

Background: Cellular iron efflux occurs via cooperation of ferroportin and a ferroxidase.
Results: Depletion of ferroxidase and ferroportin from human brain microvascular endothelial cells decreases their ability to efflux iron.
Conclusion: Iron efflux from brain microvascular endothelial cell ferroportin requires exocytoplasmic ferroxidase activity.
Significance: The mechanism of iron efflux from endothelial cells of the blood-brain barrier is fundamental to how iron enters the brain.
Keywords: Brain Metabolism, Cell Biology, Iron Metabolism, Neurochemistry, Transport Metals, Blood Brain Barrier, Ceruloplasmin, Ferroportin, Ferroxidase, Hephaestin
Abstract
The mechanism(s) of iron flux across the brain microvasculature endothelial cells (BMVEC) of the blood-brain barrier remains unknown. Although both hephaestin (Hp) and the ferrous iron permease ferroportin (Fpn) have been identified in BMVEC, their roles in iron efflux have not been examined. Using a human BMVEC line (hBMVEC), we have demonstrated that these proteins are required for iron efflux from these cells. Expression of both Hp and Fpn protein was confirmed in hBMVEC by immunoblot and indirect immunofluorescence; we show that hBMVEC express soluble ceruloplasmin (Cp) transcript as well. Depletion of endogenous Hp and Cp via copper chelation leads to the reduction of hBMVEC Fpn protein levels as well as a complete inhibition of 59Fe efflux. Both hBMVEC Fpn protein and 59Fe efflux activity are restored upon incubation with 6.6 nm soluble plasma Cp. These results are independent of the source of cell iron, whether delivered as transferrin- or non-transferrin-bound 59Fe. Our results demonstrate that iron efflux from hBMVEC Fpn requires the action of an exocytoplasmic ferroxidase, which can be either endogenous Hp or extracellular Cp.
Introduction
Several neuronal processes require iron such as myelination, mitochondrial energy generation, cell division, and neurotransmission (1, 2). When not properly regulated, iron can readily form cell-damaging reactive oxygen species via Fenton chemistry (Equation 1) and thus must be tightly regulated by both cellular and extracellular mechanisms.
To prevent such free radical formation in blood or the interstitial spaces of the brain, mammals have developed ways to maintain iron in its less reactive FeIII form. To accomplish this, coordination between several iron-related proteins is required to safely export FeIII from cells. Cellular iron efflux occurs through the FeII transporter ferroportin (Fpn)2 (3, 4); Fpn has been identified in the brain microvascular endothelial cells (BMVEC) of the blood-brain barrier (BBB) (5, 6). Upon cellular iron efflux through Fpn, FeII is oxidized by a multicopper oxidase; two such multicopper oxidases exist in mammals, hephaestin (Hp) and ceruloplasmin (Cp) (7–9). Hephaestin is found co-localized and in functional complex with Fpn on the surface of mammalian epithelial cells lining the gut (8, 10, 11). The ferroxidase-generated FeIII becomes bound by a variety of iron-chelators and proteins found in blood, e.g. transferrin (Tf), ferritin, and citrate; this same cohort of ferric iron ligands are found in the brain (12). In the brains of sex-linked anemic mice (sla−/−), which contain mutations in the Hp gene, iron accumulation has been shown in oligodendrocytes of the gray matter, suggesting a role for Hp in iron efflux from cells of the CNS (9, 13). Although these mice still accumulate brain iron, they likely do so via soluble Cp (sCp) present in the interstitial fluid localized to the abluminal surface of the BMVEC. This sCp can be provided by astrocytes (14) or, as we demonstrate here, by hBMVEC. Hp but not Cp has been reported previously in the BMVEC of the BBB (6, 15, 16).
Brain iron accumulation has been correlated with several neurological diseases, including Alzheimer and Parkinson disease (17, 18). Understanding the mechanism of iron transport across the endothelial cells of the BBB is crucial in developing an understanding of the pathogenesis of these neurological diseases. Currently, the mechanism of iron transport across the endothelial cells of the BBB is poorly understood. In one model, serum Tf-iron is transcytosed across the BMVEC followed by release of Tf-iron or iron alone at the abluminal membrane (18, 19). This model, which has not been experimentally verified, does not account for the uptake by these cells of non-transferrin-bound iron (20). Our goal in this work was to delineate the mechanism of iron efflux from BMVEC; we specifically compared efflux of iron obtained from either Tf-iron (TBI or non-Tf-bound iron (NTBI).
We demonstrate that Fpn and Hp proteins are expressed by human BMVEC (hBMVEC). Furthermore, we report here the presence of the soluble Cp transcript. This is the first example of the existence of the Cp message in the endothelial cells of the BBB. Depletion of endogenous multi-copper oxidase proteins via treatment with the copper chelating agent bathrocuproinedisulfonic acid disodium salt (BCS) demonstrated that hBMVEC iron efflux is dependent upon endogenous Fpn and exocytoplasmic ferroxidase activity. Our results indicate that endogenous Hp and/or sCp can fulfill the role of ferroxidase in this iron efflux pathway. The positive correlation between hBMVEC ferroxidase expression and iron efflux is quantitatively the same irrespective of cell iron loading by TBI or NTBI suggesting that Fpn-dependent efflux predominates in this monolayer system.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
hBMVEC were cultured in RPMI 1640 with 10% FBS and 10% NuSerum as described previously (20). Experiments were performed in 33-mm tissue culture dishes or 24-well tissue culture dishes unless otherwise specified. Human soluble plasma ceruloplasmin was purchased from GenWay Biotech, Inc. (San Diego, CA).
