

Background: ER stress induces apoptosis.
Results: TMEM214 is localized at the ER and is required for ER stress-induced caspase 4 activation and apoptosis.
Conclusion: TMEM214 mediates ER stress-induced apoptosis.
Significance: This study helps to understand the molecular mechanisms of ER stress-induced apoptosis.
Keywords: Apoptosis, Caspase, Cell Signaling, ER Stress, Unfolded Protein Response, TMEM214, Caspase 4
Abstract
Endoplasmic reticulum (ER) stress caused by excessive aggregation of misfolded proteins induces apoptosis. Although ER stress-induced apoptosis has been implicated in many diseases, the detailed mechanisms are not well understood. Here, we identified human transmembrane protein 214 (TMEM214) as a critical mediator of ER stress-induced apoptosis. Overexpression of TMEM214 induced apoptosis, whereas knockdown of TMEM214 inhibited ER stress-induced apoptosis. TMEM214 was localized on the outer membrane of the ER and constitutively associated with procaspase 4, which was also critical for ER stress-induced apoptosis. TMEM214-induced apoptosis was abolished by a dominant negative mutant of procaspase 4, whereas caspase 4-induced apoptosis was inhibited by knockdown of TMEM214. Furthermore, knockdown of TMEM214 inhibited the activation and cleavage of procaspase 4 by impairing its recruitment to the ER. Our findings suggest that TMEM214 is essential for ER stress-induced apoptosis by acting as an anchor for recruitment of procaspase 4 to the ER and its subsequent activation.
Introduction
Endoplasmic reticulum (ER)2 functions in the folding of secretory and membrane proteins, calcium homeostasis, and lipid biosynthesis (1). ER stress, which disturbs ER function, occurs in response to a wide variety of cellular stressors that perturb cellular energy equilibrium, calcium concentration, and the redox state. These cellular stressors often lead to the accumulation of unfolded and misfolded proteins in the ER. The unfolded protein response is then activated to relieve the stress (2). However, if chronic or unresolved ER stress and excessive aggregation of misfolded proteins cannot be resolved, cells undergo apoptosis (3).
Prolonged ER stress triggers apoptosis by activating the CCAAT/enhancer-binding protein homologous protein (CHOP)-, JNK-, and caspase-dependent pathways (4–6). Activation of the transcription factor CHOP negatively regulates expression of the antiapoptotic proteins Bcl-2 and Bnip3 and mediates transcriptional induction of proapoptotic proteins, including p53 up-regulated modulator of apoptosis (PUMA), Bcl2-interacting mediator of cell death (BIM), Tribbles homolog 3, and death receptor 5 (DR5) (7–11). ER stress activates the JNK signaling pathway through apoptosis signal-regulating kinase 1 (ASK1) and, subsequently, promotes Bcl2-interacting mediator of cell death- and BAX-dependent apoptosis (12, 13). Moreover, ER stress also disturbs the mobilization of intracellular calcium ions, leading to activation of the cysteine protease calpain and subsequent caspase-dependent apoptosis (14).
Mouse caspase 12 is an ER-associated proximal effector that activates procaspase 9 to cleave procaspase 3, which leads directly to apoptosis (15). However, human caspase 12 has no similar function because its gene is disrupted by a frameshift, which results in a premature stop codon. In addition, human caspase 12 also contains amino acid substitutions in the critical sites for caspase activity (16, 17). Instead, human caspase 4 is specifically cleaved under ER stress, suggesting that it may be a functional ortholog of mouse caspase 12 in ER stress-induced apoptosis (17, 18). So far, little is known about how caspase 4 is activated in ER stress-triggered apoptosis.
Transmembrane protein 214 (TMEM214), also known as FLJ20254, has been reported to play roles in dengue virus replication and cellular mRNA degradation (19). In this study, we found that TMEM214 was localized on the ER and mediated ER stress-induced apoptosis. Overexpression of TMEM214 led to apoptosis, which required the activity of caspase 4. Conversely, caspase 4-induced apoptosis was blocked by knockdown of TMEM214. Furthermore, we found that TMEM214 recruited caspase 4 to the ER, which was essential for ER stress-induced procaspase 4 activation and apoptosis. Our findings suggest that TMEM214 mediates stress-induced apoptosis by acting as an anchor for the recruitment of procaspase 4 to the ER and its subsequent activation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Rabbit monoclonal antibodies against Bip, CHOP, PARP-1, and AIF (Santa Cruz Biotechnology); mouse monoclonal antibodies against caspase 4 (MBL); phosphorylated JNK, JNK, and caspase 7 (Cell Signaling Technology); FLAG and β-actin (Sigma); TG, BFA, and etoposide (Tocris Bioscience); and actinomycin D and TNFα (Sigma) were purchased from the indicated companies. Rabbit anti-TMEM214 antiserum was raised against recombinant human TMEM214 (amino acids 1–110).
