from Invitrogen). Cortices were isolated and digested at 37 °C in papain (Worthington), diluted 1:500 in the dissection solution, for 1 h, during which time the solution was twice triturated 20–30 times. The digested tissue was collected by centrifugation (5 min at 100 × g), the papain solution was removed, and the cells were resuspended in plating media consisting of Neurobasal supplemented with B27, Glutamax, sodium pyruvate, 1 mm HEPES, 1% penicillin-streptomycin, and 10% FBS (all from Invitrogen). Cells were counted in Trypan Blue using a hemocytometer, and 0.7 × 106 cells were plated per well on poly-l-lysine-coated 6-well plates. On the following day, the media was replaced with Neurobasal containing all of above supplements except FBS. Neurons were maintained in this media and experiments were performed after 14–15 days in vitro. Forskolin (10 μm) and BDNF (Millipore, 100 ng/ml in water) were added to the conditioned media for 3 or 6 h, and cells were lysed as above. MG132 (25 μm) was added 30 min prior to the start of stimulation.
Plasmids and Transfection
GST-F-box, FLAG-DN-Cul1, and shβTRCP constructs were generous gifts from J. W. Harper. The GFP-EF2K construct was generated by sub-cloning rat EF2K into pEGFP-N1 (Clontech) and point mutants were generated using standard PCR procedures and validated by DNA sequencing. For transfections, DNA was incubated with Lipofectamine 2000 (Invitrogen) in Opti-MEM media (Invitrogen) and added to cells. Protein expression was examined 24–48 h later.
GST Pull-downs
HEK cells were transfected with GFP-EF2K and GST-tagged F-box constructs and lysed in Nonidet P-40 buffer as above. Lysates were cleared by centrifugation (14,000 rpm for 20 min), and protein concentration was assayed by BCA (Pierce). For each pull-down, 2 mg of protein was used, diluted in 1 ml of lysis buffer, and 50 μl of glutathione-Sepharose beads (50% slurry, GE Healthcare) were added to the lysates, which were rotated for 30 min at 4 °C. The beads were washed three times with lysis buffer, and bound proteins were eluted by boiling in 100 μl of 2× SDS-PAGE sample buffer.
EF2K Ubiquitylation Experiments
PC12 cells were transfected with GFP-EF2K, with or without GST-βTRCP-1 and treated with MG132 (25 μm) for 2 h to accumulate polyubiquitylated proteins. Lysates were boiled in 1% SDS for 10 min to both inactivate de-ubiquitylating enzymes and disociate any ubiquitylated contaminates that could otherwise co-immunoprecipitate with EF2K. The lysates were diluted to 0.1% SDS prior to the addition of monoclonal GFP antibodies (19C8 and 19F7 from Memorial Sloan-Kettering), and were rotated overnight at 4 °C. Protein G beads (50% slurry, GE Healthcare) were added, and the mixture was rotated for 1 h at 4 °C. The beads were washed once each in a series of stringent buffers: 1) 10 mm Tris, pH 8.0, 500 mm NaCl, 0.5% Nonidet P-40, 0.05% SDS; 2) 10 mm Tris, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40, 0.05% SDS, 0.5% deoxycholate; 3) 10 mm Tris, pH 8.0, 0.05% SDS (22). Bound proteins were eluted by boiling in 2× SDS-PAGE sample buffer.
Statistics
Data were analyzed by one-sample t test, one-way ANOVA with Tukey's post-hoc tests, or two-way ANOVA with Bonferroni or Tukey's post-hoc tests, as appropriate. Significance was considered to be p < 0.05. All data are representative of at least three independent experiments. All graphs are displayed with error bars representing the standard error of the mean.
RESULTS
Proteasome-dependent Degradation of EF2K during Treatment of PC12 Cells with Forskolin or IGF-1
To test whether the reduced EF2K activity that we previously observed during cAMP elevation in PC12 cells is caused by proteasomal degradation of EF2K, we treated PC12 cells for 6 h with forskolin (Fsk, 10 μm) in the absence or presence of the proteasome inhibitor, MG132 (25 μm). Fsk significantly reduced EF2K protein levels relative to control (49.38 ± 6.17, n = 7), and caused a corresponding decrease in eEF-2 phosphorylation at Thr-56 (Fig. 1, A and B). Building on our earlier results with NGF, we found that IGF-1 treatment (100 ng/ml) for 6 h also reduced EF2K protein levels (62.46 ± 7.24, n = 4) and eEF-2 phosphorylation (Fig. 1, A and B). Both the Fsk- and IGF-1-mediated decreases in EF2K levels were blocked by co-application of MG132 (Fsk+MG132: 84.53 ± 6.187, n = 5; IGF-1+MG132: 93.47 ± 4.35, n = 2), indicating they are proteasome-dependent. Interestingly, while co-application of Fsk and MG132 restored p-eEF-2 to control levels, co-application of IGF-1 and MG132 decreased p-eEF-2 relative to control samples, which may be caused by the accumulation of EF2K that has been inhibited by phosphorylation by p70 S6 kinase (16).
FIGURE 1.

Stimulation of PC12 cells with Fsk or IGF-1 causes proteasome-dependent degradation of EF2K. A, PC12 cells were stimulated for 6 h with Fsk (10 μm) or IGF-1 (100 ng/ml) with or without MG132 (25 μm), and EF2K and GAPDH, and eEF-2 phosphorylation levels were assessed by SDS-PAGE and immunoblot. B, graph of summary data. A two-way ANOVA revealed significant main effects of stimulation and MG132, as well as a significant interaction between stimulation and MG132 treatment. Bonferroni post-hoc tests revealed that both Fsk and IGF-1 treatment reduced EF2K levels relative to control (Fsk: 49.38 ± 6.17, n = 7; IGF-1: 62.46 ± 7.24, n = 4, ***, p < 0.001), and this effect was blocked by the addition of MG132 (Fsk+MG132: 84.53 ± 6.187, n = 5; IGF-1+MG132: 93.47 ± 4.35, n = 2). C, PC12 cells were stimulated with Fsk (10 μm) in the presence of Chx (30 μg/ml) for 0–6 h and EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. D, graph of summary data. Dotted lines represent single exponential decay curve fits of the data. A two-way ANOVA revealed a significant main effect of Fsk treatment on the degradation of EF2K (***, p < 0.0001).