Immunoblots
Immunoblots were performed as described previously with minor changes (20). Briefly, hBMVEC were harvested with radioimmune precipitation assay buffer plus protease inhibitors. SDS-PAGE (8%) was used to separate 20 μg of protein per lane. The membrane was blocked 1 h with PBS 3% BSA, incubated with mouse anti-hephaestin (ab56729) or rabbit anti-SLC40A1 (ferroportin) (ab78066) (Abcam, Inc., Cambridge, MA) overnight at 4 °C and then incubated with secondary anti-HRP antibody (Santa Cruz Biotechnology). Blots were adjusted for brightness and contrast using Adobe Photoshop software (version 7.0).
Indirect Immunofluorescence
hBMVEC were fixed for 10 min with 3.7% paraformaldehyde and 4% sucrose in PBS containing 1 mm CaCl2 and 0.5 mm MgCl2. hBMVEC were then blocked and permeabilized for 1 h in PBS 0.1% Tween 20 with 1% BSA and 0.3 m glycine. Cells were then incubated for 30 min with anti-hephaestin (1:100) or anti-SLC40A1 (1:100) in PBS 1% BSA and then incubated for 30 min with Alexa Fluor 488-conjugated donkey anti-mouse or goat anti-rabbit (1:1000, Invitrogen) in PBS 1% BSA. Coverslips were mounted onto glass slides using SlowFade gold antifade reagent with DAPI (Invitrogen). Images from at least five different fields of view were obtained using a Zeiss AxioImager Z1 Axiophot wide-field fluorescence microscope and Zeiss AxioVision software (Zeiss, Thornwood, NY). Analyses of relevant fluorescent intensities were performed using Zeiss AxioVision software and Prism (version 4.0, GraphPad Software). All fluorescent intensities were normalized for their respective secondary antibody control fluorescent intensities.
59Fe Accumulation and Efflux Assays
All 59Fe uptake and efflux assays were performed using confluent monolayers of hBMVEC grown in 24-well tissue culture dishes. Assays were performed at 37 °C 5% CO2 at 75 rpm. The radionuclide 59FeCl3 was purchased from Perkin-Elmer Life Sciences. HBMVEC were loaded with 59FeII citrate using a reductase-independent uptake protocol based on the presence of 5 mm dihydroascorbate and 250 μm citrate. Reactions were quenched with ice-cold quench buffer as described previously (20) and lysed with radioimmune precipitation assay buffer. Lysates were assayed for 59Fe and protein concentration. For efflux assays, hBMVEC were loaded for 24 h, washed with RPMI 1640 containing 250 μm citrate, and incubated with reagents as noted in each experiment for 0–24 h. The medium was collected, and the cells were quenched and processed as described above.
RT-PCR
Total RNA was extracted from hBMVEC, C6 glioma cells, or HepG2 cells using the TRIzol reagent (Invitrogen) as per the manufacturer's instructions. After DNase treatment, RNA was reverse-transcribed, and PCR was performed using the Qiagen OneStep RT-PCR kit. Primers used for RT-PCR are listed in Table 1.
TABLE 1.
Primer list used for RT-PCR
| Transcript | Forward primer | Reverse primer |
|---|---|---|
| Hephaestin | GGATGCATGCAATCAATGG | CATCTTGAAAGGCTTCATATCG |
| Ferroportin | CAGGGACTGAGTGGTTCCAT | GGAGATTATGGGGACGGATT |
| Secreted (soluble) ceruloplasmin | TCCCTGGAACATACCAAACC | CCAATTTATTTCATTCAGCCGA |
Ferroxidase Assays
hBMVEC were lysed with 0.2% Triton X-100 for 1 h at 37 °C 5% CO2 at 75 rpm. Lysates were concentrated, and 300 μg total protein was used per condition unless indicated otherwise. Assays were performed in 100 mm MES buffer, pH 6, 10 μm freshly prepared ferrous ammonium sulfate for 1 h at room temperature. Ferrozine (100 μm) was used as the colorimetric indicator producing an absorbance at 550 nm when in complex with ferrous iron. All assays were blanked for iron-only controls.
Other Procedures
Apo-Tf (Sigma-Aldrich) was loaded with 59Fe as described previously (20). Efflux of 59Fe resulting from 59Fe-Tf uptake were performed as described for 59FeII citrate. Copper chelation was performed with 500 μm BCS (Sigma-Aldrich) for 24 to 48 h. All statistical analyses were performed by using Prism software (version 4.0). All counts were normalized for protein concentration obtained at time 0 h.
RESULTS
hBMVEC Efflux Iron Accumulated from59FeII citrate (NTBI) or 59Fe-transferrin (TBI)
We have demonstrated previously that hBMVEC readily import 59FeII citrate (20). Prior to investigating the mechanism of hBMVEC iron efflux, we monitored the accumulation of 59FeII citrate in an effort to establish an end point. The accumulation of 59FeII citrate by hBMVEC appears to plateau after 20 min (Fig. 1A, inset). However, data collected for up to 24 h indicate that the accumulation of 59FeII citrate by hBMVEC is biphasic in nature (Fig. 1A). As the end point for hBMVEC iron accumulation was reached by 24 h, we used this time frame for hBMVEC iron loading in our subsequent experiments.
FIGURE 1.