Constructs
Mammalian expression plasmids for human FLAG- or cherry-tagged TMEM214 and its truncated mutants were constructed by standard molecular biology techniques. GFP-tagged organelle markers were purchased from Origene Inc.
Screening
Approximately 10,000 human cDNA expression plasmids (obtained from Origene Inc.) were transfected individually into 293 cells seeded in a 48-well plate by using a standard calcium phosphate precipitation method. Twenty hours later, transfected cells were observed under a microscope. The clones that caused a round-up morphology of transfected cells were selected for further characterization.
Cell Lines and Retrovirally Mediated Gene Transfer
The RNAi plasmids were transduced to 293 or HeLa cells by retrovirally mediated gene transfer. Briefly, 293 or HeLa cells plated on 100-mm dishes were transiently transfected with the indicated RNAi retroviral plasmids (10 μg each) together with the pGag-pol (10 μg) and the pVSV-G (3 μg) plasmids for 48 h. The recombinant retroviruses were harvested for infection of the indicated cells in the presence of polybrene (4 μg/ml). The stable RNAi-transduced cells were selected with puromycin (1 μg/ml) for at least 5 days.
Apoptosis Assays
HeLa or 293 cells were collected and stained by annexin V-FITC in PBS. The apoptotic cells were assessed by flow cytometry analysis using a FACSCalibur flow cytometer (XDP, Beckman Coulter).
Statistical Analysis
The data were analyzed by two tailed Student's t test. p < 0.05 was considered significant.
RNAi Experiments
Double-strand oligonucleotides corresponding to the target sequences were cloned into the pSUPER.Retro RNAi vector (Oligoengine). The target sequences for human TMEM214 cDNA were as follows: #1, GGTGGGAGGTAGTGAAGAA; #2, CAGCAAAGTGTCTCACCAT; and #3, GGGAGTCACTACATGGTTA. The target sequence for human procaspase 4 cDNA was as follows: GGACTATAGTGTAGATGTA. The target sequence for human CHOP cDNA was as follows: ACAGGAGAATGAAAGGAAA.
Subcellular Fractionation
HeLa cells (1 × 107) were washed with PBS and lysed by douncing 30 times in 0.5 ml homogenization buffer (10 mm Tris-HCl (pH 7.4), 2 mm MgCl2, 10 mm KCl, and 250 mm sucrose). The homogenate was centrifuged at 500 × g for 10 min, and the pellet (P5) was saved as crude nuclei. The supernatant (S5) was centrifuged at 5000 × g for 10 min to precipitate crude mitochondria (P5K). The supernatant (S5K) was further centrifuged at 20,000 × g for 30 min for preparation of S50K and P50K.
Coimmunoprecipitation and Immunoblot Analysis
For transient transfection and coimmunoprecipitation experiments, 293 cells (1 × 106) were transfected for 18 h. The transfected cells were lysed in 0.8 ml of lysis buffer (20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton, 1 mm EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride). For each immunoprecipitation, a 0.4-ml aliquot of lysate was incubated with 0.5 μg of the indicated antibody or control IgG and 25 μl of a 1:1 slurry of GammaBind G Plus-Sepharose for 2 h. The Sepharose beads were washed three times with 1 ml of lysis buffer containing 500 mm NaCl. The precipitates were analyzed by immunoblotting procedures. For endogenous coimmunoprecipitation experiments, HeLa cells (5 × 107) were treated with apoptotic inducer, TG, BFA, or TNFα for the indicated times. Coimmunoprecipitation and immunoblot experiments were performed as described above.
RESULTS
TMEM214 Mediates ER Stress-induced Apoptosis
During a functional screening for proteins that can induce cell death, TMEM214 was identified from ∼10,000 independent human cDNA expression clones. Overexpression of TMEM214 induced morphological changes characteristic of apoptosis, such as round-up morphology and detachment from the culture dishes (Fig. 1A). Overexpression also caused positive staining of annexin V (Fig. 1A) and DNA fragmentation (B), which are hallmarks of apoptosis. TRIF (Toll/IL-1 receptor domain containing adaptor-inducing interferon β), a critical adaptor protein that mediates Toll-like receptor 3-induced apoptosis (20), has been used as a positive control for these experiments (Fig. 1, A and B). Overexpression of TMEM214 induced apoptosis in a dose-dependent manner (supplemental Fig. S1). These results suggest that overexpression of TMEM214 induces apoptosis.