To confirm that Fsk causes degradation of EF2K per se, rather than activating a proteasome-dependent process that decreases EF2K levels by another means (e.g. reduced transcription or translation), we performed a chase experiment in PC12 cells to selectively examine the effect of Fsk on EF2K stability in the presence of the protein synthesis inhibitor, cycloheximide. We found that Fsk stimulation significantly hastened EF2K degradation (Fig. 1, C and D), shortening its half-life from 4.3 h to 2.0 h. Together, these data indicate that EF2K itself is targeted for proteasomal degradation in response to Fsk or IGF-1 stimulation.
We examined whether a more physiological means of increasing cAMP- activation of the Gs-coupled adenosine A2A receptor also induced EF2K degradation. PC12 cells were treated for 3 or 6 h with the A2A receptor agonist, CGS 21680 (100 nm), in the presence or absence of MG132. CGS 21680 application for either 3 or 6 h significantly reduced EF2K levels relative to control (CGS 3 h: 65.95 ± 9.89, n = 3; CGS 6 h: 49.31 ± 12.02, n = 3) and concomitantly increased PKA activity, as assessed by phosphorylation of TH at Ser-40 (Fig. 2, A and B). The addition of MG132 significantly blocked the CGS 21680-mediated decrease in EF2K levels at both time points (CGS+MG132 3 h: 126.44 ± 13.87, n = 3; CGS+MG132 6 h: 106.45 ± 16.35, n = 3).
FIGURE 2.
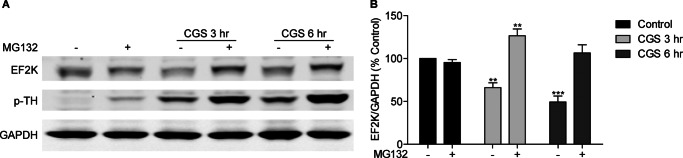
Activation of adenosine A2A receptors in PC12 cells causes proteasome-dependent degradation of EF2K. A, PC12 cells were stimulated for 3 or 6 h with CGS 21680 (100 nm) with or without MG132 (25 μm), and EF2K, p-tyrosine hydroxylase (p-TH), which served as a positive control for PKA activation downstream of A2A receptors, and GAPDH levels were assessed by SDS-PAGE and immunoblot. B, graph of summary data. A two-way ANOVA revealed significant main effects of CGS 21680 treatment and MG132, as well as a significant interaction between CGS 21680 and MG132. Bonferroni post-hoc tests revealed that either 3 or 6 h treatment with CGS 21680 reduced EF2K levels relative to control (CGS 3 h: 65.95 ± 9.89, n = 3, **, p < 0.01; CGS 6 h: 49.31 ± 12.02, n = 3, ***, p < 0.001), and this effect was blocked by the addition of MG132 at both time points (CGS+MG132 3 h: 126.44 ± 13.87, n = 3; CGS+MG132 6 h: 106.45 ± 16.35, n = 3).
EF2K Degradation Is Induced by cAMP/Growth Factor Signaling in Neurons and HEK Cells
To assess whether the degradation of EF2K we described in PC12 cells represents a more general translational control mechanism, we investigated whether similar phenomena may be engaged in neurons and HEK cells. Primary cortical neurons (DIV 14–15) were treated with Fsk (10 μm) and BDNF (100 ng/ml) in the absence or presence of MG132. In contrast to our findings in PC12 cells, treatment with either Fsk or BDNF alone for 6 h was not sufficient to change EF2K levels in neurons (data not shown), but co-application of Fsk and BDNF significantly reduced EF2K levels after 3 or 6 h (3 h: 86.00 ± 3.39, n = 5; 6 h: 72.23 ± 2.548, n = 7) (Fig. 3, A and B). This decrease in EF2K levels was accompanied by a decrease in p-eEF-2 (data not shown). As in PC12 cells, the effect of co-application Fsk and BDNF was attenuated by administration of MG132 at both 3 and 6 h time points (3 h+MG132: 99.26 ± 0.67, n = 4; 6 h+MG132: 86.79 ± 3.48, n = 4) (Fig. 3, A and B), indicating that the effect is proteasome-dependent.
FIGURE 3.