hBMVEC, which express Hp, sCp, and Fpn, readily accumulate and efflux iron. A, hBMVEC accumulation of 0.5 μm 59FeII citrate plus ascorbate over a 24-h period. Inset illustrates the first 1 h of accumulation. Time 0 h is subtracted from all data points (n = 6, experimental replicates). B, 59Fe efflux assays were performed using hBMVEC cell pellets loaded for 24 h with 59FeII citrate plus ascorbate (open) or 59Fe-Tf (filled). hBMVEC pellets were harvested after allowing 24 h for 59Fe efflux. Data are represented as the percent loss of cell-associated 59Fe with respect to time 0 h. Data are represented as means ± S.D. (n = 4–8, experimental replicates). C, immunoblots from whole cell lysates of C6 glioma, HepG2, Caco-2 cells, and hBMVEC were probed for either Hp or Fpn. Respective molecular weights are indicated to the right of the blots. Each lane was loaded with 20 μg of total protein. D, total RNA was collected from hBMVEC cells, and RT-PCR was performed to amplify Hp, Fpn, and Cp transcripts. Isoform-specific primers were used to amplify human soluble Cp from hBMVEC total RNA.
To establish an end point for iron efflux from hBMVEC, cells were 59Fe-loaded with 59Fe-citrate (NTBI), washed, and incubated with efflux media for 0, 24, or 48 h. hBMVEC exported 59Fe over time, reaching an apparent end point during the first 24 h of the assay (data not shown). hBMVEC lost 35.4 ± 3.2% of their cell-associated 59Fe after 24 h (Fig. 1B, open bar). Efflux assays were performed also after loading hBMVEC with 59Fe-Tf (TBI). A similar loss of cell-associated 59Fe after 24 h was observed (34.9 ± 0.2%) (Fig. 1B, filled bar). These data show that hBMVEC accumulate and efflux 59Fe presented in both transferrin-bound and non-transferrin-bound forms.
Hp, Fpn, and sCp Are Expressed by hBMVEC
The mechanism of cellular iron efflux from intestinal (21) and lens epithelial cells (22), macrophages (23), and oligodendrocytes (13) involves the functional cooperation of Fpn and an exocytoplasmic ferroxidase, either Hp or Cp. We propose that the same mechanism of iron efflux is true for hBMVEC. To test this, we first used Western blot to demonstrate expression of both Hp and Fpn as well as to establish the specificity of the antibodies, which we used throughout. The Caco-2 intestinal epithelial cell line was used as a positive control in our immunoblots. Caco-2 cells express both Hp and Fpn and have been established as a model for iron transport across the intestinal barrier (21, 24, 25). A distinct band at ∼130 kDa is observed on immunoblots of Caco-2 and hBMVEC extracts probed for Hp; two distinct bands are observed at ∼63 and 70 kDa when probed for Fpn (Fig. 1C). HepG2 cells were used as a negative control for Fpn antibody specificity in our immunoblots. HepG2 cells express minimal Fpn (26). As a negative control for Hp antibody, C6 glioma cells were used (Fig. 1C). Transcripts for both Hp and Fpn were also identified in hBMVEC via RT-PCR (Fig. 1D). Two isoforms of Cp have been identified in mammals, glycophosphatidylinositol-anchored Cp (GPI-Cp) and sCp. Further examination of the hBMVEC Cp message revealed that the transcript encoding sCp is expressed in this cell type (Fig. 1D). Primers used for RT-PCR are given in Table 1.
Hp and Fpn Are Co-localized to the Plasma Membrane of hBMVEC
We utilized indirect immunofluorescence with confocal microscopy to demonstrate subcellular localization of Hp and Fpn in hBMVEC monolayers. Both Fpn and Hp appear to be localized to the plasma membrane of hBMVEC; however, significant cytosolic staining of Fpn was observed in these cells as well (Fig. 2A). Merged images of Hp and Fpn demonstrated detectable co-localization of the two antigens in hBMVEC with this co-localization principally confined to the plasma membrane (Fig. 2A). High resolution images demonstrated that Hp presented with strict basolateral localization in the hBMVEC monolayers; in contrast, Fpn was visualized on both the cell surface and in the cytosol (Fig. 2B). Given the apparent diffuse Fpn localization in hBMVEC (Fig. 2B), we utilized HepG2 cells as a negative control for these indirect immunofluorescence studies. Indeed, compared with the HepG2 control, the Fpn antibody specifically binds antigen on the hBMVEC plasma membrane (Fig. 2C).
FIGURE 2.

Hp and Fpn are co-localized to the cell surface of hBMVEC. A, hBMVEC monolayers were grown on glass coverslips and were processed for indirect immunofluorescence by confocal microscopy probing for Hp and Fpn. DAPI (blue) stains the nucleus. Circles highlight areas of Hp and Fpn co-localization in the merged image of hBMVEC. B, confocal microscopy performed on hBMVEC probed for Hp or Fpn was used to delineate polarized localization of these proteins. Side views demonstrate polarity in the case of Hp but not Fpn. Apical and basal surfaces are indicated to the right of the images. DAPI (blue) stains the nucleus. C, hBMVEC and HepG2 (negative control) monolayers were processed for indirect immunofluorescence probing for Fpn. DAPI (blue) stains the nucleus.