FIGURE 1.

TMEM214 mediates ER stress-induced apoptosis. A, overexpression of TMEM214 induces apoptosis. 293 cells (1 × 105) were transfected with the indicated expression plasmids (1 μg each) for 24 h. Transfected cells were observed under a microscope (upper panels), digested by trypsin, and stained with annexin V-FITC. The annexin V-positive cells were quantified by flow cytometry (lower panels). Results are representative of three independent experiments. Con, control; TRIF, Toll/IL-1 receptor domain containing adaptor-inducing interferon β. B, TMEM214 induces DNA fragmentation. 293 cells (1 × 105) were transfected with the indicated expression plasmids (1 μg each) for 24 h before DNA fragmentation assays were performed. C, effects of TMEM214-RNAi on the expression of TMEM214. Lysates from HeLa cells stably transfected with the control, TMEM214-RNAi-#1, TMEM214-RNAi-#2, or TMEM214-RNAi-#3 plasmids were analyzed by immunoblot analysis with anti-TMEM214. D, effects of TMEM214-RNAi on ER stress-induced apoptosis. HeLa cells stably transfected with the indicated RNAi plasmids were untreated or treated with TG (3 μm), BFA (10 μm), or TNFα plus cycloheximide (CHX) (50 ng/ml+10 μm) for 24 h. The apoptotic cells were quantified by annexin V-FITC staining and flow cytometry analysis (left panel). The flow cytometry data are also presented quantitatively (right panel). The graphs show mean ± S.D., n = 3. *, p < 0.01. E, effects of TMEM214-RNAi on apoptosis induced by actinomycin D and etoposide. HeLa cells stably transfected with the TMEM214-RNAi #3 plasmid were untreated or treated with actinomycin D (1 μg/ml) or etoposide (100 μm) for 36 h. The cells were then stained by annexin V-FITC and analyzed by flow cytometry (left panel). The flow cytometry data are presented quantitatively (right panel). The graphs show mean ± S.D., n = 3.
To determine the roles of TMEM214 in various apoptotic pathways, we constructed three RNAi plasmids targeting different sites of the human TMEM214 mRNA. The RNAi plasmids were stably transduced into HeLa cells by retroviral-mediated gene transfer. As shown in Fig. 1C, the #2 and #3 TMEM214-RNAi plasmids could markedly inhibit TMEM214 expression in HeLa cells. We then examined the effects of TMEM214 knockdown on apoptosis triggered by divergent stimuli. As shown in Fig. 1D, knockdown of TMEM214 significantly inhibited apoptosis induced by two ER stress inducers, TG and BFA, but not by the external apoptosis inducer TNFα. In similar experiments, knockdown of TMEM214 did not markedly inhibit apoptosis induced by actinomycin D and etoposide (Fig. 1E), which induce mitochondrion-dependent apoptosis. Moreover, it seems that levels of TMEM214 in several examined cell lines, including HeLa, HCT116, HepG2, and A549, were correlated with the abilities of TG to induce apoptosis in these cells (supplemental Fig. S2). These results suggest that TMEM214 specifically mediates ER stress-induced apoptosis.
It has been reported previously that TMEM214 plays a role in cellular mRNA degradation (19). Because TMEM214 plays a role in ER stress-induced apoptosis, we examined whether TMEM214 is involved in unfolded protein response. We found that overexpression of TMEM214 had no marked effect on both protein and mRNA levels of unfolded protein response markers, including GRP78, GRP94, and PDI (supplemental Figs. S1 and S3). These results suggest that TMEM214 is not involved in unfolded protein response.
TMEM214-mediated Apoptosis Is Independent of CHOP and JNK
Our earlier experiments indicated that TMEM214 mediated ∼30% of ER stress-induced apoptosis (Fig. 1). It has been reported previously that ER stress triggers apoptosis by several independent mechanisms, including CHOP induction, JNK phosphorylation, or caspase activation (4–6). Therefore, we examined the relationship between TMEM214-mediated apoptosis and CHOP induction or JNK phosphorylation. We found that knockdown of TMEM214 had no marked effects on TG-induced JNK phosphorylation and CHOP induction (Fig. 2A). Conversely, knockdown of CHOP had no marked effect on TMEM214-mediated apoptosis (Fig. 2B). These results suggest that TMEM214-mediated apoptosis is independent of CHOP induction or JNK phosphorylation.
FIGURE 2.