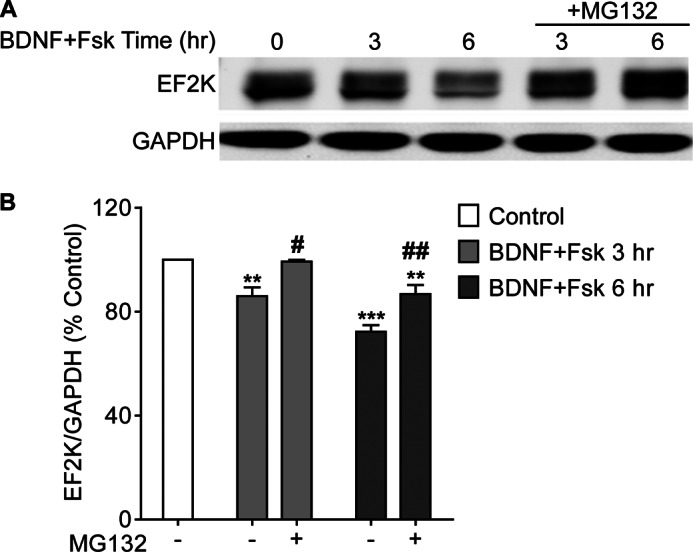
Co-stimulation with BDNF and Fsk causes proteasome-dependent EF2K degradation in cortical neurons. A, cortical neurons (14–15 DIV) were treated with Fsk (10 μm) plus BDNF (100 ng/ml) with or without MG132 (25 μm) for 3 or 6 h and EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. B, graph of summary data. A one-way ANOVA with Tukey's post-hoc tests showed that BDNF and Fsk co-stimulation for 3 or 6 h reduced EF2K levels relative to control (3 h: 86.00 ± 3.39, n = 5; 6 h: 72.23 ± 2.548, n = 7, ** or *** p < 0.01 or p < 0.001, respectively, relative to control). Addition of MG132 significantly blocked this effect (3 h+MG132: 99.26 ± 0.67, n = 4; 6 h+MG132: 86.79 ± 3.48, n = 4, # or ##, p < 0.05 or p < 0.01, respectively, relative to stimulation without MG132).
In HEK cells, the level of endogenous EF2K is low compared with PC12 cells, and therefore we transiently transfected GFP-EF2K and examined the effect of Fsk (10 or 50 μm) treatment on its stability during a 48 h chase with Chx (30 μg/ml). As in PC12 cells, application of Fsk dose-dependently accelerated GFP-EF2K turnover in HEK cells (Fig. 4, A and B), shortening its half-life from 24.8 h under control conditions to 9.4 and 5.9 h for the 10 μm and 50 μm doses, respectively, during the initial 24 h stimulation period.
FIGURE 4.
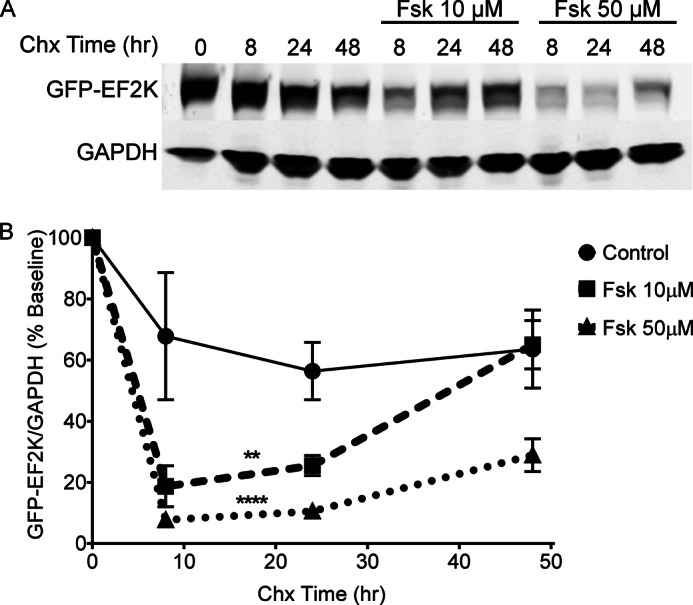
Fsk treatment accelerates EF2K turnover in HEK cells. A, HEK cells were transfected with GFP-EF2K and treated with 10 or 50 μm Fsk in the presence of Chx (30 μg/ml) to examine the rate of EF2K degradation. GFP-EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. B, graph of summary data. A two-way ANOVA revealed significant main effects of chase time and Fsk dose, as well as a significant interaction between time and dose. Tukey's post-hoc tests revealed that both 10 and 50 μm Fsk significantly decreased GFP-EF2K levels relative to control (n = 3, ** or ****, p < 0.01 or p < 0.0001, respectively).
Mechanism of cAMP-dependent Regulation of EF2K Turnover
We hypothesized that activation of PKA mediates the effects of Fsk and A2A receptor activation because EF2K is known to be a PKA substrate (13–15), and we previously observed that a mutant strain of PC12 cells (A126–1B2) that is deficient in PKA does not exhibit reduced EF2K activity during Fsk treatment (19). Treatment of PC12 cells with the selective PKA activator, 6-BNZ cAMP (100 μm) was found to be sufficient to significantly decrease EF2K levels relative to control (68.84 ± 6.35, n = 3) after 1 h (Fig. 5, A and B). EF2K levels returned to baseline after continued stimulation with 6-BNZ cAMP for up to 6 h. Activation of a parallel cAMP-dependent signaling module, exchange protein activated by cAMP (Epac), with 8-CPT cAMP (100 μm) did not affect EF2K levels in PC12 cells (data not shown).
FIGURE 5.
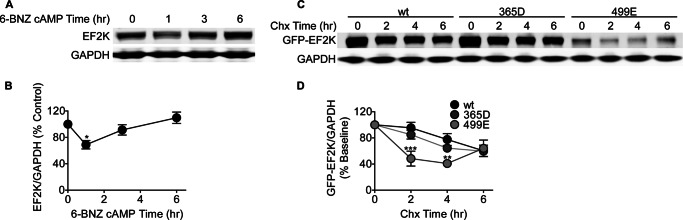
EF2K stability is regulated by PKA signaling. A, PC12 cells were treated with the selective PKA activator 6-BNZ cAMP (100 μm) for 0–6 h, and EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. B, graph of summary data. A one-way ANOVA with Tukey's post-hoc tests demonstrated that EF2K levels were significantly reduced after 1 h of 6-BNZ cAMP treatment (68.84 ± 6.35, n = 3, *, p < 0.05), and these levels subsequently recovered to baseline. C, PC12 cells were transfected with either wild-type GFP-EF2K or 365D or 499E mutant forms and treated with Chx (30 μg/ml) for 0–6 h to assess their stability. EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. D, graph of summary data. A two-way ANOVA revealed significant main effects of the form of GFP-EF2K transfected and chase time, as well as a significant interaction between transfection and time. Bonferroni post-hoc tests indicated a significantly increased degradation rate of the 499E mutant relative to that of wild-type GFP-EF2K at 2 and 4 h (2 h: 48.32 ± 11.35, n = 3, ***, p < 0.001; 4 h: 40.81 ± 5.25, n = 3, **, p < 0.01), and this rate later recovered to wild-type levels. The 365D mutant did not exhibit a different level of degradation than wild-type GFP-EF2K at any time point.