Copper Chelation Depletes hBMVEC Hp Protein Abundance
To assess the contribution of Hp and/or sCp to hBMVEC iron efflux, we first stimulated the loss of ferroxidase in vitro by incubating hBMVEC with the copper chelating agent BCS (500 μm). Cellular copper depletion causes the loss of endogenous multicopper oxidase activity (8, 27). Comparison of indirect immunofluorescence images of BCS-treated to those of control cells indicated that the overall abundance of Hp in BCS-treated hBMVEC was decreased (Fig. 3A). To quantify the effect of BCS treatment on hBMVEC Hp expression, indirect immunofluorescence images taken from five separate fields of view were analyzed for their average fluorescent intensity. This analysis demonstrated that BCS-treated hBMVEC displayed decreased average fluorescence intensity when probed for Hp (Fig. 3B). As shown, there was a partial recovery of Hp after 48 h of BCS treatment. This pattern is addressed under the “Discussion.”
FIGURE 3.

Copper chelation depletes hBMVEC of endogenous Hp. A, indirect immunofluorescence was used to investigate the abundance of Hp protein in BCS-treated and untreated hBMVEC. hBMVEC were treated with 500 μm BCS for 24–48 h. Secondary antibody only controls are shown. DAPI (blue) stains the nucleus. Scale bar represents 10 μm. B, average (Avg) fluorescent intensity from at least five separate fields of view were taken from each condition at each time point and were normalized for their respective secondary only control average fluorescent intensities. A series of paired t tests were used to analyze the data from each time point. *, p < 0.05 and **, p < 0.005. Data are represented as means ± S.D. (n = 5, technical replicates).
hBMVEC Fpn Expression Is Decreased upon Loss of Endogenous Ferroxidase via BCS Treatment
In the absence of ferroxidase activity, e.g. with BCS treatment, Fpn is lost from the plasma membrane of cells (8, 27). Thus, we hypothesized that Fpn expression also would be diminished upon BCS treatment. BCS-treated and -untreated hBMVEC were probed for Fpn and analyzed via indirect immunofluorescence. Qualitative assessment of the images as above revealed a decrease in overall Fpn abundance in BCS-treated hBMVEC (Fig. 4A); quantitatively, the average fluorescent intensity of hBMVEC Fpn was significantly diminished as well (Fig. 4D). As observed for Hp, Fpn partially recovered 48h after treatment (Fig. 4D). The decrease in hBMVEC Fpn abundance upon treatment with BCS was confirmed by Western blot (Fig. 4B). In using BCS, of course, we cannot exclude the possibility that the effects on Fpn abundance are, to some extent, unrelated to the loss of cell Cp and Hp.
FIGURE 4.

Exocytoplasmic ferroxidase activity stabilizes hBMVEC Fpn but not Hp abundance in the presence of BCS. A, indirect immunofluorescence was used to investigate the abundance of Fpn protein in BCS-treated and untreated hBMVEC. hBMVEC were treated with 500 μm BCS for 24–48 h. Additionally, recovery of Fpn protein abundance in BCS-treated hBMVEC by ferroxidase-active sCp (6.6 nm) was investigated. Fresh sCp was added to media during the final 24 h of BCS treatment. Insets depict secondary antibody only controls. DAPI (blue) stains the nucleus. Scale bar represents 10 μm. B, immunoblot depicting Fpn abundance in hBMVEC lysates (20 μg of protein per lane). hBMVEC were incubated for 24 h without BCS, with BCS, or with BCS plus sCp (6.6 nm) for an additional 24 h before lysis. C, ferroxidase assays were used to investigate the abundance of endogenous ferroxidase activity in hBMVEC lysates. hBMVEC were treated for 24 h without BCS, with BCS, or with BCS plus sCp (6.6 nm) for an additional 24 h. Ferroxidase activity is indicated by the fraction of FeII remaining as detected by formation of the Fe(II)-ferrozine complex (n = 3, experimental replicates). D and E, samples from A were used to quantify average fluorescent intensities from at least five separate fields of view from each condition at each time point and were normalized for their respective secondary only control average fluorescent intensities. Significance of the data (C) was assessed using one-way analysis of variance parameters. A series of paired t tests were used to analyze the data from each time point in D and E. *, p < 0.005; **, p < 0.001; ***, p < 0.0001. Data are represented as means ± S.D. (A, D, and E, n = 5, technical replicates).
Addition of 6.6 nm soluble human plasma Cp (sCp) in the presence of BCS stabilized Fpn protein abundance in hBMVEC (Figs. 4A and 4E); this positive effect on Fpn due to added sCp was confirmed by Western blot (Fig. 4B). This concentration of sCp has been used previously to effectively stimulate iron efflux from cells depleted of their endogenous ferroxidase activity (7). In these experiments, sCp was added 24 h after BCS treatment was initiated to limit a possible thermal loss of sCp activity due to an extended incubation at 37 °C (28). The endogenous ferroxidase activity that was present in hBMVEC lysates was lost when hBMVEC were treated with BCS or BCS plus sCp for 24 h (Fig. 4C). These data suggest that the positive effect on Fpn due to sCp addition was correlated with the added sCp and not to a sCp-induced recovery of hBMVEC Hp.
Iron Efflux from hBMVEC Fpn Requires the Action of an Exocytoplasmic Ferroxidase
We hypothesized that depletion of Hp and sCp from hBMVEC due to BCS treatment would result in a reduced capacity for iron efflux. BCS has been used previously to knock down ferroxidase-catalyzed cellular iron mobilization (8). hBMVEC treated with 500 μm BCS and 0.5 μm 59FeII citrate plus ascorbate retained more 59Fe over 24 h than did BCS-untreated hBMVEC (Fig. 5A). Increased retention of 59Fe in BCS-treated hBMVEC as compared with untreated hBMVEC was also observed when 59Fe-Tf was used as substrate for loading (Fig. 5B). The increased cellular retention of 59Fe in the presence of BCS could be due to either increased 59Fe uptake or decreased 59Fe efflux.