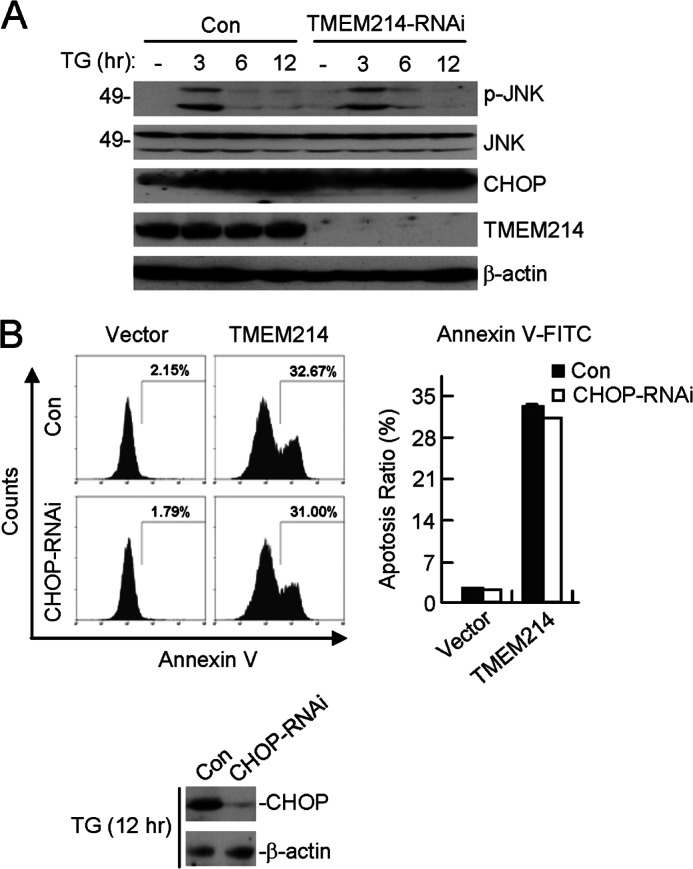
TMEM214-mediated apoptosis is independent of CHOP and JNK. A, effects of TMEM214-RNAi on TG-triggered CHOP induction and JNK phosphorylation. TMEM214-knockdown HeLa cells were untreated (-) or treated with TG for the indicated times. Cell lysates were analyzed by immunoblot analyses with the indicated antibodies. Con, control. B, effects of CHOP-RNAi on TMEM214-induced apoptosis. CHOP-knockdown cells were transfected with TMEM214. The transfected cells were stained with annexin V-FITC and analyzed by flow cytometry (left panel). The flow cytometry data are presented quantitatively (right panel). CHOP levels in control and knockdown cells after TG induction for 12 h were examined by immunoblot analyses (bottom panel).
TMEM214 Is Localized to the Outer Membrane of the ER
TMEM214 is a 689-amino acid protein. Structural analysis with several programs indicated that TMEM214 contains two putative transmembrane domains at amino acids 480–500 and 616–636, respectively. Because TMEM214 mediates ER stress-induced apoptosis, we determined whether TMEM214 is localized to the ER by immunofluorescent staining and cell fractionation analysis. Confocal microscopy analysis indicated that TMEM214 was mostly colocalized with the ER marker Sec61β but with little overlapping with the Golgi body or mitochondrial marker (Fig. 3A). Cell fractionation and immunoblot analysis indicated that endogenous TMEM214 existed mostly in the ER-containing membrane fraction, with a low level in the mitochondrion and not detectable in the cytosol (Fig. 3B). In these experiments, endogenous procaspase-4 was found in all cellular fractions (Fig. 3B). Results from trypsin-protection assays with purified membrane fraction indicated that both TMEM214 and procaspase 4 were sensitive to trypsin treatment, whereas the ER lumen protein Bip was protected (Fig. 3C). These data suggest that most TMEM214 and a portion of procaspase 4 are localized on the outer membrane of the ER.
FIGURE 3.

TMEM214 and procaspase 4 are localized to the ER. A, confocal microscopy study of TMEM214 localization. 293 (a–l) or HeLa (m–p) cells were transfected with the indicated expression plasmids for 24 h and subsequently stained with DAPI. Cells were observed under a confocal microscope. Mito, mitochondrion. B, cellular fractionation and immunoblot analysis of TMEM214 and procaspase 4 localization. HeLa cells were fractionated, and cellular fractions were analyzed by immunoblot analyses with the indicated antibodies. C, TMEM214 and procaspase 4 in the membrane fraction were sensitive to trypsin treatment. The purified ER-enriched membrane fraction was treated with trypsin (5 μm) and analyzed by immunoblot analyses with the indicated antibodies.