We next investigated whether PKA phosphorylation of EF2K affected its degradation. Two EF2K residues, Ser-365 and Ser-499, have previously been identified as PKA phosphorylation sites (15), and we generated GFP-tagged EF2K constructs bearing mutations of either of these sites to putatively phospho-mimetic residues (365D and 499E) and examined the stability of these mutants in PC12 cells during a cycloheximide chase (Fig. 5, C and D). The 499E mutant form of EF2K, but not 365D, exhibited lower steady-state expression and accelerated turnover relative to wild-type GFP-EF2K. Together, these data suggest that cAMP-PKA signaling accelerates EF2K degradation via phosphorylation of Ser-499.
EF2K Is a Substrate of SCFβTRCP
To further elucidate the mechanism by which phosphorylated EF2K is targeted to the proteasome, we examined whether phosphorylated EF2K is selectively polyubiquitylated by a Skp1-Cul1-F-box-protein (SCF)-type ubiquitin E3 ligase. SCF complexes comprise the scaffolding protein, Cul1, which recruits an E2 ubiquitin-conjugating enzyme via its interaction with the RING-finger protein, Rbx1, and substrate proteins via its interaction with the adaptor protein, Skp1, which in turn binds to an F-box protein, which serves as the substrate receptor. F-box proteins give rise to the selectivity of SCF-type ligases, in many cases binding to substrates in a manner dependent on phosphorylation of a specific motif, termed a phospho-degron (23, 24).
To evaluate the potential role of SCF complexes in regulating EF2K levels, we co-transfected HEK cells with GFP-EF2K in the presence or absence of a FLAG-tagged dominant-negative truncation mutant of Cul1 (amino acids 1–452) that abrogates its binding to Rbx1, and thus to the machinery for substrate ubiquitylation (25, 26). Co-expression of DN-Cul1 increased GFP-EF2K levels nearly 10-fold (982 ± 229, n = 4) relative to cells expressing GFP-EF2K alone (Fig. 6, A and B). These data strongly suggest that a SCF-type E3 ubiquitin ligase negatively regulates EF2K stability.
FIGURE 6.
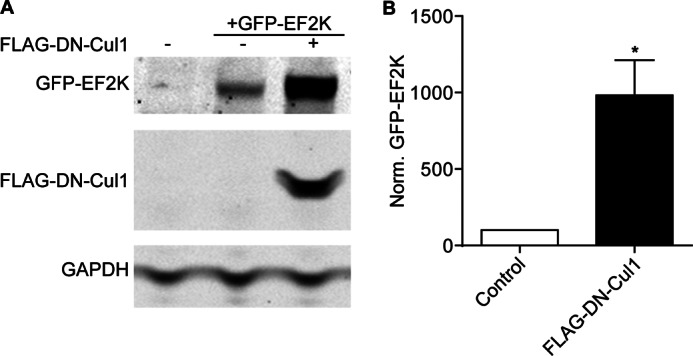
Cul1 regulates EF2K levels. A, HEK cells were transfected with GFP-EF2K with or without FLAG-DN-Cul1 and GFP-EF2K, FLAG-DN-Cul1, and GAPDH levels in lysates were assessed 48 h later by SDS-PAGE and immunoblot. B, graph of summary data. Co-expression of FLAG-DN-Cul1 significantly increased steady-state levels of GFP-EF2K to 982% ± 229 of cells expressing only GFP-EF2K as determined by t test, n = 4, *, p < 0.05.
We next sought to identify the F-box protein that recruits EF2K to the SCF complex. HEK cells were co-transfected with GFP-EF2K and one of a panel of GST-tagged F-box proteins (26, 27). Lysates were incubated with glutathione beads to pull-down GST-F-box proteins and eluates were immunoblotted for GFP-EF2K to assess binding. GFP-EF2K was found to bind to βTRCP1 and βTRCP2, but not to any of the other F-box proteins tested (Fig. 7A). While EF2K contains a consensus sequence for the phospho-degron domain recognized by the βTRCP proteins (28), the sequence DSGYPS containing Ser-440 and Ser-444, neither mutation of both Ser residues nor of Tyr-442 appeared sufficient to disrupt EF2K binding to βTRCP-1 (Fig. 7B).
FIGURE 7.

EF2K binds to βTRCP1 and βTRCP2. A, HEK cells were co-transfected with GFP-EF2K and one of a panel of GST-tagged F-box proteins. Cell lysates were incubated with glutathione beads, and binding was assessed in inputs (upper panel) and eluates (lower panel) by performing SDS-PAGE and immunoblot for GFP and GST. * indicates GST-Fbw7a, which migrates at a similar but distinct molecular weight as GFP-EF2K by SDS-PAGE. B, HEK cells were co-transfected with GST-βTRCP1, and either wild-type GFP-EF2K, or S440/S444 AA, or Y442F mutant forms and pull-downs were performed as above. Binding was assessed in inputs (upper panel) and eluates (lower panel) by performing SDS-PAGE and immunoblot for GFP and GST.