FIGURE 5.

Copper chelation increases hBMVEC iron retention. A, hBMVEC-associated 59Fe after loading for 24 h with 0.5 μm 59FeII citrate plus ascorbate in the presence or absence of 500 μm BCS. B, hBMVEC-associated 59Fe after loading for 24 h with 59Fe-Tf plus citrate in the presence or absence of 500 μm BCS. A series of paired t tests were used to analyze the data. *, p < 0.05 and **, p < 0.005. Data are represented as means ± S.D. (n = 4–8, experimental replicates).
To delineate between these two potential mechanisms of hBMVEC 59Fe retention, we utilized 59Fe efflux assays along with BCS treatment. Treatment of hBMVEC with BCS resulted in the complete inhibition of 59Fe efflux (Fig. 6A). Soluble Cp partially restored 59Fe efflux from BCS-treated hBMVEC (Fig. 6A). BCS-treated hBMVEC loaded with 59Fe-Tf exhibited a similar pattern of 59Fe efflux; efflux was knocked down by BCS treatment with sCp supporting a 78% recovery (Fig. 6B). We infer from these data that BCS reduced the rate of 59Fe efflux from hBMVEC rather than inhibiting iron uptake. Taken together, these data suggest that 59Fe efflux from hBMVEC Fpn is potentiated by an exocytoplasmic ferroxidase regardless of its association with the plasma membrane, an effect likely associated with the restoration/stabilization of Fpn in the membrane. This pattern was the same whether TBI or NTBI was the 59Fe source used to load the cells.
FIGURE 6.
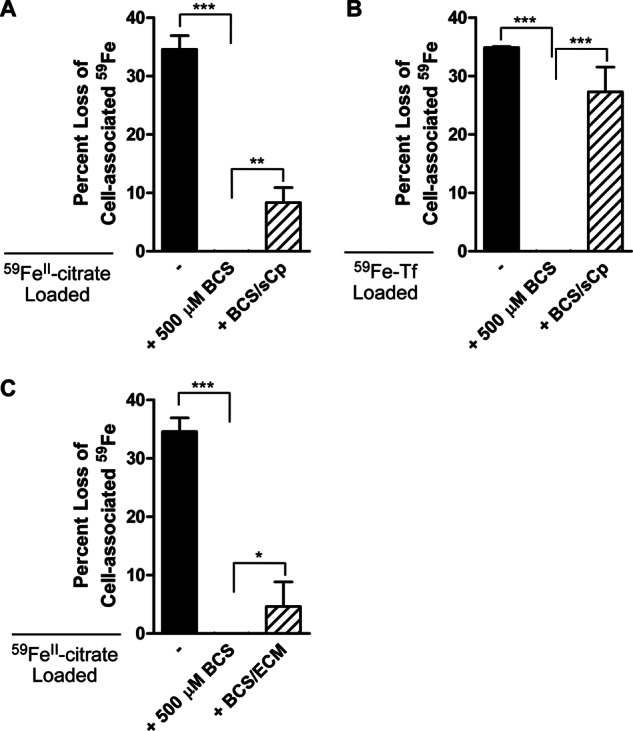
hBMVEC iron efflux inhibited by BCS treatment can be restored via the addition of exogenous sCp or hBMVEC-conditioned media. A, 59Fe efflux assays were performed on hBMVEC loaded with 59FeII citrate plus ascorbate. 59Fe efflux assays were also performed using BCS-treated hBMVEC in the presence or absence of ferroxidase active sCp (6.6 nm) added at the beginning of the 24 h efflux assay. B, 59Fe efflux assays were performed on hBMVEC loaded with 59Fe-Tf plus citrate. 59Fe efflux assays were also performed using BCS-treated hBMVEC in the presence or absence of ferroxidase active sCp (6.6 nm) added at the beginning of the 24 h efflux assay. C, 59Fe efflux assays were performed on hBMVEC loaded with 59FeII citrate plus ascorbate. Efflux activity was partially recovered via the addition of hBMVEC-conditioned media (ECM) to BCS-treated hBMVEC. All BCS values were not statistically different than zero. Significance was determined for each set of data using one-way analysis of variance analysis. *, p < 0.05; **, p < 0.01; and **, p < 0.001. Data are represented as means ± S.D. (n = 4–8, experimental replicates).
We next assessed the possible contribution of sCp secreted by hBMVEC to the stimulation of Fpn-dependent 59Fe efflux. To do this, we applied fresh medium to a hBMVEC monolayer and incubated for 24 h to condition the media with the components of the hBMVEC secretome, including sCp. We collected the hBMVEC-conditioned media and applied it to BCS-treated hBMVEC, which had been loaded with 59FeII citrate for 24 h. Efflux was monitored over 24 h in the continued presence of BCS. Indeed, although sCp restored efflux by 24% (Fig. 6A) hBMVEC-conditioned media also had a positive effect on iron efflux (13% recovery, Fig. 6C). These results suggest the likelihood that hBMVEC sCp does contribute to overall Fpn-dependent iron efflux from these cells.