Caspase 4 Is Required for ER Stress-induced Apoptosis
Previous studies reveal that procaspase 4 is cleaved and activated during tunicamycin- or TG-induced ER stress in several cell types, including SK-N-SH, SH-SY5Y, and human retinal pigment epithelial cells (17, 18, 21). As shown in Fig. 4A, treatment with the specific caspase 4 inhibitor Z-LEVD-FMK markedly decreased TG-induced annexin V-FITC-stained cells from 87.31 to 51.98% and BFA-induced annexin V-FITC-stained cells from 73.90 to 46.10%. As a control, the percentage of TNFα-induced annexin V-FITC-stained cells was not markedly affected by treatment of Z-LEVD-FMK (Fig. 4A). Consistently, treatment of HeLa cells with Z-LEVD-FMK inhibited the cleavage of procaspase 4 and the caspase substrate poly(ADP-ribose) polymerase 1 (PARP-1) induced by TG and BFA but not TNFα (Fig. 4B). These results demonstrate that caspase 4 mediates apoptosis induced by the ER stress agents TG and BFA in HeLa cells.
FIGURE 4.

Caspase 4 and TMEM214 mediate ER stress-induced apoptosis. A, effects of the caspase 4 inhibitor on ER stress-induced apoptosis. HeLa cells (2 × 105) were pretreated with Z-LEVD-FMK (5 μm) for 6 h. The pretreated cells were treated further with TG (3 μm), BFA (10 μm), or TNFα plus cycloheximide (CHX) (50 ng/ml+10 μm) for 24 h. The cells were stained with annexin V-FITC and analyzed by flow cytometry. The graphs show mean ± S.D., n = 3. *, p < 0.01. Con, control. B, effects of the caspase 4 inhibitor on ER stress- and TNFα-induced cleavages of PARP-1 and procaspase 4. HeLa cells were treated as in A, and the cell lysates were analyzed by immunoblot analyses with the indicated antibodies. C, effects of procaspase 4 RNAi on the expression of procaspase 4. HeLa cells were stably transfected with a procaspase 4 RNAi or control plasmid. Cell lysates were analyzed by immunoblot analyses with the indicated antibodies. D, effects of the TMEM214 RNAi and procaspase 4 RNAi plasmids on apoptosis induced by ER stressors or TNFα. HeLa cells stably transfected with the indicated RNAi plasmids were untreated or treated with TG (3 μm), BFA (10 μm), or TNFα plus CHX (50 ng/ml +10 μm) for 24 h. The cells were then stained by annexin V-FITC and analyzed by flow cytometry. The graphs show mean ± S.D., n = 3. *, p < 0.01.
Caspase 4 and TMEM214 Are Mutually Involved in ER Stress-induced Apoptosis
Because procaspase 4 is localized at the ER and activated in ER stress-induced apoptosis, we determined whether caspase 4 is required for ER stress-induced apoptosis. We made a procaspase 4 RNAi plasmid that could markedly knock down the level of procaspase 4 (Fig. 4C). As shown in Fig. 4D, knockdown of either procaspase 4 or TMEM214 markedly inhibited apoptosis induced by TG and BFA, whereas simultaneous knockdown of both procaspase 4 and TMEM214 had an even better inhibitory effect on apoptosis induced by these stimuli. In these experiments, knockdown of procaspase 4 and TMEM214 individually or simultaneously had no marked effects on apoptosis induced by TNFα (Fig. 4D). These results suggest that both TMEM214 and caspase 4 are required for ER stress-induced apoptosis.
We then further determined the relationship between TMEM214 and procaspase 4 in ER stress-induced apoptosis. As shown in Fig. 5A, the apoptotic morphological change induced by overexpression of procaspase 4 was mostly inhibited in TMEM214 knockdown cells in comparison to the control cells. In these experiments, the apoptotic morphological change induced by procaspase 8, a caspase required for death receptor-induced apoptosis, was not inhibited by knockdown of TMEM214 (Fig. 5A). Similarly, annexin V staining indicated that the percentage of apoptotic cells induced by overexpression of procaspase 4 but not procaspase 8 was decreased significantly in TMEM214 knockdown cells (Fig. 5B). These results suggest that caspase 4- but not caspase 8-mediated apoptosis requires TMEM214. Conversely, a caspase-inactive mutant of procaspase 4, procaspase 4(C258S), but not a caspase-inactive mutant of procaspase 8, procaspase 8(C360S), markedly inhibited cell death induced by overexpression of TMEM214 (Fig. 5C). In these experiments, crmA, a caspase inhibitor from Vaccinia virus (22, 23), also markedly inhibited apoptosis induced by overexpression of TMEM214 (Fig. 5C). These results suggest that TMEM214 and caspase 4 mediate ER stress-induced apoptosis in a reciprocally dependent manner.