To determine whether βTRCP regulates EF2K expression, HEK cells were co-transfected with GFP-EF2K and a previously characterized shRNA that targets both βTRCP1 and βTRCP2 (26). Knock-down of βTRCP caused a >14-fold increase (1441 ± 276; n = 3) in steady-state EF2K levels relative to cells transfected with GFP-EF2K alone (Fig. 8, A and B).
FIGURE 8.
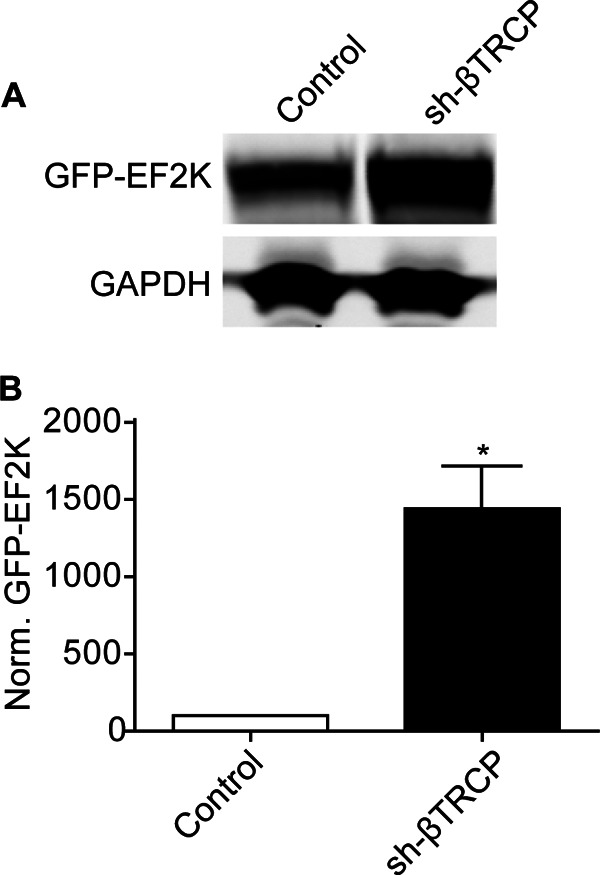
βTRCP regulates EF2K levels. A, HEK cells were transfected with GFP-EF2K with or without sh-βTRCP and levels of GFP-EF2K and GAPDH in lysates were assessed by SDS-PAGE and immunoblot 48 h later. B, graph of summary data. A t test demonstrated that knock-down of βTRCP significantly increased steady-state GFP-EF2K levels to 1441% ± 276 of control, n = 3, *, p < 0.05.
We also investigated whether co-transfection with sh-βTRCP or DN-Cul1 affected Fsk-mediated turnover of GFP-EF2K during a cycloheximide chase. As in our earlier experiments, both sh-βTRCP and DN-Cul1 significantly increased the steady-state expression of GFP-EF2K and Fsk treatment significantly hastened GFP-EF2K degradation (Fig. 9, A and B). After 8 h treatment with Chx and Fsk, levels of GFP-EF2K normalized to the control baseline were higher in both sh-βTRCP- (124% ± 15) and DN-Cul1- (115% ± 13) transfected cells than control-transfected cells (59% ± 5), suggesting that SCFβTRCP is involved in both steady-state and Fsk-stimulated EF2K turnover in HEK cells.
FIGURE 9.
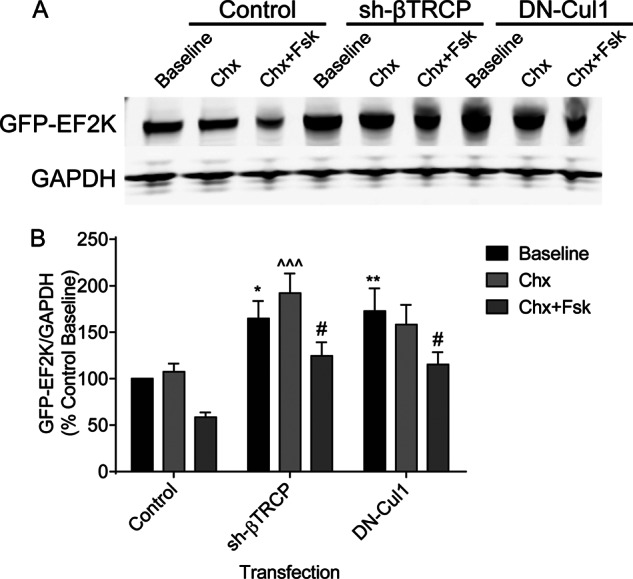
Interference with βTRCP or Cul1 function reduces the degradation of GFP-EF2K in the presence of Fsk. A, HEK cells were co-transfected with GFP-EF2K and either a control plasmid, sh-βTRCP, or DN-Cul1. Cells were treated with Chx (30 μg/ml) for 8 h in the presence or absence of Fsk (10 μm), and GFP-EF2K and GAPDH levels in lysates were assessed by SDS-PAGE and immunoblot. B, graph of summary data normalized to the control baseline. A two-way ANOVA revealed significant main effects of both Fsk treatment and plasmid transfected. Tukey's post-hoc tests revealed significant differences in GFP-EF2K levels between control-transfected and both sh-βTRCP- and DN-Cul1-transfected cells at baseline (sh-βTRCP: 164% ± 19, n = 14, *, p < 0.05; DN-Cul1: 173% ± 25, n = 15, **, p < 0.01), between control- and sh-βTRCP-transfected cells after 8 h treatment with Chx (sh-βTRCP: 192% ± 21, n = 15, ^^^, p < 0.001), and between control-transfected and both sh-βTRCP- and DN-Cul1-transfected cells after 8 h treatment with Chx and Fsk (sh-βTRCP: 124% ± 15, n = 15, #, p < 0.05; DN-Cul1: 115% ± 13, n = 15, #, p < 0.05).