DISCUSSION
In this work, we focused on the roles of Fpn and Hp in iron efflux from hBMVEC. Our first finding was that quantitatively, efflux and its dependence on a ferroxidase and Fpn are independent of the iron substrate (59Fe-citrate or 59Fe-Tf) used for hBMVEC 59Fe loading. The data indicate that the 59Fe obtained from these two substrates appears to follow the same efflux pathway in this model system. The most significant result shown is that efflux from BCS-treated cells loaded with 59Fe-Tf exhibit no iron efflux. Thus, at least in the experimental paradigm described here, we found no evidence for detectable 59Fe-Tf recycling.
Iron efflux in mammals generally occurs via the cooperation of Fpn and a multicopper oxidase. Both Fpn and the ferroxidase Hp have been identified at the transcript and protein level in BMVEC (5, 6). We confirmed these observations in our hBMVEC model system (Figs. 1 and 2). Furthermore, we demonstrated that Hp and Fpn are colocalized to the plasma membrane of hBMVEC, an observation that correlates with the bulk of the literature regarding Hp and Fpn subcellular localization (11).
Cooperation of Fpn and Hp in supporting iron export from cells endogenous to the CNS has been demonstrated previously (13, 29). Here, we have presented evidence that Fpn and Hp cooperate in the process by which iron is released from the barrier cells of the brain, the BMVEC; note that the polarity of this efflux could not be determined in our monolayer system. We have investigated the roles of Hp, sCp, and Fpn in hBMVEC iron efflux using the copper-chelating agent BCS. Decreased intracellular copper levels results in an increase in the turnover of mature endogenous Hp due to an abrogation of copper incorporation into the newly synthesized protein (30). Examination of rats with perinatal copper deficiency revealed decreased plasma and brain iron concentrations as compared with copper sufficient controls, indicating a role for multicopper oxidases in systemic iron trafficking (31).
In the absence of a ferroxidase (via BCS treatment), there is a reported hepcidin-independent internalization of Fpn from the cell surface of both HeLa and C6 glioma cells (8, 27). In glioma cells, the ferroxidase lost is GPI-linked Cp. This effect has been paired with a loss in iron efflux from these cells (8). Here, we have shown Hp and Fpn depletion from BCS-treated hBMVEC via quantification of the relative fluorescent intensity of indirect immunofluorescent images (Figs. 3 and 4); a loss in BCS-treated hBMVEC 59Fe-efflux was also observed (Fig. 6).
Ferroportin mRNA contains a 5′-UTR iron-responsive element (32). Binding of the iron regulatory protein 1 to the iron-responsive element under low cytosolic iron conditions post-transcriptionally regulates the expression of Fpn by repressing the translation initiation of the transcript (33). Under conditions of high intracellular iron, as created by the inhibition of iron efflux activity via BCS treatment, the iron regulatory protein no longer associates with the Fpn iron-responsive element, allowing for translation initiation of the transcript. Although Fpn protein is turned over in the absence of a ferroxidase (8, 27), the increased translation initiation of the transcript may account for the subtle recovery of hBMVEC Fpn seen after 48 h of BCS treatment (Fig. 4). Increased turnover of Fpn in the absence of a ferroxidase does not mean that newly synthesized protein is not trafficked to the membrane; the increased turnover results from internalization due to increased ubiquitination (34).
The loss of Fpn protein in C6 glioma cells depleted of ferroxidase activity (GPI-Cp) can be reversed via the addition of exogenous ferroxidase activity (8). The most likely candidate ferroxidase in the blood and interstitial spaces of the brain is sCp. Soluble Cp in the blood is secreted primarily by hepatocytes (35). We have demonstrated that sCp transcript is present in hBMVEC (Fig. 1D). This is the first example of Cp message in the endothelial cells of the BBB; this result suggests that BMVEC may be a source of brain sCp.
Soluble Cp exists in concentrations ranging from 1.6–3.4 μm in the blood and 5.8–6.8 nm in the cerebrospinal fluid (12). A concentration of 6.6 nm sCp, the concentration we have used here, effectively stimulates iron efflux from cells depleted of their endogenous ferroxidase activity (7). At this concentration, sCp significantly increased the abundance of Fpn protein in BCS-treated hBMVEC (Fig. 4). The restoration of hBMVEC Fpn protein abundance in conjunction with the ferroxidase activity provided by sCp significantly increased the ability of BCS-treated hBMVEC to efflux 59Fe (Fig. 6); such activity was also provided by hBMVEC-secreted sCp (Fig. 6C). Our data suggest that Fpn and exocytoplasmic ferroxidase activity are required for 59Fe efflux from hBMVEC. This ferroxidase activity can be provided by either a cell endogenous (Hp) or exogenous (sCp) ferroxidase protein.
Soluble Cp more effectively stimulated iron efflux from hBMVEC loaded with iron-Tf than with iron citrate (Fig. 6). This difference may be due to the fact that more iron is accumulated and retained in hBMVEC when loaded with Tf-iron than iron citrate, thus creating an iron gradient within the cells (Fig. 5). This gradient provides a stronger driving force for efflux initiated upon the recovery of Fpn due to the addition of exogenous sCp.