FIGURE 5.

TMEM214 and caspase 4 mediate apoptosis in a reciprocally dependent manner. A and B, effects of TMEM214 knockdown on caspase 4-induced apoptosis. TMEM214 knockdown cells were transfected with procaspase 4 or procaspase 8. The morphology of the cells was observed under a microscope (A, left panel). The effect of TMEM214-RNAi-#3 on the expression of TMEM214 was analyzed by immunoblot analyses with the indicated antibodies (A, right panel). The transfected cells were also stained with annexin V-FITC and analyzed by flow cytometry (B). The graphs show mean ± S.D., n = 3. *, p < 0.01. Con, control. C, effects of caspase-inactive mutants of procaspase 4 and procaspase 8 on TMEM214-induced apoptosis. 293 cells (1 × 105) were transiently transfected with TMEM214 and procaspase 4(C258S), procaspase 8(C360S), or crmA for 24 h. The morphology of cells was observed under a microscope (upper panel). The transfected cells were also stained with annexin V-FITC and analyzed by flow cytometry (lower panel). The flow cytometry data are also presented quantitatively (right panel). The graphs show mean ± S.D., n = 3. *, p < 0.01.
Anchorage of Procaspase 4 to the ER by TMEM214 Is Important for ER Stress-induced Apoptosis
Because TMEM214 and caspase 4 are linked in ER stress-induced apoptosis, we determined whether they physically interact with each other. Coimmunoprecipitation experiments indicated that TMEM214 was constitutively associated with procaspase 4 in the presence or absence of TG or BFA stimulation (Fig. 6, A and B).
FIGURE 6.

TMEM214 interacts with procaspase 4 and anchors procaspase 4 to the ER. A and B, TMEM214 interacts with procaspase 4. HeLa cells (5 × 107) were treated with TG (3 μm) (A) or BFA (10 μm) (B) or left untreated (-) for 3 h. Cell lysates were immunoprecipitated (IP) with anti-TMEM214 (αTM) or preimmune serum (Pre). The immunoprecipitates (upper two panels) and cell lysates (lower three panels) were analyzed by immunoblot analyses with the indicated antibodies (Ab). C, knockdown of TMEM214 impairs the localization of procaspase 4 to the ER. TMEM214 knockdown HeLa cells (1 × 107) were untreated (-) or treated with TG for the indicated times. Cell fractionation was performed. The ER-enriched membrane fraction and whole cell lysate (WCL) were analyzed by immunoblot analyses with the indicated antibodies. Con, control. D, knockdown of TMEM214 inhibits ER stress-induced cleavages of procaspase 4 and PARP-1. TMEM214 knockdown HeLa cells were untreated (-) or treated with TG for the indicated times. Cell lysates were analyzed by immunoblot analyses with the indicated antibodies.
Because TMEM214 interacts with procaspase 4 and this interaction is important for ER stress-induced apoptosis, we determined whether the localization of procaspase 4 on the ER is dependent on TMEM214. We isolated the ER fractions from TMEM214 knockdown and control HeLa cells for immunoblot analysis. The results showed that the level of ER-associated procaspase 4 in both unstimulated and TG-stimulated cells was decreased dramatically by knockdown of TMEM214 (Fig. 6C). In these experiments, the level of total procaspase 4 in cells was not changed markedly by TMEM214 knockdown or TG simulation (Fig. 6C). These results suggest that TMEM214 is required for the localization of procaspase 4 to the ER.
Furthermore, knockdown of TMEM214 markedly inhibited the cleavage of procaspase 4 and PARP-1 induced by TG stimulation (Fig. 6D), suggesting that TMEM214 is critical for the activation of caspase 4 following ER stress. In these experiments, induction of Bip and CHOP by TG was not affected markedly by TMEM214 knockdown, suggesting that TMEM214-mediated apoptosis is independent of Bip and CHOP.
Domain mapping indicated that the N-terminal cytoplasmic region of TMEM214, amino acids 176–354, was required for its binding with procaspase 4 (Fig. 7, A and B). Interestingly, either of the two transmembrane domains (amino acids 480–500 or 616–636) of TMEM214 was sufficient to localize it to the ER (Fig. 7A and supplemental Fig. S4), whereas both the N-terminal cytoplasmic region and either one of the transmembrane domains were required for its ability to induce apoptosis (Fig. 7C). Taken together, these results suggest that the TMEM214-procaspase 4 interaction and the ER localization of TMEM214 are critical for ER stress-induced apoptosis.