Finally, we examined whether βTRCP1 regulated EF2K ubiquitylation in PC12 cells. We expressed GFP-EF2K in PC12 cells in the absence or presence of GST-βTRCP1 expression and treated cells with MG132 for 2 h to allow the accumulation of polyubiquitylated proteins. GFP-EF2K was then immunoprecipitated from lysates, and its ubiquitylation status was assessed by immunoblot (Fig. 10). Expression of βTRCP1 increased the polyubiquitylation of EF2K, indicated both by the accumulation of heavy molecular weight species of EF2K and by an increase in the ubiquitin signal in immunoprecipitates.
FIGURE 10.
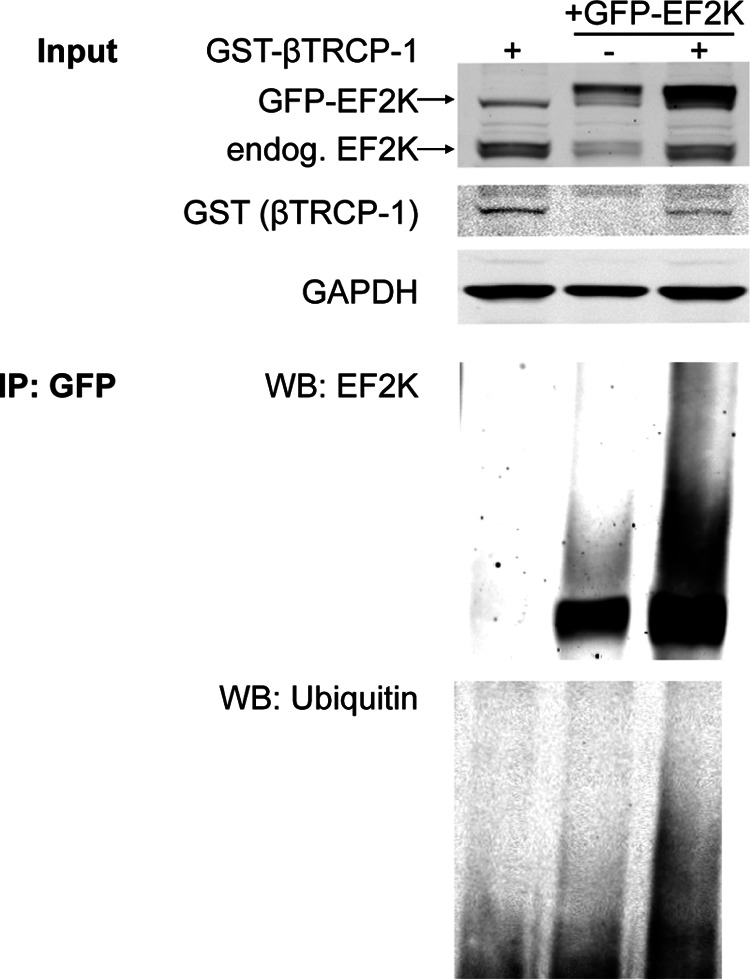
βTRCP drives EF2K polyubiquitylation in PC12 cells. PC12 cells were transfected with GFP-EF2K, with or without GST-βTRCP-1 and treated with MG132 (25 μm) for 2 h to accumulate polyubiquitylated proteins. Above: total cell lysates used as input for the immunoprecipitation were subjected to SDS-PAGE and immunoblot to assess levels of GFP-tagged and endogenous EF2K, GST-βTRCP, and GAPDH. Below: GFP-EF2K was immunoprecipitated, and eluates were subjected to SDS-PAGE and immunoblot for EF2K and ubiquitin.
DISCUSSION
Our data demonstrate that EF2K is degraded by the proteasome in response to cAMP signaling in PC12 cells, neurons, and HEK cells. Adenosine A2A receptor or PKA activation and phosphorylation of EF2K on Ser-499 appear to be sufficient to engage this degradation, presumably by recruiting the ubiquitin E3 ligase, SCFβTRCP, which binds to EF2K and promotes its polyubiquitylation. As EF2K is expressed fairly ubiquitously across many cell types (as are PKA and βTRCP), we believe that our data describe a cAMP-dependent mechanism that is likely to be employed in diverse cellular contexts.
Notably, a recent study found that DNA damage resulted in proteosomal degradation of EF2K in U2OS cells, and this also required SCFβTRCP (21). Under these conditions, AMPK phosphorylated EF2K at Ser-398, apparently causing intramolecular phosphorylation at the Ser-440/Ser-444 degron residues, which in turn was necessary and sufficient for EF2K to bind to βTRCP. It is possible that phosphorylation at Ser-499 promotes βTRCP binding and EF2K degradation in a similar manner as Ser-398 phosphorylation. However, while we found no obvious effect of mutation of Ser-440/Ser-444 on the interaction with βTRCP, phospho-proteomic analysis of EF2K in PC12 cells and HEK cells identified phosphorylation of 20 sites (data not shown), including Ser-398 and Ser-444, suggestive of complex control of EF2K activity and turnover. Thus, EF2K degradation by the proteasome may be mediated in response to different stimuli or in different cell types, by independent or concurrent phosphorylation events. In support of the idea of distinct pathways, our results indicated that while inhibitors of PI3K were able to block IGF-1-mediated degradation of EF2K, these inhibitors had no effect on Fsk-mediated decreases in EF2K levels in PC12 cells.