Functionally, Cp protein in the CNS and in the peripheral circulation is similar; however, in the brain and a few other tissues, a post-translational modification yields a GPI anchor that attaches the protein to the extracellular surface of the cell in which it is expressed (36–38). In the CNS, GPI-Cp is highly concentrated to astrocytes (15, 36, 39) where an interaction with Fpn has been observed (7); the majority of these astrocytes neighbor BMVEC (39, 40). We demonstrated that sCp can stimulate hBMVEC 59Fe efflux (Fig. 6). Our data suggest the possibility that in the intact brain, astrocyte GPI-Cp or BMVEC sCp could support iron efflux through Fpn even in the absence of BMVEC Hp as seen in the sla−/− mouse in which iron accumulation is observed in oligodendrocytes (9, 13). This hypothesis is the subject of ongoing research.
In addition to suggesting a mechanism for iron efflux from hBMVEC, we have presented several interesting observations that contribute broadly to the field of brain iron accumulation. First, we have presented data that suggest the main iron substrate for hBMVEC is Tf-iron, which is retained by hBMVEC in nearly a 5-fold excess to non-Tf bound iron (Fig. 5). Second, our data strongly suggest that iron is released from Tf-iron, likely via the canonical transferrin receptor cycling pathway, before exiting the cell as “free” iron. Thus, our results are not easily reconciled with the proposition that a major fraction of brain iron uptake is supported by a holo-Tf transcytosis pathway through these endothelial cells (18, 19). Experiments in a Transwell model of the BBB will be required to specifically examine this transcytosis mechanism.
Acknowledgments
We thank Dr. Supriya Mahajan for generous donation of hBMVEC. We acknowledge the assistance of Dr. Wade Sigurdson at the Confocal Microscope and Flow Cytometry Facility in the School of Medicine and Biomedical Sciences (University at Buffalo).
This work was supported by Grant DK053820 from the National Institutes of Health (to D. J. K.) and a predoctoral fellowship from the American Heart Association (to R. C. M.).
- Fpn
- ferroportin
- BMVEC
- brain microvascular endothelial cell(s)
- Cp
- ceruloplasmin
- Hp
- hephaestin
- GPI-Cp
- glycophosphatidylinositol-anchored Cp
- hBMVEC
- human brain microvascular cell line
- BBB
- blood-brain barrier
- Tf
- transferrin
- sCp
- soluble plasma ceruloplasmin
- TBI
- transferrin-bound iron
- NTBI
- non-transferrin-bound iron
- BCS
- bathrocuproinedisulfonic acid disodium salt.
REFERENCES
- 1. Salvador G. A. (2010) Iron in neuronal function and dysfunction. BioFactors 36, 103–110 [DOI] [PubMed] [Google Scholar]
- 2. Todorich B., Pasquini J. M., Garcia C. I., Paez P. M., Connor J. R. (2009) Oligodendrocytes and myelination: The role of iron. Glia 57, 467–478 [DOI] [PubMed] [Google Scholar]
- 3. Ganz T. (2005) Cellular iron: ferroportin is the only way out. Cell Metab. 1, 155–157 [DOI] [PubMed] [Google Scholar]
- 4. Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., Paw B. H., Drejer A., Barut B., Zapata A., Law T. C., Brugnara C., Lux S. E., Pinkus G. S., Pinkus J. L., Kingsley P. D., Palis J., Fleming M. D., Andrews N. C., Zon L. I. (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 [DOI] [PubMed] [Google Scholar]
- 5. Wu L. J., Leenders A. G., Cooperman S., Meyron-Holtz E., Smith S., Land W., Tsai R. Y., Berger U. V., Sheng Z. H., Rouault T. A. (2004) Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. 1001, 108–117 [DOI] [PubMed] [Google Scholar]
- 6. Yang W., Jung K., Lee M., Lee Y., Nakagawa S., Niwa M., Cho S., Kim D. (2011) Transient expression of iron transport proteins in the capillary of the developing rat brain. Cell Mol. Neurobiol. 31, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeong S. Y., David S. (2003) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 278, 27144–27148 [DOI] [PubMed] [Google Scholar]
- 8. De Domenico I., Ward D. M., di Patti M. C., Jeong S. Y., David S., Musci G., Kaplan J. (2007) Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 26, 2823–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vulpe C. D., Kuo Y. M., Murphy T. L., Cowley L., Askwith C., Libina N., Gitschier J., Anderson G. J. (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 21, 195–199 [DOI] [PubMed] [Google Scholar]
- 10. De Domenico I., Ward D. M., Langelier C., Vaughn M. B., Nemeth E., Sundquist W. I., Ganz T., Musci G., Kaplan J. (2007) The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 18, 2569–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han O., Kim E. Y. (2007) Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J. Cell Biochem. 101, 1000–1010 [DOI] [PubMed] [Google Scholar]
- 12. Gaasch J. A., Lockman P. R., Geldenhuys W. J., Allen D. D., Van der Schyf C. J. (2007) Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 32, 1196–1208 [DOI] [PubMed] [Google Scholar]
- 13. Schulz K., Vulpe C. D., Harris L. Z., David S. (2011) Iron efflux from oligodendrocytes is differentially regulated in gray and white matter. J. Neurosci. 31, 13301–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Greco T. M., Seeholzer S. H., Mak A., Spruce L., Ischiropoulos H. (2010) Quantitative mass spectrometry-based proteomics reveals the dynamic range of primary mouse astrocyte protein secretion. J. Proteome Res. 9, 2764–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel B. N., David S. (1997) A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 272, 20185–20190 [DOI] [PubMed] [Google Scholar]