FIGURE 7.

Domain mapping of TMEM214. A, schematic presentation of TMEM214 truncations and their interaction with procaspase 4, ER localization, and ability to induce apoptosis. B, interactions between TMEM214 truncations and procaspase 4. 293 cells (1 × 107) were transfected with FLAG-tagged TMEM214 truncation mutants. Cell lysates were immunoprecipitated (IP) with anti-FLAG or control mouse lgG. The immunoprecipitates and cell lysates were analyzed by immunoblot analyses with the indicated antibodies (Ab). C, apoptosis induced by TMEM214 truncations. 293 cells (1 × 105) were transfected with TMEM214 truncations for 24 h. The cells were stained with annexin V-FITC and analyzed by flow cytometry. The graphs show mean ± S.D., n = 3.
Caspase 7 Plays a Minor Role in TMEM214-mediated Apoptosis
Previous studies have demonstrated that overexpressed GRP78 binds to cytoplasmic caspase 7 and reduces apoptotic cell death (24). We determined whether caspase 7 also plays a role in TMEM214-mediated apoptosis. We found that knockdown of TMEM214 only had a minor inhibitory effect on TG-induced procaspase 7 cleavage (supplemental Fig. S5A). We also examined the effect of procaspase 7(C186S) on TMEM214-induced apoptosis. The results indicated that procaspase 7(C186S) partially inhibited apoptosis induced by overexpression of TMEM214 (supplemental Fig. S5B). However, the degree of inhibition of TMEM214-induced apoptosis by procaspase 7(C186S) is markedly lower in comparison to procaspase 4(C284S). These data suggest that caspase 7 plays a minor role in ER stress-induced apoptosis.
DISCUSSION
ER stress-induced apoptosis has been investigated extensively in the past years. However, the detailed mechanisms are still unclear. In this study, we identified TMEM214 as an ER-associated membrane protein that mediates ER stress-induced apoptosis.
Immunofluorescent staining and cellular fractionation indicated that TMEM214 was mostly localized at the ER. Trypsin digestion experiments indicated that TMEM214 was localized at the outer membrane of the ER. Overexpression of TMEM214 induced apoptosis, whereas knockdown of TMEM214 inhibited apoptosis induced by ER stressors such as TG and BFA. Several lines of evidence suggest that TMEM214 signals through caspase 4 in ER stress-induced apoptosis. Firstly, TMEM214 interacts with procaspase 4 constitutively, and both are located at the outer membrane of the ER. Knockdown of TMEM214 abolished the association of procaspase 4 with the ER. These results suggest that TMEM214 anchors procaspase 4 to the outer membrane of the ER. Second, the caspase 4 inhibitor Z-LEVD inhibited TG- and BFA- but not TNFα- or DNA damage-induced apoptosis as well as cleavage of procaspase 4, suggesting that caspase 4 specifically mediates ER stress-induced apoptosis. Third, knockdown of TMEM214 inhibited apoptosis induced by overexpression of procaspase 4 but not procaspase 8, whereas a caspase-inactive mutant of procaspase 4 but not procaspase 8 inhibited apoptosis induced by overexpression of TMEM214. These results suggest that TMEM214 and caspase 4 are mutually dependent on each other in ER stress-induced apoptosis.
TMEM214 contains two transmembrane domains at its C terminus and a large N terminus expected to extend to the cytosol. Domain mapping experiments indicated that a fragment within the N-terminal cytoplasmic domain of TMEM214 (amino acids 176–354) is required for its interaction with procaspase 4, which does not have a recognizable transmembrane domain. Interestingly, deletion of either of the two transmembrane domains of TMEM214 did not impair its ER localization and ability to induce apoptosis. However, deletion of either the N-terminal procaspase 4-binding region or the C-terminal two-transmembrane domains of TMEM214 abolished its ability to induce apoptosis. Taken together, these results suggest that proper localization of TMEM214 at the outer membrane of the ER and its interaction with procaspase 4 are essential for its ability to mediate ER stress-induced apoptosis.
In our experiments, we found that knockdown of TMEM214 and procaspase 4 did not fully abolish ER stress-induced apoptosis. This is explained by our observations that TMEM214-mediated apoptosis is independent of CHOP induction and JNK phosphorylation. In light of these observations, it has been demonstrated previously that ER stress-induced apoptosis can be independently mediated by CHOP induction, JNK phosphorylation, and caspase activation (4–6).