EF2K activity is known to be regulated by increases in intracellular [Ca2+], which occurs within seconds, and phosphorylation, which occurs within minutes. Here, we demonstrate that degradation by the UPS represents an additional mode of regulation of EF2K, one which occurs over the course of a few hours. These modes of regulation are not mutually exclusive and likely interact. For example, we observed that while eEF-2 phosphorylation levels in Fsk-stimulated cells treated with MG132 were similar to those of control cells, in IGF-1-stimulated cells, MG132 treatment led to a decrease of p-eEF-2, which may reflect an accumulation of EF2K bearing inhibitory phosphorylation downstream of p70 S6 kinase (16).
Together, our data suggest an cAMP-dependent mechanism by which receptor activation can engage both the ubiquitin-proteasome system and regulation of protein translation, which in turn may enable protein synthesis and degradation to be balanced or coordinated to effect cellular changes. Such a coordination appears to be necessary for long-term synaptic plasticity based on studies showing that simultaneous inhibition of protein synthesis and UPS-mediated degradation rescues deficits in LTP caused by inhibition of either alone (5, 6). A balance between protein synthesis and degradation may also play a role in homeostatic synaptic plasticity, as inhibiting and driving action potential firing in cultured neurons produces reciprocal changes in synaptic protein levels that are mediated in part by the UPS (29). Interestingly, regulation of EF2K activity has been shown to play a role in both these types of plasticity (10, 30), but it remains to be determined whether proteasomal degradation of EF2K is involved in some way in long-term forms of plasticity.
Activation of EF2K has been shown not only to decrease overall protein synthesis by inhibiting eEF-2, but to paradoxically activate the translation of some messages. For example, NMDA receptor stimulation in synaptic preparations resulted in rapid increases in p-eEF-2 concomitant with decreased overall protein synthesis, and yet an increase in the translation of α-CaMKII (31). This form of EF2K-mediated translational control appears to be required for hippocampal metabotropic glutamate receptor-mediated long-term depression (mGluR-LTD), in this case by activating the translation of Arc, as this form of plasticity is abolished in EF2K knock-out mice (30). Thus, degradation of EF2K by the UPS is expected to not only facilitate protein synthesis overall, but to decrease the translation of such anomalously regulated transcripts, thereby remodeling the repertoire of actively translating messages and contributing to long-term cellular changes.
Our findings build on a growing body of literature describing UPS-mediated degradation of proteins involved in translational control. In particular, SCFBTRCP has been shown to mediate the phosphorylation-dependent degradation of several such proteins, including the eIF4A inhibitor, PDCD4 (32), the mTOR inhibitor, DEPTOR (33–35), and the RNA-binding protein, CPEB (36). Additionally, in neuronal systems, plasticity-inducing stimulation has been shown to cause the degradation of the RNA-induced silencing complex (RISC) protein, Armitage/MOV10 (37, 38), and the RNA-binding protein, FMRP (39). It is striking that, as is the case with EF2K, all of the examples above involve the degradation of a negative regulator of protein synthesis, suggesting that engagement of the UPS may play a general role in activating translation.
Acknowledgment
DN-Cul, sh-βTRCP, and GST-F-box constructs were kindly provided by J. Wade Harper.
This work was supported, in whole or in part, by National Institutes of Health Grants DA018343 and DA10044 (to A. C. N.) and 1F31 DA029361 (to S. L. W.). Support was also obtained from the State of Connecticut, Department of Mental Health and Addiction Services.
- UPS
- ubiquitin-proteasome system
- LTP
- long-term potentiation
- EF2K
- eukaryotic elongation factor-2 kinase
- BDNF
- brain-derived neurotrophic growth factor
- eEF
- eukaryotic elongation factor
- Fsk
- forskolin
- SCF
- Skp1-Cul1-F-box-protein
- Chx
- cycloheximide.
REFERENCES
- 1. Davis H. P., Squire L. R. (1984) Protein synthesis and memory: a review. Psychol. Bull. 96, 518–559 [PubMed] [Google Scholar]
- 2. Lopez-Salon M., Alonso M., Vianna M. R., Viola H., Mello e Souza T., Izquierdo I., Pasquini J. M., Medina J. H. (2001) The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 14, 1820–1826 [DOI] [PubMed] [Google Scholar]
- 3. Artinian J., McGauran A. M., De Jaeger X., Mouledous L., Frances B., Roullet P. (2008) Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. Eur. J. Neurosci. 27, 3009–3019 [DOI] [PubMed] [Google Scholar]
- 4. Costa-Mattioli M., Sossin W. S., Klann E., Sonenberg N. (2009) Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karpova A., Mikhaylova M., Thomas U., Knöpfel T., Behnisch T. (2006) Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J. Neurosci. 26, 4949–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fonseca R., Vabulas R. M., Hartl F. U., Bonhoeffer T., Nägerl U. V. (2006) A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245 [DOI] [PubMed] [Google Scholar]
- 7. Nairn A. C., Matsushita M., Nastiuk K., Horiuchi A., Mitsui K., Shimizu Y., Palfrey H. C. (2001) Elongation factor-2 phosphorylation and the regulation of protein synthesis by calcium. Prog. Mol. Subcell. Biol. 27, 91–129 [DOI] [PubMed] [Google Scholar]
- 8. Wiseman S. L., Wei F. Y., Nairn A. C. (2009) in Handbook of Cell Signaling (Bradshaw R. A., Dennis E. A., eds), pp. 587–599, Elsevier, San Francisco [Google Scholar]
- 9. Marin P., Nastiuk K. L., Daniel N., Girault J. A., Czernik A. J., Glowinski J., Nairn A. C., Prémont J. (1997) Glutamate-dependent phosphorylation of elongation factor-2 and inhibition of protein synthesis in neurons. J. Neurosci. 17, 3445–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sutton M. A., Taylor A. M., Ito H. T., Pham A., Schuman E. M. (2007) Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55, 648–661 [DOI] [PubMed] [Google Scholar]