- 16. Qian Z. M., Chang Y. Z., Zhu L., Yang L., Du J. R., Ho K. P., Wang Q., Li L. Z., Wang C. Y., Ge X., Jing N. L., Li L., Ke Y. (2007) Development and iron-dependent expression of hephaestin in different brain regions of rats. J. Cell Biochem. 102, 1225–1233 [DOI] [PubMed] [Google Scholar]
- 17. Greenough M. A., Camakaris J., Bush A. I. (2013) Metal dyshomeostasis and oxidative stress in Alzheimer's disease. Neurochem. Int. 62, 540–555 [DOI] [PubMed] [Google Scholar]
- 18. Skjørringe T., Møller L. B., Moos T. (2012) Impairment of interrelated iron- and copper homeostatic mechanisms in brain contributes to the pathogenesis of neurodegenerative disorders. Front. Pharmacol. 3, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moos T., Rosengren Nielsen T., Skjørringe T., Morgan E. H. (2007) Iron trafficking inside the brain. J. Neurochem. 103, 1730–1740 [DOI] [PubMed] [Google Scholar]
- 20. McCarthy R. C., Kosman D. J. (2012) Mechanistic analysis of iron accumulation by endothelial cells of the BBB. Biometals 25, 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H., Attieh Z. K., Dang T., Huang G., van der Hee R. M., Vulpe C. (2009) Decreased hephaestin expression and activity leads to decreased iron efflux from differentiated Caco2 cells. J. Cell Biochem. 107, 803–808 [DOI] [PubMed] [Google Scholar]
- 22. Wolkow N., Song D., Song Y., Chu S., Hadziahmetovic M., Lee J. C., Iacovelli J., Grieco S., Dunaief J. L. (2012) Ferroxidase hephaestin's cell-autonomous role in the retinal pigment epithelium. Am. J. Pathol. 180, 1614–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson G. J., Vulpe C. D. (2009) Mammalian iron transport. Cell Mol. Life Sci. 66, 3241–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas C., Oates P. S. (2004) Ferroportin/IREG-1/MTP-1/SLC40A1 modulates the uptake of iron at the apical membrane of enterocytes. Gut. 53, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung B., Chaston T., Marks J., Srai S. K., Sharp P. A. (2009) Hepcidin decreases iron transporter expression in vivo in mouse duodenum and spleen and in vitro in THP-1 macrophages and intestinal Caco-2 cells. J. Nutr. 139, 1457–1462 [DOI] [PubMed] [Google Scholar]
- 26. Marques L., Auriac A., Willemetz A., Banha J., Silva B., Canonne-Hergaux F., Costa L. (2012) Immune cells and hepatocytes express glycosylphosphatidylinositol-anchored ceruloplasmin at their cell surface. Blood Cells Mol. Dis. 48, 110–120 [DOI] [PubMed] [Google Scholar]
- 27. Kono S., Yoshida K., Tomosugi N., Terada T., Hamaya Y., Kanaoka S., Miyajima H. (2010) Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 1802, 968–975 [DOI] [PubMed] [Google Scholar]
- 28. Sedlák E., Žoldák G., Wittung-Stafshede P. (2008) Role of copper in thermal stability of human ceruloplasmin. Biophys. J. 94, 1384–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J., Jiang H., Xie J. X. (2007) Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Eur. J. Neurosci. 25, 2766–2772 [DOI] [PubMed] [Google Scholar]
- 30. Nittis T., Gitlin J. D. (2004) Role of copper in the proteosome-mediated degradation of the multicopper oxidase hephaestin. J. Biol. Chem. 279, 25696–25702 [DOI] [PubMed] [Google Scholar]
- 31. Prohaska J. R., Gybina A. A. (2005) Rat brain iron concentration is lower following perinatal copper deficiency. J. Neurochem. 93, 698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abboud S., Haile D. J. (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 275, 19906–19912 [DOI] [PubMed] [Google Scholar]
- 33. Leipuviene R., Theil E. (2007) The family of iron responsive RNA structures regulated by changes in cellular iron and oxygen. Cell Mol. Life Sci. 64, 2945–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiao B., Sugianto P., Fung E., Del-Castillo-Rueda A., Moran-Jimenez M. J., Ganz T., Nemeth E. (2012) Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 15, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slany A., Haudek V. J., Zwickl H., Gundacker N. C., Grusch M., Weiss T. S., Seir K., Rodgarkia-Dara C., Hellerbrand C., Gerner C. (2010) Cell characterization by proteome profiling applied to primary hepatocytes and hepatocyte cell lines Hep-G2 and Hep-3B. J. Proteome Res. 9, 6–21 [DOI] [PubMed] [Google Scholar]
- 36. Patel B. N., Dunn R. J., David S. (2000) Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J. Biol. Chem. 275, 4305–4310 [DOI] [PubMed] [Google Scholar]
- 37. Rouault T. A., Cooperman S. (2006) Brain iron metabolism. Semin. Pediatr. Neurol. 13, 142–148 [DOI] [PubMed] [Google Scholar]
- 38. Dringen R., Bishop G. M., Koeppe M., Dang T. N., Robinson S. R. (2007) The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 32, 1884–1890 [DOI] [PubMed] [Google Scholar]
- 39. Klomp L. W., Farhangrazi Z. S., Dugan L. L., Gitlin J. D. (1996) Ceruloplasmin gene expression in the murine central nervous system. J. Clin. Invest. 98, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klomp L. W., Gitlin J. D. (1996) Expression of the ceruloplasmin gene in the human retina and brain: Implications for a pathogenic model in Aceruloplasminemia. Hum. Mol. Genet. 5, 1989–1996 [DOI] [PubMed] [Google Scholar]