The major conclusion of this manuscript is that TMEM214 mediates one of the ER stress-induced apoptotic pathways. This study does not exclude the possibility that TMEM214 also plays roles in other cellular functions. Because the TMEM214 protein level is not markedly changed during ER stress, other mechanisms may account for its activation during ER stress-induced apoptosis. For example, ER stress may lead to conformational changes, oligomerization, or posttranslational modifications of TMEM214 and its subsequent activation. No matter what the mechanism is, our study establishes TMEM214 as an ER protein that can mediate ER stress-induced apoptosis. TMEM214 acts by anchoring procaspase 4 to the ER and mediating caspase 4 activation. Our study provides important insights into the molecular mechanisms of ER stress-induced apoptosis.
Acknowledgments
We thank Dr. Bo Zhong for critical reading of the manuscript.
This work was supported by Chinese Ministry of Science and Technology Grant 2012CB910201, by National Science Foundation of China Grants 91029302, 31130020, and 31221061, and by Chinese 111 Project Grant B06018.

This article contains supplemental Figs. S1–S5.
- CHOP
- CCAAT/enhancer-binding protein homologous protein
- TG
- thapsigargin
- BFA
- brefeldin A.
REFERENCES
- 1. Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 2. Walter P., Ron D. (2011) The unfolded protein response. From stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 3. Tabas I., Ron D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 5. Szegezdi E., Logue S. E., Gorman A. M., Samali A. (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li B., Yi P., Zhang B., Xu C., Liu Q., Pi Z., Xu X., Chevet E., Liu J. (2011) Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell. Signal. 23, 35–45 [DOI] [PubMed] [Google Scholar]
- 7. Moon D. O., Park S. Y., Choi Y. H., Ahn J. S., Kim G. Y. (2011) Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5. Involvement of ROS-dependent ER-stress. Biochem. Pharmacol. 82, 1641–1650 [DOI] [PubMed] [Google Scholar]
- 8. Yamaguchi H., Wang H. G. (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 [DOI] [PubMed] [Google Scholar]
- 9. Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 10. Ohoka N., Yoshii S., Hattori T., Onozaki K., Hayashi H. (2005) TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santos C. X., Tanaka L. Y., Wosniak J., Laurindo F. R. (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response. Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 11, 2409–2427 [DOI] [PubMed] [Google Scholar]
- 12. Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H. (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marani M., Tenev T., Hancock D., Downward J., Lemoine N. R. (2002) Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol. Cell. Biol. 22, 3577–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakagawa T., Yuan J. (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150, 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B. A., Yuan J. (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103 [DOI] [PubMed] [Google Scholar]
- 16. Fischer H., Koenig U., Eckhart L., Tschachler E. (2002) Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun. 293, 722–726 [DOI] [PubMed] [Google Scholar]
- 17. Hitomi J., Katayama T., Eguchi Y., Kudo T., Taniguchi M., Koyama Y., Manabe T., Yamagishi S., Bando Y., Imaizumi K., Tsujimoto Y., Tohyama M. (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 165, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bian Z. M., Elner S. G., Elner V. M. (2009) Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 50, 6006–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lehner B., Sanderson C. M. (2004) A protein interaction framework for human mRNA degradation. Genome Res. 14, 1315–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han K. J., Su X., Xu L. G., Bin L. H., Zhang J., Shu H. B. (2004) Mechanisms of the TRIF-induced interferon-stimulated response element and NF-κB activation and apoptosis pathways. J. Biol. Chem. 279, 15652–15661 [DOI] [PubMed] [Google Scholar]
- 21. Yamamuro A., Kishino T., Ohshima Y., Yoshioka Y., Kimura T., Kasai A., Maeda S. (2011) Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J. Pharmacol. Sci. 115, 239–243 [DOI] [PubMed] [Google Scholar]
- 22. Kamada S., Funahashi Y., Tsujimoto Y. (1997) Caspase-4 and caspase-5, members of the ICE/CED-3 family of cysteine proteases, are CrmA-inhibitable proteases. Cell Death Differ. 4, 473–478 [DOI] [PubMed] [Google Scholar]
- 23. Zhou Q., Snipas S., Orth K., Muzio M., Dixit V. M., Salvesen G. S. (1997) Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 272, 7797–7800 [DOI] [PubMed] [Google Scholar]
- 24. Reddy R. K., Mao C., Baumeister P., Austin R. C., Kaufman R. J., Lee A. S. (2003) Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors. Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 278, 20915–20924 [DOI] [PubMed] [Google Scholar]