- 11. Mackie K. P., Nairn A. C., Hampel G., Lam G., Jaffe E. A. (1989) Thrombin and histamine stimulate the phosphorylation of elongation factor 2 in human umbilical vein endothelial cells. J. Biol. Chem. 264, 1748–1753 [PubMed] [Google Scholar]
- 12. Palfrey H. C., Nairn A. C., Muldoon L. L., Villereal M. L. (1987) Rapid activation of calmodulin-dependent protein kinase III in mitogen-stimulated human fibroblasts. Correlation with intracellular Ca2+ transients. J. Biol. Chem. 262, 9785–9792 [PubMed] [Google Scholar]
- 13. Mitsui K., Brady M., Palfrey H. C., Nairn A. C. (1993) Purification and characterization of calmodulin-dependent protein kinase III from rabbit reticulocytes and rat pancreas. J. Biol. Chem. 268, 13422–13433 [PubMed] [Google Scholar]
- 14. Redpath N. T., Proud C. G. (1993) Cyclic AMP-dependent protein kinase phosphorylates rabbit reticulocyte elongation factor-2 kinase and induces calcium-independent activity. Biochem. J. 293, 31–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diggle T. A., Subkhankulova T., Lilley K. S., Shikotra N., Willis A. E., Redpath N. T. (2001) Phosphorylation of elongation factor-2 kinase on serine 499 by cAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem. J. 353, 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Li W., Williams M., Terada N., Alessi D. R., Proud C. G. (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Browne G. J., Finn S. G., Proud C. G. (2004) Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J. Biol. Chem. 279, 12220–12231 [DOI] [PubMed] [Google Scholar]
- 18. Nairn A. C., Nichols R. A., Brady M. J., Palfrey H. C. (1987) Nerve growth factor treatment or cAMP elevation reduces Ca2+/calmodulin-dependent protein kinase III activity in PC12 cells. J. Biol. Chem. 262, 14265–14272 [PubMed] [Google Scholar]
- 19. Brady M. J., Nairn A. C., Wagner J. A., Palfrey H. C. (1990) Nerve growth factor-induced down-regulation of calmodulin-dependent protein kinase III in PC12 cells involves cyclic AMP-dependent protein kinase. J. Neurochem. 54, 1034–1039 [DOI] [PubMed] [Google Scholar]
- 20. Arora S., Yang J. M., Hait W. N. (2005) Identification of the ubiquitin-proteasome pathway in the regulation of the stability of eukaryotic elongation factor-2 kinase. Cancer Res. 65, 3806–3810 [DOI] [PubMed] [Google Scholar]
- 21. Kruiswijk F., Yuniati L., Magliozzi R., Low T. Y., Lim R., Bolder R., Mohammed S., Proud C. G., Heck A. J., Pagano M., Guardavaccaro D. (2012) Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Science Signaling 5, ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Picciotto M. R., Cohn J. A., Bertuzzi G., Greengard P., Nairn A. C. (1992) Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 267, 12742–12752 [PubMed] [Google Scholar]
- 23. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell. Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 24. Petroski M. D., Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 25. Donzelli M., Squatrito M., Ganoth D., Hershko A., Pagano M., Draetta G. F. (2002) Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 21, 4875–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin J., Shirogane T., Xu L., Nalepa G., Qin J., Elledge S. J., Harper J. W. (2003) SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 17, 3062–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shirogane T., Jin J., Ang X. L., Harper J. W. (2005) SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 280, 26863–26872 [DOI] [PubMed] [Google Scholar]
- 28. Winston J. T., Strack P., Beer-Romero P., Chu C. Y., Elledge S. J., Harper J. W. (1999) The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and beta-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 13, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ehlers M. D. (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242 [DOI] [PubMed] [Google Scholar]
- 30. Park S., Park J. M., Kim S., Kim J. A., Shepherd J. D., Smith-Hicks C. L., Chowdhury S., Kaufmann W., Kuhl D., Ryazanov A. G., Huganir R. L., Linden D. J., Worley P. F. (2008) Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheetz A. J., Nairn A. C., Constantine-Paton M. (2000) NMDA receptor-mediated control of protein synthesis at developing synapses. Nat. Neurosci. 3, 211–216 [DOI] [PubMed] [Google Scholar]
- 32. Dorrello N. V., Peschiaroli A., Guardavaccaro D., Colburn N. H., Sherman N. E., Pagano M. (2006) S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 [DOI] [PubMed] [Google Scholar]
- 33. Duan S., Skaar J. R., Kuchay S., Toschi A., Kanarek N., Ben-Neriah Y., Pagano M. (2011) mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol. Cell 44, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao D., Inuzuka H., Tan M. K., Fukushima H., Locasale J. W., Liu P., Wan L., Zhai B., Chin Y. R., Shaik S., Lyssiotis C. A., Gygi S. P., Toker A., Cantley L. C., Asara J. M., Harper J. W., Wei W. (2011) mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao Y., Xiong X., Sun Y. (2011) DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Setoyama D., Yamashita M., Sagata N. (2007) Mechanism of degradation of CPEB during Xenopus oocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 18001–18006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ashraf S. I., McLoon A. L., Sclarsic S. M., Kunes S. (2006) Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell 124, 191–205 [DOI] [PubMed] [Google Scholar]
- 38. Banerjee S., Neveu P., Kosik K. S. (2009) A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron 64, 871–884 [DOI] [PubMed] [Google Scholar]
- 39. Hou L., Antion M. D., Hu D., Spencer C. M., Paylor R., Klann E. (2006) Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454 [DOI] [PubMed] [Google Scholar]